
The Muscle-Brain Axis and Neurodegenerative Diseases: The Key Role of Mitochondria in Exercise-Induced Neuroprotection

 , , ,
, , ,  and
and 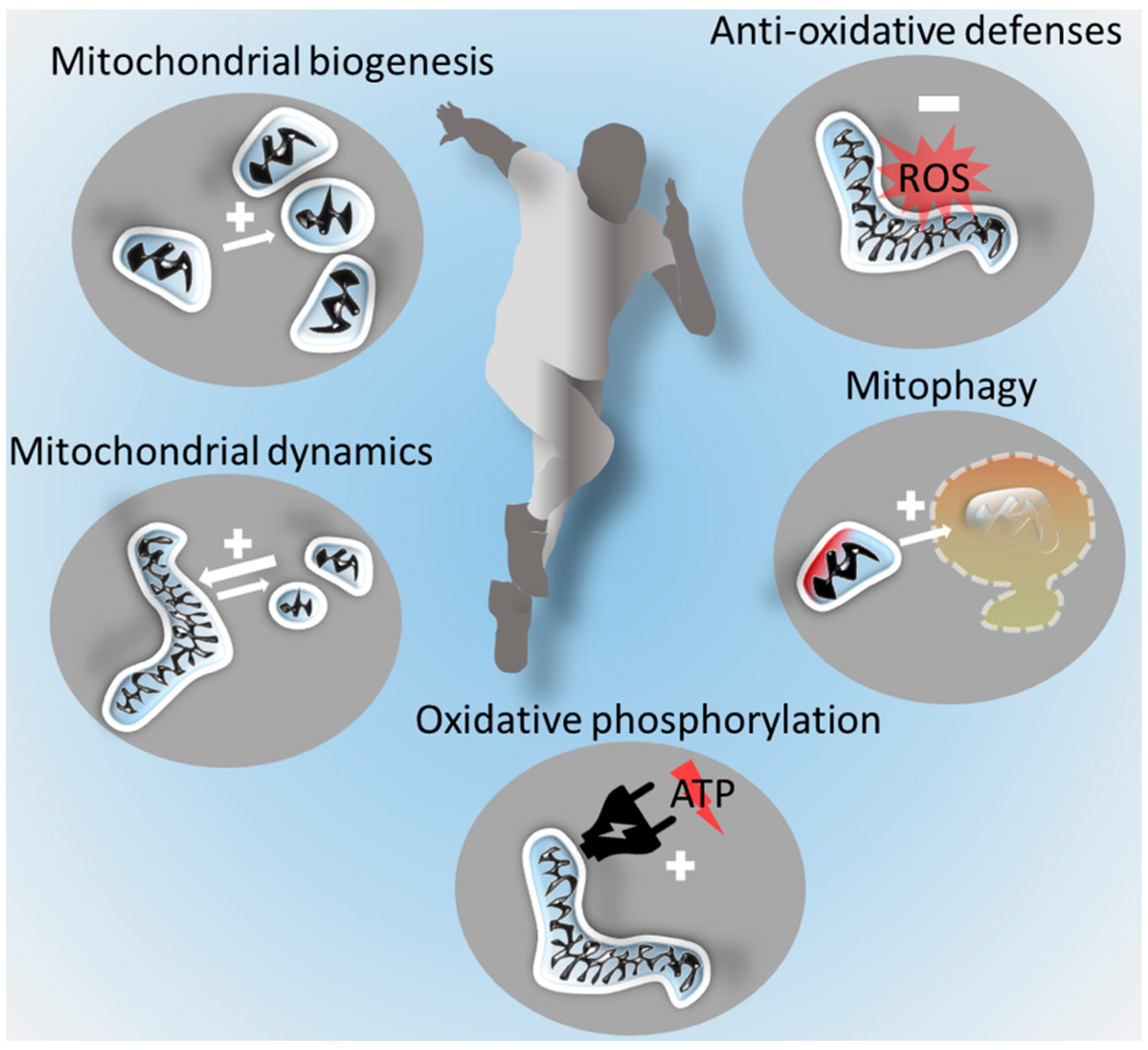{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mitochondrial Dysfunction in Neurodegenerative Diseases
