
Ras Isoforms from Lab Benches to Lives—What Are We Missing and How Far Are We?



Abstract
:1. Introduction
1.1. The History of Ras Isoforms—From Viral Oncogenes to Pivotal Cellular Genes
1.2. Ras the Crucial Signal Relay Protein
2. Ras in Cancers
3. One Switch Many Outputs
To Divide or Not—Ras Decides the Cellular Fate
4. Four Decades of Research and Why Ras Remains Undruggable?
5. Missing Pieces in the Ras Puzzle