2.1. Mitochondrial Respiration and Energy Provision
2.2. Mitochondrial ROS and Oxidative Stress
2.3. Mitochondrial Biogenesis
2.4. Mitochondrial Dynamics and Mitophagy
2.5. Mitochondrial Control of Cell Death and Survival
3. Improving Mitochondrial Functions by Exercise in Skeletal Muscle
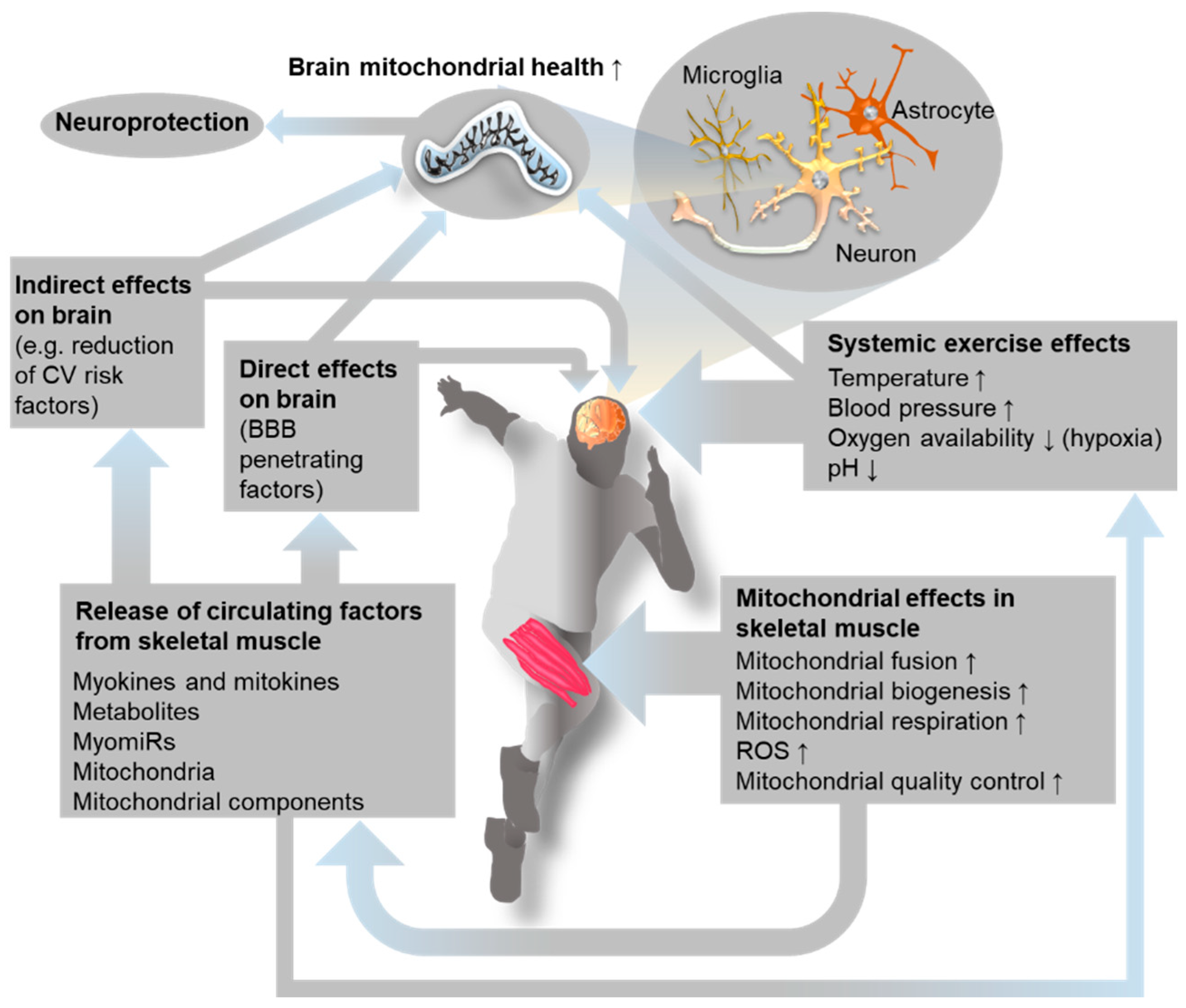
4. Exercise Effects on the Brain: A Focus on Mitochondria and Oxidative Stress
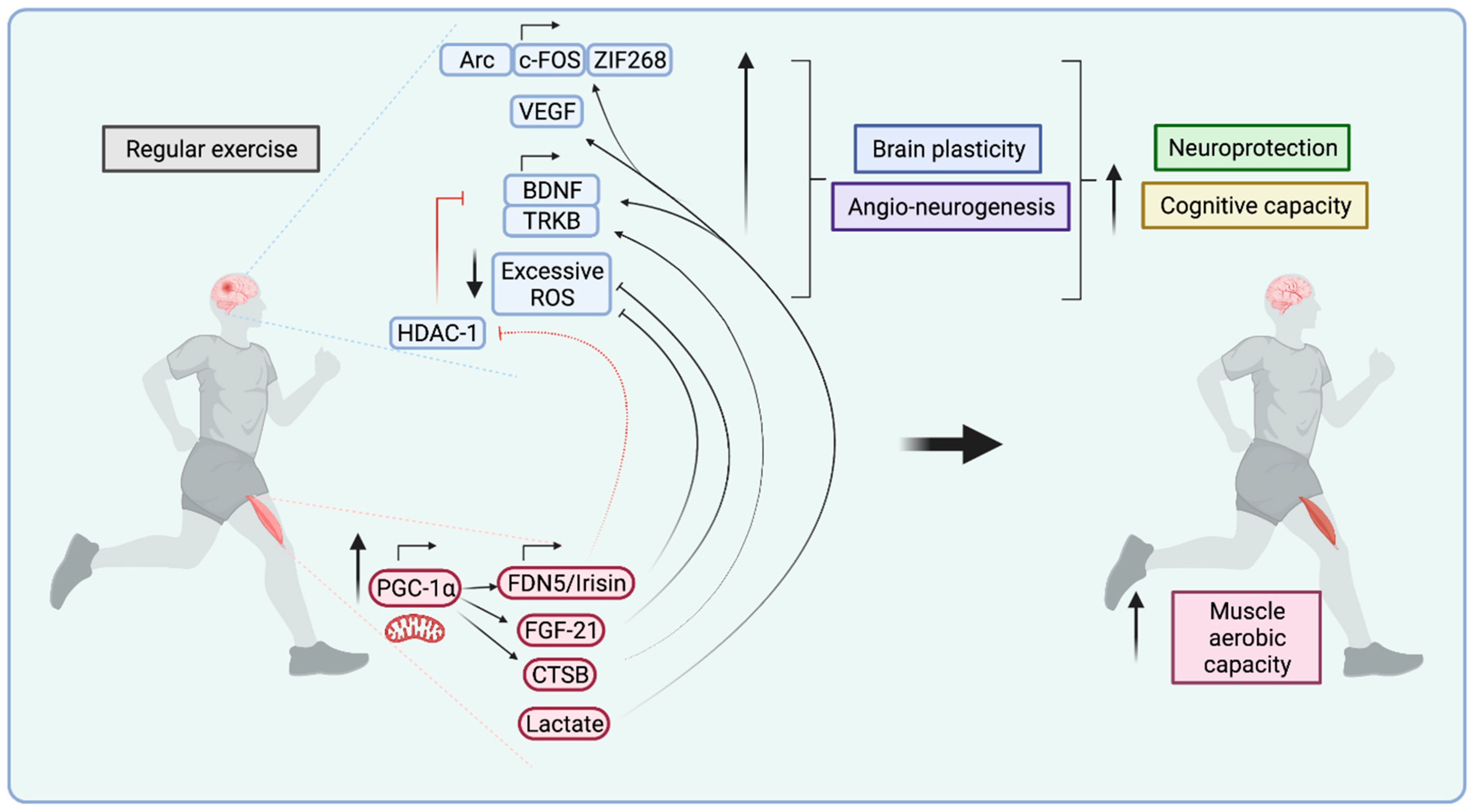
5. How Do Muscles Communicate with the Brain?
5.1. Exercise-Induced Alteration of Systemic Parameters
5.2. Myokines
5.2.1. Irisin
5.2.2. Cathepsin B
5.2.3. BDNF
5.2.4. FGF21
5.2.5. Humanin
5.2.6. Cytokines
5.3. Metabolites
5.4. MicroRNAs
5.5. Direct Neuroprotective Signaling from Skeletal Muscle Mitochondria?
6. Strategies to Boost Brain Mitochondria by Exercise
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alzheimer’s Association. 2019 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Dorsey, E.R.; Elbaz, A.; Nichols, E.; Abd-Allah, F.; Abdelalim, A.; Adsuar, J.C.; Ansha, M.G.; Brayne, C.; Choi, J.-Y.J.; Collado-Mateo, D. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Alzheimer’s Disease International. World Alzheimer Report 2019: Attitudes to Dementia; Alzheimer’s Disease International: London, UK, 2019. [Google Scholar]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, P.L.; Castillo-García, A.; Morales, J.S.; de la Villa, P.; Hampel, H.; Emanuele, E.; Lista, S.; Lucia, A. Exercise benefits on Alzheimer’s disease: State-of-the-science. Ageing Res. Rev. 2020, 62, 101108. [Google Scholar] [CrossRef]
- Ellis, T.; Rochester, L. Mobilizing Parkinson’s disease: The future of exercise. J. Parkinson’s Dis. 2018, 8, S95–S100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques-Aleixo, I.; Beleza, J.; Sampaio, A.; Stevanović, J.; Coxito, P.; Gonçalves, I.; Ascensão, A.; Magalhães, J. Preventive and therapeutic potential of physical exercise in neurodegenerative diseases. Antioxid. Redox Signal. 2020, 34, 674–693. [Google Scholar] [CrossRef] [PubMed]
- Caspersen, C.J.; Powell, K.E.; Christenson, G.M. Physical activity, exercise, and physical fitness: Definitions and distinctions for health-related research. Public Health Rep. 1985, 100, 126. [Google Scholar]
- Burtscher, J.; Burtscher, M. Run for Your Life: Tweaking the Weekly Physical Activity Volume for Longevity; BMJ Publishing Group Ltd.: London, UK; British Association of Sport and Exercise Medicine: Edinburgh, UK, 2020. [Google Scholar]
- Memme, J.M.; Erlich, A.T.; Phukan, G.; Hood, D.A. Exercise and mitochondrial health. J. Physiol. 2021, 599, 803–817. [Google Scholar] [CrossRef]
- Oliveira, A.N.; Richards, B.J.; Slavin, M.; Hood, D.A. Exercise is muscle mitochondrial medicine. Exerc. Sport Sci. Rev. 2021, 49, 67–76. [Google Scholar] [CrossRef]
- Monzio Compagnoni, G.; Di Fonzo, A.; Corti, S.; Comi, G.P.; Bresolin, N.; Masliah, E. The role of mitochondria in neurodegenerative diseases: The lesson from Alzheimer’s disease and Parkinson’s disease. Mol. Neurobiol. 2020, 57, 2959–2980. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Spires-Jones, T.L.; Attems, J.; Thal, D.R. Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathol. 2017, 134, 187–205. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Fu, H.; Hardy, J.; Duff, K.E. Selective vulnerability in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1350–1358. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989, 1, 1269. [Google Scholar] [CrossRef]
- Johnson, A.B.; Blum, N.R. Nucleoside phosphatase activities associated with the tangles and plaques of alzheimer’s disease: A histochemical study of natural and experimental neurofibrillary tangles. J. Neuropathol. Exp. Neurol. 1970, 29, 463–478. [Google Scholar] [CrossRef] [PubMed]
- Goebel, H.H.; Heipertz, R.; Scholz, W.; Iqbal, K.; Tellez-Nagel, I. Juvenile Huntington chorea: Clinical, ultrastructural, and biochemical studies. Neurology 1978, 28, 23–31. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Allaman, I. Lactate in the brain: From metabolic end-product to signalling molecule. Nat. Rev. Neurosci. 2018, 19, 235. [Google Scholar] [CrossRef]
- Browne, S.E. Mitochondria and Huntington’s disease pathogenesis: Insight from genetic and chemical models. Ann. N. Y. Acad. Sci. 2008, 1147, 358–382. [Google Scholar] [CrossRef] [PubMed]
- Damiano, M.; Diguet, E.; Malgorn, C.; D’Aurelio, M.; Galvan, L.; Petit, F.; Benhaim, L.; Guillermier, M.; Houitte, D.; Dufour, N.; et al. A role of mitochondrial complex II defects in genetic models of Huntington’s disease expressing N-terminal fragments of mutant huntingtin. Hum. Mol. Genet. 2013, 22, 3869–3882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; McDonald, D.; Blain, A.; Sachdeva, A.; Bone, L.; Smith, A.L.M.; Warren, C.; Pickett, S.J.; Hudson, G.; Filby, A.; et al. Imaging mass cytometry reveals generalised deficiency in OXPHOS complexes in Parkinson’s disease. NPJ Parkinson’s Dis. 2021, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef] [Green Version]
- Hood, D.A.; Memme, J.M.; Oliveira, A.N.; Triolo, M. Maintenance of skeletal muscle mitochondria in health, exercise, and aging. Ann. Rev. Physiol. 2019, 81, 19–41. [Google Scholar] [CrossRef]
- Ristow, M.; Zarse, K.; Oberbach, A.; Klöting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Blüher, M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, J.; Simon, D.K. Transcribe to survive: Transcriptional control of antioxidant defense programs for neuroprotection in Parkinson’s disease. Antioxid. Redox Signal. 2009, 11, 509–528. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Chung, H.Y.; Koltai, E.; Taylor, A.W.; Goto, S. Exercise, oxidative stress and hormesis. Ageing Res. Rev. 2008, 7, 34–42. [Google Scholar] [CrossRef]
- Pesta, D.; Roden, M. The Janus head of oxidative stress in metabolic diseases and during physical exercise. Curr. Diabetes Rep. 2017, 17, 41. [Google Scholar] [CrossRef]
- Dexter, D.T.; Carter, C.J.; Wells, F.R.; Javoy-Agid, F.; Agid, Y.; Lees, A.; Jenner, P.; Marsden, C.D. Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. J. Neurochem. 1989, 52, 381–389. [Google Scholar] [CrossRef]
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popov, L.-D. Mitochondrial biogenesis: An update. J. Cell Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar]
- Salminen, A.; Kaarniranta, K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res. Rev. 2012, 11, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Burkewitz, K.; Zhang, Y.; Mair, W.B. AMPK at the nexus of energetics and aging. Cell Metab. 2014, 20, 10–25. [Google Scholar] [CrossRef] [Green Version]
- Golpich, M.; Amini, E.; Mohamed, Z.; Azman Ali, R.; Mohamed Ibrahim, N.; Ahmadiani, A. Mitochondrial dysfunction and biogenesis in neurodegenerative diseases: Pathogenesis and treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef]
- Sheng, B.; Wang, X.; Su, B.; Lee, H.G.; Casadesus, G.; Perry, G.; Zhu, X. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J. Neurochem. 2012, 120, 419–429. [Google Scholar] [CrossRef]
- Ryan, S.D.; Dolatabadi, N.; Chan, S.F.; Zhang, X.; Akhtar, M.W.; Parker, J.; Soldner, F.; Sunico, C.R.; Nagar, S.; Talantova, M. Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1α transcription. Cell 2013, 155, 1351–1364. [Google Scholar] [CrossRef] [Green Version]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; García-Prieto, J.; Baker, M.J.; Merkwirth, C.; Benit, P.; Rustin, P.; Rupérez, F.J.; Barbas, C.; Ibañez, B.; Langer, T. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 2015, 350, aad0116. [Google Scholar] [CrossRef]
- Schrepfer, E.; Scorrano, L. Mitofusins, from mitochondria to metabolism. Mol. Cell 2016, 61, 683–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Horn, A.; Raavicharla, S.; Shah, S.; Cox, D.; Jaiswal, J.K. Mitochondrial fragmentation enables localized signaling required for cell repair. J. Cell Biol. 2020, 219, e201909154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batista, A.F.; Rody, T.; Forny-Germano, L.; Cerdeiro, S.; Bellio, M.; Ferreira, S.T.; Munoz, D.P.; De Felice, F.G. Interleukin-1β mediates alterations in mitochondrial fusion/fission proteins and memory impairment induced by amyloid-β oligomers. J. Neuroinflamm. 2021, 18, 54. [Google Scholar] [CrossRef]
- Gao, G.; Wang, Z.; Lu, L.; Duan, C.; Wang, X.; Yang, H. Morphological analysis of mitochondria for evaluating the toxicity of α-synuclein in transgenic mice and isolated preparations by atomic force microscopy. Biomed. Pharmacother. 2017, 96, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Carelli, V.; Musumeci, O.; Caporali, L.; Zanna, C.; La Morgia, C.; Del Dotto, V.; Porcelli, A.M.; Rugolo, M.; Valentino, M.L.; Iommarini, L.; et al. Syndromic parkinsonism and dementia associated with OPA1 missense mutations. Ann. Neurol. 2015, 78, 21–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Srivastava, S. The mitochondrial basis of aging and age-related disorders. Genes 2017, 8, 398. [Google Scholar] [CrossRef] [Green Version]
- Chistiakov, D.A.; Sobenin, I.A.; Revin, V.V.; Orekhov, A.N.; Bobryshev, Y.V. Mitochondrial aging and age-related dysfunction of mitochondria. BioMed Res. Int. 2014, 2014, 7. [Google Scholar] [CrossRef] [Green Version]
- Conley, K.E.; Amara, C.E.; Jubrias, S.A.; Marcinek, D.J. Mitochondrial function, fibre types and ageing: New insights from human muscle in vivo. Exp. Physiol. 2007, 92, 333–339. [Google Scholar] [CrossRef]
- Liu, Y.J.; McIntyre, R.L.; Janssens, G.E.; Houtkooper, R.H. Mitochondrial fission and fusion: A dynamic role in aging and potential target for age-related disease. Mech. Ageing Dev. 2020, 186, 111212. [Google Scholar] [CrossRef]
- Montgomery, M.K.; Turner, N. Mitochondrial dysfunction and insulin resistance: An update. Endocr. Connect. 2015, 4, R1–R15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booth, F.W.; Roberts, C.K.; Laye, M.J. Lack of exercise is a major cause of chronic diseases. Compr. Physiol. 2011, 2, 1143–1211. [Google Scholar]
- Valenzuela, P.L.; Maffiuletti, N.A.; Joyner, M.J.; Lucia, A.; Lepers, R. Lifelong endurance exercise as a countermeasure against age-related VO2max decline: Physiological overview and insights from masters athletes. Sports Med. 2020, 50, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Capelli, C.; Rittveger, J.; Bruseghini, P.; Calabria, E.; Tam, E. Maximal aerobic power and anaerobic capacity in cycling across the age spectrum in male master athletes. Eur. J. Appl. Physiol. 2016, 116, 1395–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radak, Z.; Torma, F.; Berkes, I.; Goto, S.; Mimura, T.; Posa, A.; Balogh, L.; Boldogh, I.; Suzuki, K.; Higuchi, M.; et al. Exercise effects on physiological function during aging. Free Radic. Biol. Med. 2019, 132, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Romanello, V.; Sandri, M. The connection between the dynamic remodeling of the mitochondrial network and the regulation of muscle mass. Cell Mol. Life Sci. 2020, 78, 1305–1328. [Google Scholar] [CrossRef]
- Huertas, J.R.; Casuso, R.A.; Agustín, P.H.; Cogliati, S. Stay fit, stay young: Mitochondria in movement: The role of exercise in the new mitochondrial paradigm. Oxidative Med. Cell. Longev. 2019, 2019, 7058350. [Google Scholar] [CrossRef] [Green Version]
- Granata, C.; Caruana, N.J.; Botella, J.; Jamnick, N.A.; Huynh, K.; Kuang, J.; Janssen, H.A.; Reljic, B.; Mellett, N.A.; Laskowski, A. Multi-omics reveal intricate network of mitochondrial adaptations to training in human skeletal muscle. bioRxiv 2021. [Google Scholar] [CrossRef]
- Larsen, F.J.; Schiffer, T.A.; Zinner, C.; Willis, S.J.; Morales-Alamo, D.; Calbet, J.A.L.; Boushel, R.; Holmberg, H.C. Mitochondrial oxygen affinity increases after sprint interval training and is related to the improvement in peak oxygen uptake. Acta Physiol. 2020, 229, e13463. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Youle, R.J.; Finkel, T. The mitochondrial basis of aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holloszy, J.O. Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J. Biol. Chem. 1967, 242, 2278–2282. [Google Scholar] [CrossRef]
- Bishop, D.J.; Botella, J.; Genders, A.J.; Lee, M.J.; Saner, N.J.; Kuang, J.; Yan, X.; Granata, C. High-intensity exercise and mitochondrial biogenesis: Current controversies and future research directions. Physiology 2019, 34, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Granata, C.; Jamnick, N.A.; Bishop, D.J. Training-induced changes in mitochondrial content and respiratory function in human skeletal muscle. Sports Med. 2018, 48, 1809–1828. [Google Scholar] [CrossRef]
- Hood, D.A. Invited review: Contractile activity-induced mitochondrial biogenesis in skeletal muscle. J. Appl. Physiol. 2001, 90, 1137–1157. [Google Scholar] [CrossRef]
- Granata, C.; Oliveira, R.S.; Little, J.P.; Renner, K.; Bishop, D.J. Mitochondrial adaptations to high-volume exercise training are rapidly reversed after a reduction in training volume in human skeletal muscle. FASEB J. 2016, 30, 3413–3423. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, R.A.; Rasmussen, P.; Siebenmann, C.; Díaz, V.; Gassmann, M.; Pesta, D.; Gnaiger, E.; Nordsborg, N.B.; Robach, P.; Lundby, C. Determinants of time trial performance and maximal incremental exercise in highly trained endurance athletes. J. Appl. Physiol. 2011, 111, 1422–1430. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, R.A.; Lundby, C. Mitochondria express enhanced quality as well as quantity in association with aerobic fitness across recreationally active individuals up to elite athletes. J. Appl. Physiol. 2013, 114, 344–350. [Google Scholar] [CrossRef] [Green Version]
- Broskey, N.T.; Greggio, C.; Boss, A.; Boutant, M.; Dwyer, A.; Schlueter, L.; Hans, D.; Gremion, G.; Kreis, R.; Boesch, C. Skeletal muscle mitochondria in the elderly: Effects of physical fitness and exercise training. J. Clin. Endocrinol. Metab. 2014, 99, 1852–1861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merry, T.L.; Ristow, M. Mitohormesis in exercise training. Free Radic. Biol. Med. 2016, 98, 123–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arumugam, T.V.; Gleichmann, M.; Tang, S.C.; Mattson, M.P. Hormesis/preconditioning mechanisms, the nervous system and aging. Ageing Res. Rev. 2006, 5, 165–178. [Google Scholar] [CrossRef]
- Flockhart, M.; Nilsson, L.C.; Tais, S.; Ekblom, B.; Apró, W.; Larsen, F.J. Excessive exercise training causes mitochondrial functional impairment and decreases glucose tolerance in healthy volunteers. Cell Metab. 2021, 33, 957–970. [Google Scholar] [CrossRef]
- Hawley, J.A.; Bishop, D.J. High-intensity exercise training—Too much of a good thing? Nat. Rev. Endocrinol. 2021, 17, 385–386. [Google Scholar] [CrossRef] [PubMed]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brierley, E.J.; Johnson, M.A.; James, O.F.; Turnbull, D.M. Mitochondrial involvement in the ageing process. Facts and controversies. Mol. Cell Biochem. 1997, 174, 325–328. [Google Scholar] [CrossRef]
- Holloway, G.P. Nutrition and training influences on the regulation of mitochondrial adenosine diphosphate sensitivity and bioenergetics. Sports Med. 2017, 47 (Suppl. S1), 13–21. [Google Scholar] [CrossRef] [Green Version]
- Bouzid, M.A.; Filaire, E.; Matran, R.; Robin, S.; Fabre, C. Lifelong voluntary exercise modulates age-related changes in oxidative stress. Int. J. Sports Med. 2018, 39, 21–28. [Google Scholar] [CrossRef]
- Lancaster, G.I.; Febbraio, M.A. The immunomodulating role of exercise in metabolic disease. Trends Immunol. 2014, 35, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Valacchi, G.; Virgili, F.; Cervellati, C.; Pecorelli, A. OxInflammation: From subclinical condition to pathological biomarker. Front. Physiol. 2018, 9, 858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baar, K.; Wende, A.R.; Jones, T.E.; Marison, M.; Nolte, L.A.; Chen, M.; Kelly, D.P.; Holloszy, J.O. Adaptations of skeletal muscle to exercise: Rapid increase in the transcriptional coactivator PGC-1. FASEB J. 2002, 16, 1879–1886. [Google Scholar] [CrossRef]
- Eisele, P.S.; Furrer, R.; Beer, M.; Handschin, C. The PGC-1 coactivators promote an anti-inflammatory environment in skeletal muscle in vivo. Biochem. Biophys. Res. Commun. 2015, 464, 692–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casuso, R.A.; Huertas, J.R. The emerging role of skeletal muscle mitochondrial dynamics in exercise and ageing. Ageing Res. Rev. 2020, 58, 101025. [Google Scholar] [CrossRef]
- Knott, A.B.; Perkins, G.; Schwarzenbacher, R.; Bossy-Wetzel, E. Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 2008, 9, 505–518. [Google Scholar] [CrossRef] [Green Version]
- Picard, M.; Gentil, B.J.; McManus, M.J.; White, K.; St. Louis, K.; Gartside, S.E.; Wallace, D.C.; Turnbull, D.M. Acute exercise remodels mitochondrial membrane interactions in mouse skeletal muscle. J. Appl. Physiol. 2013, 115, 1562–1571. [Google Scholar] [CrossRef]
- Halling, J.F.; Ringholm, S.; Olesen, J.; Prats, C.; Pilegaard, H. Exercise training protects against aging-induced mitochondrial fragmentation in mouse skeletal muscle in a PGC-1α dependent manner. Exp. Gerontol. 2017, 96, 1–6. [Google Scholar] [CrossRef]
- Arribat, Y.; Broskey, N.T.; Greggio, C.; Boutant, M.; Conde Alonso, S.; Kulkarni, S.S.; Lagarrigue, S.; Carnero, E.A.; Besson, C.; Cantó, C. Distinct patterns of skeletal muscle mitochondria fusion, fission and mitophagy upon duration of exercise training. Acta Physiol. 2019, 225, e13179. [Google Scholar] [CrossRef]
- Castells-Sánchez, A.; Roig-Coll, F.; Dacosta-Aguayo, R.; Lamonja-Vicente, N.; Sawicka, A.K.; Torán-Monserrat, P.; Pera, G.; Montero-Alía, P.; Heras-Tebar, A.; Domènech, S.; et al. Exercise and fitness neuroprotective effects: Molecular, brain volume and psychological correlates and their mediating role in healthy late-middle-aged women and men. Front. Aging Neurosci. 2021, 13, 615247. [Google Scholar] [CrossRef] [PubMed]
- Colcombe, S.J.; Erickson, K.I.; Raz, N.; Webb, A.G.; Cohen, N.J.; McAuley, E.; Kramer, A.F. Aerobic fitness reduces brain tissue loss in aging humans. J. Gerontol. A Biol. Sci. Med. Sci. 2003, 58, 176–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colcombe, S.J.; Erickson, K.I.; Scalf, P.E.; Kim, J.S.; Prakash, R.; McAuley, E.; Elavsky, S.; Marquez, D.X.; Hu, L.; Kramer, A.F. Aerobic exercise training increases brain volume in aging humans. J. Gerontol. A Biol. Sci. Med. Sci. 2006, 61, 1166–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colcombe, S.; Kramer, A.F. Fitness effects on the cognitive function of older adults: A meta-analytic study. Psychol. Sci. 2003, 14, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Ahlskog, J.E. Does vigorous exercise have a neuroprotective effect in Parkinson disease? Neurology 2011, 77, 288–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattson, M.P.; Arumugam, T.V. Hallmarks of brain aging: Adaptive and pathological modification by metabolic states. Cell Metab. 2018, 27, 1176–1199. [Google Scholar] [CrossRef] [Green Version]
- Irving, B.A.; Lanza, I.R.; Henderson, G.C.; Rao, R.R.; Spiegelman, B.M.; Nair, K.S. Combined training enhances skeletal muscle mitochondrial oxidative capacity independent of age. J. Clin. Endocrinol. Metab. 2015, 100, 1654–1663. [Google Scholar] [CrossRef] [Green Version]
- Fiuza-Luces, C.; Santos-Lozano, A.; Joyner, M.; Carrera-Bastos, P.; Picazo, O.; Zugaza, J.L.; Izquierdo, M.; Ruilope, L.M.; Lucia, A. Exercise benefits in cardiovascular disease: Beyond attenuation of traditional risk factors. Nat. Rev. Cardiol. 2018, 15, 731–743. [Google Scholar] [CrossRef]
- Pedersen, B.K. Physical activity and muscle–brain crosstalk. Nat. Rev. Endocrinol. 2019, 15, 383–392. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Febbraio, M.A. Muscles, exercise and obesity: Skeletal muscle as a secretory organ. Nat. Rev. Endocrinol. 2012, 8, 457–465. [Google Scholar] [CrossRef]
- Steiner, J.L.; Murphy, E.A.; McClellan, J.L.; Carmichael, M.D.; Davis, J.M. Exercise training increases mitochondrial biogenesis in the brain. J. Appl. Physiol. 2011, 111, 1066–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marosi, K.; Bori, Z.; Hart, N.; Sárga, L.; Koltai, E.; Radák, Z.; Nyakas, C. Long-term exercise treatment reduces oxidative stress in the hippocampus of aging rats. Neuroscience 2012, 226, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Boveris, A.; Navarro, A. Systemic and mitochondrial adaptive responses to moderate exercise in rodents. Free Radic. Biol. Med. 2008, 44, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Laing, B.T.; Do, K.; Matsubara, T.; Wert, D.W.; Avery, M.J.; Langdon, E.M.; Zheng, D.; Huang, H. Voluntary exercise improves hypothalamic and metabolic function in obese mice. J. Endocrinol. 2016, 229, 109–122. [Google Scholar] [CrossRef] [Green Version]
- Erickson, K.I.; Voss, M.W.; Prakash, R.S.; Basak, C.; Szabo, A.; Chaddock, L.; Kim, J.S.; Heo, S.; Alves, H.; White, S.M. Exercise training increases size of hippocampus and improves memory. Proc. Natl. Acad. Sci. USA 2011, 108, 3017–3022. [Google Scholar] [CrossRef] [Green Version]
- Navarro, A.; Gomez, C.; López-Cepero, J.M.; Boveris, A. Beneficial effects of moderate exercise on mice aging: Survival, behavior, oxidative stress, and mitochondrial electron transfer. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R505–R511. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Yeo, H.C.; Overvik-Douki, E.; Hagen, T.; Doniger, S.J.; Chu, D.W.; Brooks, G.A.; Ames, B.N. Chronically and acutely exercised rats: Biomarkers of oxidative stress and endogenous antioxidants. J. Appl. Physiol. 2000, 89, 21–28. [Google Scholar] [CrossRef]
- Aguiar, A.S.; Tuon, T.; Soares, F.S.; da Rocha, L.G.C.; Silveira, P.C.; Pinho, R.A. The effect of n-acetylcysteine and deferoxamine on exercise-induced oxidative damage in striatum and hippocampus of mice. Neurochem. Res. 2008, 33, 729–736. [Google Scholar] [CrossRef]
- Quan, H.; Koltai, E.; Suzuki, K.; Aguiar, A.S.; Pinho, R.; Boldogh, I.; Berkes, I.; Radak, Z. Exercise, redox system and neurodegenerative diseases. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165778. [Google Scholar] [CrossRef]
- Radak, Z.; Taylor, A.W.; Ohno, H.; Goto, S. Adaptation to exercise-induced oxidative stress: From muscle to brain. Exerc. Immunol. Rev. 2001, 7, 90–107. [Google Scholar]
- Hawley, J.A.; Hargreaves, M.; Joyner, M.J.; Zierath, J.R. Integrative biology of exercise. Cell 2014, 159, 738–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neufer, P.D.; Bamman, M.M.; Muoio, D.M.; Bouchard, C.; Cooper, D.M.; Goodpaster, B.H.; Booth, F.W.; Kohrt, W.M.; Gerszten, R.E.; Mattson, M.P. Understanding the cellular and molecular mechanisms of physical activity-induced health benefits. Cell Metab. 2015, 22, 4–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccirillo, R. Exercise-induced myokines with therapeutic potential for muscle wasting. Front. Physiol. 2019, 10, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, R.M.; Watt, M.J.; Febbraio, M.A. Metabolic communication during exercise. Nat. Metab. 2020, 2, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Nederveen, J.P.; Warnier, G.; Di Carlo, A.; Nilsson, M.I.; Tarnopolsky, M.A. Extracellular vesicles and exosomes: Insights from exercise science. Front. Physiol. 2020, 11, 604274. [Google Scholar] [CrossRef]
- Vechetti, I.J., Jr.; Valentino, T.; Mobley, C.B.; McCarthy, J.J. The role of extracellular vesicles in skeletal muscle and systematic adaptation to exercise. J. Physiol. 2021, 599, 845–861. [Google Scholar] [CrossRef] [PubMed]
- Whitham, M.; Parker, B.L.; Friedrichsen, M.; Hingst, J.R.; Hjorth, M.; Hughes, W.E.; Egan, C.L.; Cron, L.; Watt, K.I.; Kuchel, R.P. Extracellular vesicles provide a means for tissue crosstalk during exercise. Cell Metab. 2018, 27, 237–251.e4. [Google Scholar] [CrossRef] [Green Version]
- Guescini, M.; Canonico, B.; Lucertini, F.; Maggio, S.; Annibalini, G.; Barbieri, E.; Luchetti, F.; Papa, S.; Stocchi, V. Muscle releases alpha-sarcoglycan positive extracellular vesicles carrying miRNAs in the bloodstream. PLoS ONE 2015, 10, e0125094. [Google Scholar] [CrossRef] [Green Version]
- Koelwyn, G.J.; Quail, D.F.; Zhang, X.; White, R.M.; Jones, L.W. Exercise-dependent regulation of the tumour microenvironment. Nat. Rev. Cancer 2017, 17, 620–632. [Google Scholar] [CrossRef]
- Jørgensen, L.G.; Perko, G.; Secher, N.H. Regional cerebral artery mean flow velocity and blood flow during dynamic exercise in humans. J. Appl. Physiol. 1992, 73, 1825–1830. [Google Scholar] [CrossRef]
- Secher, N.H.; Seifert, T.; Van Lieshout, J.J. Cerebral blood flow and metabolism during exercise: Implications for fatigue. J. Appl. Physiol. 2008, 104, 306–314. [Google Scholar] [CrossRef]
- Toth, P.; Tarantini, S.; Csiszar, A.; Ungvari, Z. Functional vascular contributions to cognitive impairment and dementia: Mechanisms and consequences of cerebral autoregulatory dysfunction, endothelial impairment, and neurovascular uncoupling in aging. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H1–H20. [Google Scholar] [CrossRef] [Green Version]
- Von Holstein-Rathlou, S.; Petersen, N.C.; Nedergaard, M. Voluntary running enhances glymphatic influx in awake behaving, young mice. Neurosci. Lett. 2018, 662, 253–258. [Google Scholar] [CrossRef] [Green Version]
- Jessen, N.A.; Munk, A.S.F.; Lundgaard, I.; Nedergaard, M. The glymphatic system: A beginner’s guide. Neurochem. Res. 2015, 40, 2583–2599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, T.D.; Talbot, J.S.; Douglas, A.J.M.; Richards, C.T.; Lord, R.N. Exercised state of mind: A perspective on ageing, cerebral blood flow and cognition. J. Physiol. 2021, 599, 2523–2524. [Google Scholar] [CrossRef]
- Trejo, J.L.; Llorens-Martin, M.; Torres-Alemán, I. The effects of exercise on spatial learning and anxiety-like behavior are mediated by an IGF-I-dependent mechanism related to hippocampal neurogenesis. Mol. Cell. Neurosci. 2008, 37, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Sylow, L.; Kleinert, M.; Richter, E.A.; Jensen, T.E. Exercise-stimulated glucose uptake—Regulation and implications for glycaemic control. Nat. Rev. Endocrinol. 2017, 13, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Safdar, A.; Saleem, A.; Tarnopolsky, M.A. The potential of endurance exercise-derived exosomes to treat metabolic diseases. Nat. Rev. Endocrinol. 2016, 12, 504–517. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Steensberg, A.; Keller, P.; Keller, C.; Fischer, C.; Hiscock, N.; van Hall, G.; Plomgaard, P.; Febbraio, M.A. Muscle-derived interleukin-6: Lipolytic, anti-inflammatory and immune regulatory effects. Pflug. Arch. 2003, 446, 9–16. [Google Scholar] [CrossRef]
- Whitham, M.; Febbraio, M.A. The ever-expanding myokinome: Discovery challenges and therapeutic implications. Nat. Rev. Drug Discov. 2016, 15, 719–729. [Google Scholar] [CrossRef]
- Delezie, J.; Handschin, C. Endocrine crosstalk between skeletal muscle and the brain. Front. Neurol. 2018, 9, 698. [Google Scholar] [CrossRef] [PubMed]
- Duzel, E.; van Praag, H.; Sendtner, M. Can physical exercise in old age improve memory and hippocampal function? Brain 2016, 139 Pt 3, 662–673. [Google Scholar] [CrossRef]
- Moon, H.Y.; Becke, A.; Berron, D.; Becker, B.; Sah, N.; Benoni, G.; Janke, E.; Lubejko, S.T.; Greig, N.H.; Mattison, J.A.; et al. Running-induced systemic cathepsin b secretion is associated with memory function. Cell Metab. 2016, 24, 332–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lourenco, M.V.; Frozza, R.L.; de Freitas, G.B.; Zhang, H.; Kincheski, G.C.; Ribeiro, F.C.; Goncalves, R.A.; Clarke, J.R.; Beckman, D.; Staniszewski, A.; et al. Exercise-linked FNDC5/irisin rescues synaptic plasticity and memory defects in Alzheimer’s models. Nat. Med. 2019, 25, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.M.; Sesso, H.D.; Ridker, P.M.; Mouton, C.P.; Stefanick, M.L.; Manson, J.E. Physical activity and inflammation in a multiethnic cohort of women. Med. Sci. Sports Exerc. 2012, 44, 1088–1096. [Google Scholar] [CrossRef]
- Miyamoto-Mikami, E.; Sato, K.; Kurihara, T.; Hasegawa, N.; Fujie, S.; Fujita, S.; Sanada, K.; Hamaoka, T.; Tabata, I.; Iemitsu, M. Endurance training-induced increase in circulating irisin levels is associated with reduction of abdominal visceral fat in middle-aged and older adults. PLoS ONE 2015, 10, e0120354. [Google Scholar] [CrossRef] [Green Version]
- Bostrom, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Bostrom, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef]
- Chen, K.; Xu, Z.; Liu, Y.; Wang, Z.; Li, Y.; Xu, X.; Chen, C.; Xia, T.; Liao, Q.; Yao, Y. Irisin protects mitochondria function during pulmonary ischemia/reperfusion injury. Sci. Transl. Med. 2017, 9, 418. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Chen, K.; Han, Y.; Zhu, H.; Zhou, X.; Tan, T.; Zeng, J.; Zhang, J.; Liu, Y.; Li, Y. Irisin protects heart against ischemia-reperfusion injury through a SOD2-dependent mitochondria mechanism. J. Cardiovasc. Pharmacol. 2018, 72, 259. [Google Scholar] [CrossRef]
- Bi, J.; Zhang, J.; Ren, Y.; Du, Z.; Li, Q.; Wang, Y.; Wei, S.; Yang, L.; Zhang, J.; Liu, C. Irisin alleviates liver ischemia-reperfusion injury by inhibiting excessive mitochondrial fission, promoting mitochondrial biogenesis and decreasing oxidative stress. Redox Biol. 2019, 20, 296–306. [Google Scholar] [CrossRef]
- Young, M.F.; Valaris, S.; Wrann, C.D. A role for FNDC5/Irisin in the beneficial effects of exercise on the brain and in neurodegenerative diseases. Prog. Cardiovasc. Dis. 2019, 62, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Wrann, C.D.; White, J.P.; Salogiannnis, J.; Laznik-Bogoslavski, D.; Wu, J.; Ma, D.; Lin, J.D.; Greenberg, M.E.; Spiegelman, B.M. Exercise induces hippocampal BDNF through a PGC-1alpha/FNDC5 pathway. Cell Metab. 2013, 18, 649–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppel, I.; Timmusk, T. Differential regulation of Bdnf expression in cortical neurons by class-selective histone deacetylase inhibitors. Neuropharmacology 2013, 75, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, L.; González-Alvarado, M.N.; Motalleb, R.; Blomgren, K.; Börjesson, M.; Kuhn, H.G. Constitutive PGC-1α overexpression in skeletal muscle does not protect from age-dependent decline in neurogenesis. Sci. Rep. 2019, 9, 12320. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.Z.; Nusslock, R. Exercise-mediated neurogenesis in the hippocampus via BDNF. Front. Neurosci. 2018, 12, 52. [Google Scholar] [CrossRef] [Green Version]
- Guicciardi, M.E.; Deussing, J.; Miyoshi, H.; Bronk, S.F.; Svingen, P.A.; Peters, C.; Kaufmann, S.H.; Gores, G.J. Cathepsin B contributes to TNF-α–mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J. Clin. Investig. 2000, 106, 1127–1137. [Google Scholar] [CrossRef] [Green Version]
- Chwieralski, C.; Welte, T.; Bühling, F. Cathepsin-regulated apoptosis. Apoptosis 2006, 11, 143–149. [Google Scholar] [CrossRef]
- Hook, V.; Yoon, M.; Mosier, C.; Ito, G.; Podvin, S.; Head, B.P.; Rissman, R.; O’Donoghue, A.J.; Hook, G. Cathepsin B in neurodegeneration of Alzheimer’s disease, traumatic brain injury, and related brain disorders. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2020, 1868, 140428. [Google Scholar] [CrossRef]
- Liu, J.; Amar, F.; Corona, C.; So, R.W.L.; Andrews, S.J.; Nagy, P.L.; Shelanski, M.L.; Greene, L.A. Brain-derived neurotrophic factor elevates activating transcription factor 4 (ATF4) in neurons and promotes ATF4-dependent induction of Sesn2. Front. Mol. Neurosci. 2018, 11, 62. [Google Scholar] [CrossRef]
- Neeper, S.A.; Gomez-Pinilla, F.; Choi, J.; Cotman, C. Exercise and brain neurotrophins. Nature 1995, 373, 109. [Google Scholar] [CrossRef] [PubMed]
- Vaynman, S.; Ying, Z.; Gomez-Pinilla, F. Hippocampal BDNF mediates the efficacy of exercise on synaptic plasticity and cognition. Eur. J. Neurosci. 2004, 20, 2580–2590. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, K.; Jasmin, B.J. BDNF is expressed in skeletal muscle satellite cells and inhibits myogenic differentiation. J. Neurosci. 2006, 26, 5739–5749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGregor, C.E.; English, A.W. The role of BDNF in peripheral nerve regeneration: Activity-dependent treatments and Val66Met. Front. Cell Neurosci. 2018, 12, 522. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Brobst, D.; Chan, W.S.; Tse, M.C.L.; Herlea-Pana, O.; Ahuja, P.; Bi, X.; Zaw, A.M.; Kwong, Z.S.W.; Jia, W.H.; et al. Muscle-generated BDNF is a sexually dimorphic myokine that controls metabolic flexibility. Sci. Signal. 2019, 12, 594. [Google Scholar] [CrossRef] [PubMed]
- Ferris, L.T.; Williams, J.S.; Shen, C.L. The effect of acute exercise on serum brain-derived neurotrophic factor levels and cognitive function. Med. Sci. Sports Exerc. 2007, 39, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Matthews, V.B.; Aström, M.B.; Chan, M.H.; Bruce, C.R.; Krabbe, K.S.; Prelovsek, O.; Akerström, T.; Yfanti, C.; Broholm, C.; Mortensen, O.H.; et al. Brain-derived neurotrophic factor is produced by skeletal muscle cells in response to contraction and enhances fat oxidation via activation of AMP-activated protein kinase. Diabetologia 2009, 52, 1409–1418. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.; Wan, R.; Yang, J.-L.; Kamimura, N.; Son, T.G.; Ouyang, X.; Luo, Y.; Okun, E.; Mattson, M.P. Involvement of PGC-1α in the formation and maintenance of neuronal dendritic spines. Nat. Commun. 2012, 3, 1250. [Google Scholar] [CrossRef] [Green Version]
- Michalski, B.; Fahnestock, M. Pro-brain-derived neurotrophic factor is decreased in parietal cortex in Alzheimer’s disease. Mol. Brain Res. 2003, 111, 148–154. [Google Scholar] [CrossRef]
- Ng, T.K.S.; Ho, C.S.H.; Tam, W.W.S.; Kua, E.H.; Ho, R.C.-M. Decreased serum brain-derived neurotrophic factor (BDNF) levels in patients with Alzheimer’s disease (AD): A systematic review and meta-analysis. Int. J. Mol. Sci. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.H.; Bylykbashi, E.; Chatila, Z.K.; Lee, S.W.; Pulli, B.; Clemenson, G.D.; Kim, E.; Rompala, A.; Oram, M.K.; Asselin, C. Combined adult neurogenesis and BDNF mimic exercise effects on cognition in an Alzheimer’s mouse model. Science 2018, 361, eaan8821. [Google Scholar] [CrossRef] [Green Version]
- Durieux, J.; Wolff, S.; Dillin, A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 2011, 144, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Kim, S.H.; Min, Y.-K.; Yang, H.-M.; Lee, J.-B.; Lee, M.-S. Acute exercise induces FGF21 expression in mice and in healthy humans. PLoS ONE 2013, 8, e63517. [Google Scholar] [CrossRef] [PubMed]
- Oost, L.J.; Kustermann, M.; Armani, A.; Blaauw, B.; Romanello, V. Fibroblast growth factor 21 controls mitophagy and muscle mass. J. Cachexia Sarcopenia Muscle 2019, 10, 630–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Hong, Y.; He, H.; Jiang, G.; You, W.; Liang, X.; Fu, Q.; Han, S.; Lian, Q.; Zhang, Y. FGF21 mediates mesenchymal stem cell senescence via regulation of mitochondrial dynamics. Oxidative Med. Cell. Longev. 2019, 2019, 13. [Google Scholar] [CrossRef]
- Forsström, S.; Jackson, C.B.; Carroll, C.J.; Kuronen, M.; Pirinen, E.; Pradhan, S.; Marmyleva, A.; Auranen, M.; Kleine, I.-M.; Khan, N.A. Fibroblast growth factor 21 drives dynamics of local and systemic stress responses in mitochondrial myopathy with mtDNA deletions. Cell Metab. 2019, 30, 1040–1054.e7. [Google Scholar] [CrossRef]
- Peng, H.; Wang, Q.; Lou, T.; Qin, J.; Jung, S.; Shetty, V.; Li, F.; Wang, Y.; Feng, X.-h.; Mitch, W.E. Myokine mediated muscle-kidney crosstalk suppresses metabolic reprogramming and fibrosis in damaged kidneys. Nat. Commun. 2017, 8, 1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsuchou, H.; Pan, W.; Kastin, A.J. The fasting polypeptide FGF21 can enter brain from blood. Peptides 2007, 28, 2382–2386. [Google Scholar] [CrossRef] [Green Version]
- Tezze, C.; Romanello, V.; Sandri, M. FGF21 as modulator of metabolism in health and disease. Front. Physiol. 2019, 10, 419. [Google Scholar] [CrossRef]
- Chen, J.; Hu, J.; Liu, H.; Xiong, Y.; Zou, Y.; Huang, W.; Shao, M.; Wu, J.; Yu, L.; Wang, X. FGF21 protects the blood–brain barrier by upregulating PPARγ via FGFR1/β-klotho after traumatic brain injury. J. Neurotrauma 2018, 35, 2091–2103. [Google Scholar] [CrossRef]
- Hill, C.M.; Laeger, T.; Dehner, M.; Albarado, D.C.; Clarke, B.; Wanders, D.; Burke, S.J.; Collier, J.J.; Qualls-Creekmore, E.; Solon-Biet, S.M. FGF21 signals protein status to the brain and adaptively regulates food choice and metabolism. Cell Rep. 2019, 27, 2934–2947.e3. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Chen, S.-T.; Sun, Y.; Xu, Z.; Wang, Y.; Yao, S.-Y.; Yao, W.-B.; Gao, X.-D. Fibroblast growth factor 21 ameliorates neurodegeneration in rat and cellular models of Alzheimer’s disease. Redox Biol. 2019, 22, 101133. [Google Scholar] [CrossRef]
- Chen, Y.; Shen, J.; Qi, G.; Zha, Q.; Zhang, C.; Yao, W.; Gao, X.; Chen, S. Potential therapeutic role of fibroblast growth factor 21 in neurodegeneration: Evidence for ameliorating parkinsonism via silent information regulator 2 homolog 1 and implication for gene therapy. Neuropharmacology 2020, 181, 108335. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Zhai, D.; Cabezas, E.; Welsh, K.; Nouraini, S.; Satterthwait, A.C.; Reed, J.C. Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature 2003, 423, 456–461. [Google Scholar] [CrossRef]
- Woodhead, J.S.T.; D’Souza, R.F.; Hedges, C.P.; Wan, J.; Berridge, M.V.; Cameron-Smith, D.; Cohen, P.; Hickey, A.J.R.; Mitchell, C.J.; Merry, T.L. High-intensity interval exercise increases humanin, a mitochondrial encoded peptide, in the plasma and muscle of men. J. Appl. Physiol. 2020, 128, 1346–1354. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yen, K.; Cohen, P. Humanin: A harbinger of mitochondrial-derived peptides? Trends Endocrinol. Metab. 2013, 24, 222–228. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Liu, T.; Wang, W.-X.; Xu, J.-H.; Yang, P.-B.; Lu, H.-X.; Sun, Q.-R.; Hu, H.-T. Protective effects of [Gly14]-Humanin on β-amyloid-induced PC12 cell death by preventing mitochondrial dysfunction. Neurochem. Int. 2010, 56, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, M. Humanin signal for Alzheimer’s disease. J. Alzheimer’s Dis. 2011, 24, 27–32. [Google Scholar] [CrossRef]
- Bayar, S.; Taşdemir, Ş.; Kayhan, B.; Şendemir, A.; Şengül, G. Investigation of the neuroprotective effect of humanin in an in vitro Parkinson’s disease model. Anat. Int. J. Exp. Clin. Anat. 2019, 13, S33. [Google Scholar]
- Cui, A.-L.; Zhang, Y.-H.; Li, J.-Z.; Song, T.; Liu, X.-M.; Wang, H.; Zhang, C.; Ma, G.-L.; Zhang, H.; Li, K. Humanin rescues cultured rat cortical neurons from NMDA-induced toxicity through the alleviation of mitochondrial dysfunction. Drug Des. Dev. Ther. 2017, 11, 1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieman, D.C.; Wentz, L.M. The compelling link between physical activity and the body’s defense system. J. Sport Health Sci. 2019, 8, 201–217. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Febbraio, M.A. Muscle as an endocrine organ: Focus on muscle-derived interleukin-6. Physiol. Rev. 2008, 88, 1379–1406. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, P.; de Mendonça Oliveira, L.; Brüggemann, T.R.; Sato, M.N.; Olivo, C.R.; Arantes-Costa, F.M. Physical exercise induces immunoregulation of TREG, M2, and pDCs in a lung allergic inflammation model. Front. Immunol. 2019, 10, 854. [Google Scholar] [CrossRef] [PubMed]
- Porro, C.; Cianciulli, A.; Panaro, M.A. The regulatory role of IL-10 in neurodegenerative diseases. Biomolecules 2020, 10, 1017. [Google Scholar] [CrossRef] [PubMed]
- Dowling, J.K.; Afzal, R.; Gearing, L.J.; Cervantes-Silva, M.P.; Annett, S.; Davis, G.M.; De Santi, C.; Assmann, N.; Dettmer, K.; Gough, D.J.; et al. Mitochondrial arginase-2 is essential for IL-10 metabolic reprogramming of inflammatory macrophages. Nat. Commun. 2021, 12, 1460. [Google Scholar] [CrossRef]
- Pedersen, B.; Steensberg, A.; Fischer, C.; Keller, C.; Keller, P.; Plomgaard, P.; Febbraio, M.; Saltin, B. Searching for the exercise factor: Is IL-6 a candidate? J. Muscle Res. Cell Motil. 2003, 24, 113–119. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Febbraio, M. Muscle-derived interleukin-6—a possible link between skeletal muscle, adipose tissue, liver, and brain. Brain Behav. Immun. 2005, 19, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-L.; Wang, Y.; Peng, W.-W.; Zheng, Y.-J.; Zhang, T.-N.; Wang, P.-J.; Huang, J.-D.; Zeng, Q.-Y. Effects of interleukin-6 and IL-6/AMPK signaling pathway on mitochondrial biogenesis and astrocytes viability under experimental septic condition. Int. Immunopharmacol. 2018, 59, 287–294. [Google Scholar] [CrossRef]
- Villar-Fincheira, P.; Sanhueza-Olivares, F.; Norambuena-Soto, I.; Cancino-Arenas, N.; Hernandez-Vargas, F.; Troncoso, R.; Gabrielli, L.; Chiong, M. Role of Interleukin-6 in vascular health and disease. Front. Mol. Biosci. 2021, 8, 79. [Google Scholar] [CrossRef]
- Licastro, F.; Grimaldi, L.M.E.; Bonafè, M.; Martina, C.; Olivieri, F.; Cavallone, L.; Giovanietti, S.; Masliah, E.; Franceschi, C. Interleukin-6 gene alleles affect the risk of Alzheimer’s disease and levels of the cytokine in blood and brain. Neurobiol. Aging 2003, 24, 921–926. [Google Scholar] [CrossRef]
- Arosio, B.; Trabattoni, D.; Galimberti, L.; Bucciarelli, P.; Fasano, F.; Calabresi, C.; Cazzullo, C.L.; Vergani, C.; Annoni, G.; Clerici, M. Interleukin-10 and interleukin-6 gene polymorphisms as risk factors for Alzheimer’s disease. Neurobiol. Aging 2004, 25, 1009–1015. [Google Scholar] [CrossRef] [Green Version]
- Johnston, L.C.; Su, X.; Maguire-Zeiss, K.; Horovitz, K.; Ankoudinova, I.; Guschin, D.; Hadaczek, P.; Federoff, H.J.; Bankiewicz, K.; Forsayeth, J. Human interleukin-10 gene transfer is protective in a rat model of Parkinson’s disease. Mol. Ther. 2008, 16, 1392–1399. [Google Scholar] [CrossRef]
- Bolin, L.M.; Strycharska-Orczyk, I.; Murray, R.; Langston, J.W.; Di Monte, D. Increased vulnerability of dopaminergic neurons in MPTP-lesioned interleukin-6 deficient mice. J. Neurochem. 2002, 83, 167–175. [Google Scholar] [CrossRef]
- Paulsen, G.; Ramer Mikkelsen, U.; Raastad, T.; Peake, J.M. Leucocytes, cytokines and satellite cells: What role do they play in muscle damage and regeneration following eccentric exercise? Exerc. Immunol. Rev. 2012, 18, 42–97. [Google Scholar] [PubMed]
- Zanchi, D.; Viallon, M.; Le Goff, C.; Millet, G.P.; Giardini, G.; Croisille, P.; Haller, S. Extreme mountain ultra-marathon leads to acute but transient increase in cerebral water diffusivity and plasma biomarkers levels changes. Front. Physiol. 2017, 7, 664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahangiri, Z.; Gholamnezhad, Z.; Hosseini, M.; Beheshti, F.; Kasraie, N. The effects of moderate exercise and overtraining on learning and memory, hippocampal inflammatory cytokine levels, and brain oxidative stress markers in rats. J. Physiol. Sci. 2019, 69, 993–1004. [Google Scholar] [CrossRef] [Green Version]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cui, Y.; Yu, Z.; Wang, W.; Cheng, X.; Ji, W.; Guo, S.; Zhou, Q.; Wu, N.; Chen, Y.; et al. Brain endothelial cells maintain lactate homeostasis and control adult hippocampal neurogenesis. Cell Stem Cell 2019, 25, 754–767.e9. [Google Scholar] [CrossRef] [PubMed]
- Morland, C.; Andersson, K.A.; Haugen, Ø.P.; Hadzic, A.; Kleppa, L.; Gille, A.; Rinholm, J.E.; Palibrk, V.; Diget, E.H.; Kennedy, L.H. Exercise induces cerebral VEGF and angiogenesis via the lactate receptor HCAR1. Nat. Commun. 2017, 8, 15557. [Google Scholar] [CrossRef]
- Ide, K.; Secher, N.H. Cerebral blood flow and metabolism during exercise. Prog. Neurobiol. 2000, 61, 397–414. [Google Scholar] [CrossRef]
- Coco, M.; Alagona, G.; Rapisarda, G.; Costanzo, E.; Calogero, R.A.; Perciavalle, V.; Perciavalle, V. Elevated blood lactate is associated with increased motor cortex excitability. Somat. Mot. Res. 2010, 27, 1–8. [Google Scholar] [CrossRef]
- Scandella, V.; Knobloch, M. Sensing the environment: Extracellular lactate levels control adult neurogenesis. Cell Stem Cell 2019, 25, 729–731. [Google Scholar] [CrossRef] [PubMed]
- Nicola, R.; Okun, E. Adult hippocampal neurogenesis: One lactate to rule them all. Neuromol. Med. 2021. [Google Scholar] [CrossRef]
- El Hayek, L.; Khalifeh, M.; Zibara, V.; Abi Assaad, R.; Emmanuel, N.; Karnib, N.; El-Ghandour, R.; Nasrallah, P.; Bilen, M.; Ibrahim, P.; et al. Lactate mediates the effects of exercise on learning and memory through SIRT1-dependent activation of hippocampal brain-derived neurotrophic factor (BDNF). J. Neurosci. 2019, 39, 2369–2382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winner, B.; Winkler, J. Adult neurogenesis in neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2015, 7, a021287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Ruchti, E.; Petit, J.-M.; Jourdain, P.; Grenningloh, G.; Allaman, I.; Magistretti, P.J. Lactate promotes plasticity gene expression by potentiating NMDA signaling in neurons. Proc. Natl. Acad. Sci. USA 2014, 111, 12228–12233. [Google Scholar] [CrossRef] [Green Version]
- Reddy, A.; Bozi, L.H.M.; Yaghi, O.K.; Mills, E.L.; Xiao, H.; Nicholson, H.E.; Paschini, M.; Paulo, J.A.; Garrity, R.; Laznik-Bogoslavski, D.; et al. pH-Gated succinate secretion regulates muscle remodeling in response to exercise. Cell 2020, 183, 62–75.e17. [Google Scholar] [CrossRef]
- Peruzzotti-Jametti, L.; Bernstock, J.D.; Vicario, N.; Costa, A.S.H.; Kwok, C.K.; Leonardi, T.; Booty, L.M.; Bicci, I.; Balzarotti, B.; Volpe, G.; et al. Macrophage-derived extracellular succinate licenses neural stem cells to suppress chronic neuroinflammation. Cell Stem Cell 2018, 22, 355–368.e13. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. Metazoan microRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, J.J. The MyomiR network in skeletal muscle plasticity. Exerc. Sport Sci. Rev. 2011, 39, 150. [Google Scholar] [CrossRef]
- Domańska-Senderowska, D.; Laguette, M.-J.N.; Jegier, A.; Cięszczyk, P.; September, A.V.; Brzeziańska-Lasota, E. MicroRNA profile and adaptive response to exercise training: A review. Int. J. Sports Med. 2019, 40, 227–235. [Google Scholar] [CrossRef]
- Fernández-Sanjurjo, M.; de Gonzalo-Calvo, D.; Fernández-García, B.; Díez-Robles, S.; Martínez-Canal, Á.; Olmedillas, H.; Dávalos, A.; Iglesias-Gutiérrez, E. Circulating microRNA as emerging biomarkers of exercise. Exerc. Sport Sci. Rev. 2018, 46, 160–171. [Google Scholar] [CrossRef]
- Zhang, X.; Zeng, Y. Regulation of mammalian microRNA expression. J. Cardiovasc. Transl. Res. 2010, 3, 197–203. [Google Scholar] [CrossRef]
- Davidson-Moncada, J.; Papavasiliou, F.N.; Tam, W. MicroRNAs of the immune system: Roles in inflammation and cancer. Ann. N. Y. Acad. Sci. 2010, 1183, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.H.; Liu, N.; van Rooij, E.; Olson, E.N. MicroRNA control of muscle development and disease. Curr. Opin. Cell Biol. 2009, 21, 461–469. [Google Scholar] [CrossRef] [Green Version]
- Krichevsky, A.M.; Gabriely, G. miR-21: A small multi-faceted RNA. J. Cell Mol. Med. 2009, 13, 39–53. [Google Scholar] [CrossRef]
- Chan, S.Y.; Zhang, Y.-Y.; Hemann, C.; Mahoney, C.E.; Zweier, J.L.; Loscalzo, J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009, 10, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Torma, F.; Gombos, Z.; Jokai, M.; Berkes, I.; Takeda, M.; Mimura, T.; Radak, Z.; Gyori, F. The roles of microRNA in redox metabolism and exercise-mediated adaptation. J. Sport Health Sci. 2020, 9, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Baggish, A.L.; Hale, A.; Weiner, R.B.; Lewis, G.D.; Systrom, D.; Wang, F.; Wang, T.J.; Chan, S.Y. Dynamic regulation of circulating microRNA during acute exhaustive exercise and sustained aerobic exercise training. J. Physiol. 2011, 589 Pt 16, 3983–3994. [Google Scholar] [CrossRef]
- Picard, M.; McManus, M.J.; Csordás, G.; Várnai, P.; Dorn, G.W., 2nd; Williams, D.; Hajnóczky, G.; Wallace, D.C. Trans-mitochondrial coordination of cristae at regulated membrane junctions. Nat. Commun. 2015, 6, 6259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beale, G.H.; Knowles, J.K. Interspecies transfer of mitochondria in Paramecium aurelia. Mol. Gen. Genet. 1976, 143, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.A.; Shay, J.W. Mitochondrial transformation of mammalian cells. Nature 1982, 295, 605–607. [Google Scholar] [CrossRef]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef] [Green Version]
- Kesner, E.E.; Saada-Reich, A.; Lorberboum-Galski, H. Characteristics of mitochondrial transformation into human cells. Sci. Rep. 2016, 6, 26057. [Google Scholar] [CrossRef] [PubMed]
- Katrangi, E.; D’Souza, G.; Boddapati, S.V.; Kulawiec, M.; Singh, K.K.; Bigger, B.; Weissig, V. Xenogenic transfer of isolated murine mitochondria into human rho0 cells can improve respiratory function. Rejuvenation Res. 2007, 10, 561–570. [Google Scholar] [CrossRef]
- Wu, T.-H.; Sagullo, E.; Case, D.; Zheng, X.; Li, Y.; Hong, J.S.; TeSlaa, T.; Patananan, A.N.; McCaffery, J.M.; Niazi, K.; et al. Mitochondrial transfer by photothermal nanoblade restores metabolite profile in mammalian cells. Cell Metab. 2016, 23, 921–929. [Google Scholar] [CrossRef] [Green Version]
- Davis, C.-H.O.; Kim, K.-Y.; Bushong, E.A.; Mills, E.A.; Boassa, D.; Shih, T.; Kinebuchi, M.; Phan, S.; Zhou, Y.; Bihlmeyer, N.A.; et al. Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. USA 2014, 111, 9633–9638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Nakamura, Y.; Lo, E.H.; Hayakawa, K. Astrocyte signaling in the neurovascular unit after central nervous system injury. Int. J. Mol. Sci. 2019, 20, 282. [Google Scholar] [CrossRef] [Green Version]
- Caicedo, A.; Aponte, P.M.; Cabrera, F.; Hidalgo, C.; Khoury, M. Artificial mitochondria transfer: Current challenges, advances, and future applications. Stem Cells Int. 2017, 2017, 7610414. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Zheng, X.; Wang, X.; Jin, T.; Cui, L.; Chen, Z. Mesenchymal stem/stromal cell-mediated mitochondrial transfer and the therapeutic potential in treatment of neurological diseases. Stem Cells Int. 2020, 2020, 8838046. [Google Scholar] [CrossRef]
- Todkar, K.; Chikhi, L.; Desjardins, V.; El-Mortada, F.; Pépin, G.; Germain, M. Selective packaging of mitochondrial proteins into extracellular vesicles prevents the release of mitochondrial DAMPs. Nat. Commun. 2021, 12, 1971. [Google Scholar] [CrossRef] [PubMed]
- Nitzan, K.; Benhamron, S.; Valitsky, M.; Kesner, E.E.; Lichtenstein, M.; Ben-Zvi, A.; Ella, E.; Segalstein, Y.; Saada, A.; Lorberboum-Galski, H.; et al. Mitochondrial transfer ameliorates cognitive deficits, neuronal loss, and gliosis in Alzheimer’s disease mice. J. Alzheimer’s Dis. JAD 2019, 72, 587–604. [Google Scholar] [CrossRef]
- Shi, X.; Zhao, M.; Fu, C.; Fu, A. Intravenous administration of mitochondria for treating experimental Parkinson’s disease. Mitochondrion 2017, 34, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Fuss, J.; Ben Abdallah, N.M.B.; Vogt, M.A.; Touma, C.; Pacifici, P.G.; Palme, R.; Witzemann, V.; Hellweg, R.; Gass, P. Voluntary exercise induces anxiety-like behavior in adult C57BL/6J mice correlating with hippocampal neurogenesis. Hippocampus 2010, 20, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, L.; Wilson, L.; Lucas, S.; Machado, L.; Walker, R.; Cotter, J. Indomethacin markedly blunts cerebral perfusion and reactivity, with little cognitive consequence in healthy young and older adults. J. Physiol. 2020, 599, 1097–1113. [Google Scholar] [CrossRef]
- Barnes, D.E.; Yaffe, K.; Satariano, W.A.; Tager, I.B. A longitudinal study of cardiorespiratory fitness and cognitive function in healthy older adults. J. Am. Geriatr. Soc. 2003, 51, 459–465. [Google Scholar] [CrossRef]
- Burtscher, J.; Millet, G.P.; Burtscher, M. Evaluation of a strength-training program on clinical outcomes in older adults. JAMA 2021, 325, 1110–1111. [Google Scholar] [CrossRef]
- WHO. WHO Guidelines on Physical Activity and Sedentary Behaviour. Available online: https://www.who.int/publications/i/item/9789240015128 (accessed on 16 June 2021).
- American College of Sports Medicine; Chodzko-Zajko, W.J.; Proctor, D.N.; Fiatarone Singh, M.A.; Minson, C.T.; Nigg, C.R.; Salem, G.J.; Skinner, J.S. American College of Sports Medicine position stand. Exercise and physical activity for older adults. Med. Sci. Sports Exerc. 2009, 41, 1510–1530. [Google Scholar] [CrossRef]
- Langan, S.P.; Grosicki, G.J. Exercise is medicine…and the dose matters. Front. Physiol. 2021, 12, 664. [Google Scholar] [CrossRef]
- Burtscher, J.; Burtscher, M.; Millet, G.P. The central role of mitochondrial fitness on antiviral defenses: An advocacy for physical activity during the COVID-19 pandemic. Redox Biol. 2021, 43, 101976. [Google Scholar] [CrossRef]
- Niklas, P.; Li, W.; Jens, W.; Michail, T.; Kent, S. Mitochondrial gene expression in elite cyclists: Effects of high-intensity interval exercise. Eur. J. Appl. Physiol. 2010, 110, 597–606. [Google Scholar] [CrossRef]
- Jacobs, R.A.; Flück, D.; Bonne, T.C.; Bürgi, S.; Christensen, P.M.; Toigo, M.; Lundby, C. Improvements in exercise performance with high-intensity interval training coincide with an increase in skeletal muscle mitochondrial content and function. J. Appl. Physiol. 2013, 115, 785–793. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, C.; Kayser, B. Preoperative high-intensity interval training is effective and safe in deconditioned patients with lung cancer: A randomized clinical trial. J. Rehabil. Med. 2019, 51, 712–718. [Google Scholar] [CrossRef] [Green Version]
- MacInnis, M.J.; Gibala, M.J. Physiological adaptations to interval training and the role of exercise intensity. J. Physiol. 2017, 595, 2915–2930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, B.; Zierath, J.R. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 2013, 17, 162–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granata, C.; Jamnick, N.A.; Bishop, D.J. Principles of exercise prescription, and how they influence exercise-induced changes of transcription factors and other regulators of mitochondrial biogenesis. Sports Med. 2018, 48, 1541–1559. [Google Scholar] [CrossRef] [PubMed]
- Porter, C.; Reidy, P.T.; Bhattarai, N.; Sidossis, L.S.; Rasmussen, B.B. Resistance exercise training alters mitochondrial function in human skeletal muscle. Med. Sci. Sports Exerc. 2015, 47, 1922–1931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinho, R.A.; Aguiar, A.S., Jr.; Radák, Z. Effects of resistance exercise on cerebral redox regulation and cognition: An interplay between muscle and brain. Antioxidants 2019, 8, 529. [Google Scholar] [CrossRef] [Green Version]
- Lan, Y.; Huang, Z.; Jiang, Y.; Zhou, X.; Zhang, J.; Zhang, D.; Wang, B.; Hou, G. Strength exercise weakens aerobic exercise-induced cognitive improvements in rats. PLoS ONE 2018, 13, e0205562. [Google Scholar] [CrossRef] [PubMed]
- Kettunen, J.A.; Kujala, U.M.; Kaprio, J.; Bäckmand, H.; Peltonen, M.; Eriksson, J.G.; Sarna, S. All-cause and disease-specific mortality among male, former elite athletes: An average 50-year follow-up. Br. J. Sports Med. 2015, 49, 893–897. [Google Scholar] [CrossRef]
- Rutherford, A.; Stewart, W.; Bruno, D. Heading for Trouble: Is Dementia a Game Changer for Football? BMJ Publishing Group Ltd.: London, UK; British Association of Sport and Exercise Medicine: Edinburgh, UK, 2019. [Google Scholar]
- Ling, H.; Morris, H.R.; Neal, J.W.; Lees, A.J.; Hardy, J.; Holton, J.L.; Revesz, T.; Williams, D.D. Mixed pathologies including chronic traumatic encephalopathy account for dementia in retired association football (soccer) players. Acta Neuropathol. 2017, 133, 337–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, M.E.; Ellis, T.D.; Jazayeri, D.; Heng, H.; Thomson, A.; Balasundaram, A.P.; Slade, S.C. Boxing for Parkinson’s Disease: Has implementation accelerated beyond current evidence? Front. Neurol. 2019, 10, 1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zucchella, C.; Sinforiani, E.; Tamburin, S.; Federico, A.; Mantovani, E.; Bernini, S.; Casale, R.; Bartolo, M. The multidisciplinary approach to Alzheimer’s disease and dementia. A narrative review of non-pharmacological treatment. Front. Neurol. 2018, 9, 1058. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burtscher, J.; Millet, G.P.; Place, N.; Kayser, B.; Zanou, N. The Muscle-Brain Axis and Neurodegenerative Diseases: The Key Role of Mitochondria in Exercise-Induced Neuroprotection. Int. J. Mol. Sci. 2021, 22, 6479. https://doi.org/10.3390/ijms22126479
Burtscher J, Millet GP, Place N, Kayser B, Zanou N. The Muscle-Brain Axis and Neurodegenerative Diseases: The Key Role of Mitochondria in Exercise-Induced Neuroprotection. International Journal of Molecular Sciences. 2021; 22(12):6479. https://doi.org/10.3390/ijms22126479
Chicago/Turabian StyleBurtscher, Johannes, Grégoire P. Millet, Nicolas Place, Bengt Kayser, and Nadège Zanou. 2021. "The Muscle-Brain Axis and Neurodegenerative Diseases: The Key Role of Mitochondria in Exercise-Induced Neuroprotection" International Journal of Molecular Sciences 22, no. 12: 6479. https://doi.org/10.3390/ijms22126479
APA StyleBurtscher, J., Millet, G. P., Place, N., Kayser, B., & Zanou, N. (2021). The Muscle-Brain Axis and Neurodegenerative Diseases: The Key Role of Mitochondria in Exercise-Induced Neuroprotection. International Journal of Molecular Sciences, 22(12), 6479. https://doi.org/10.3390/ijms22126479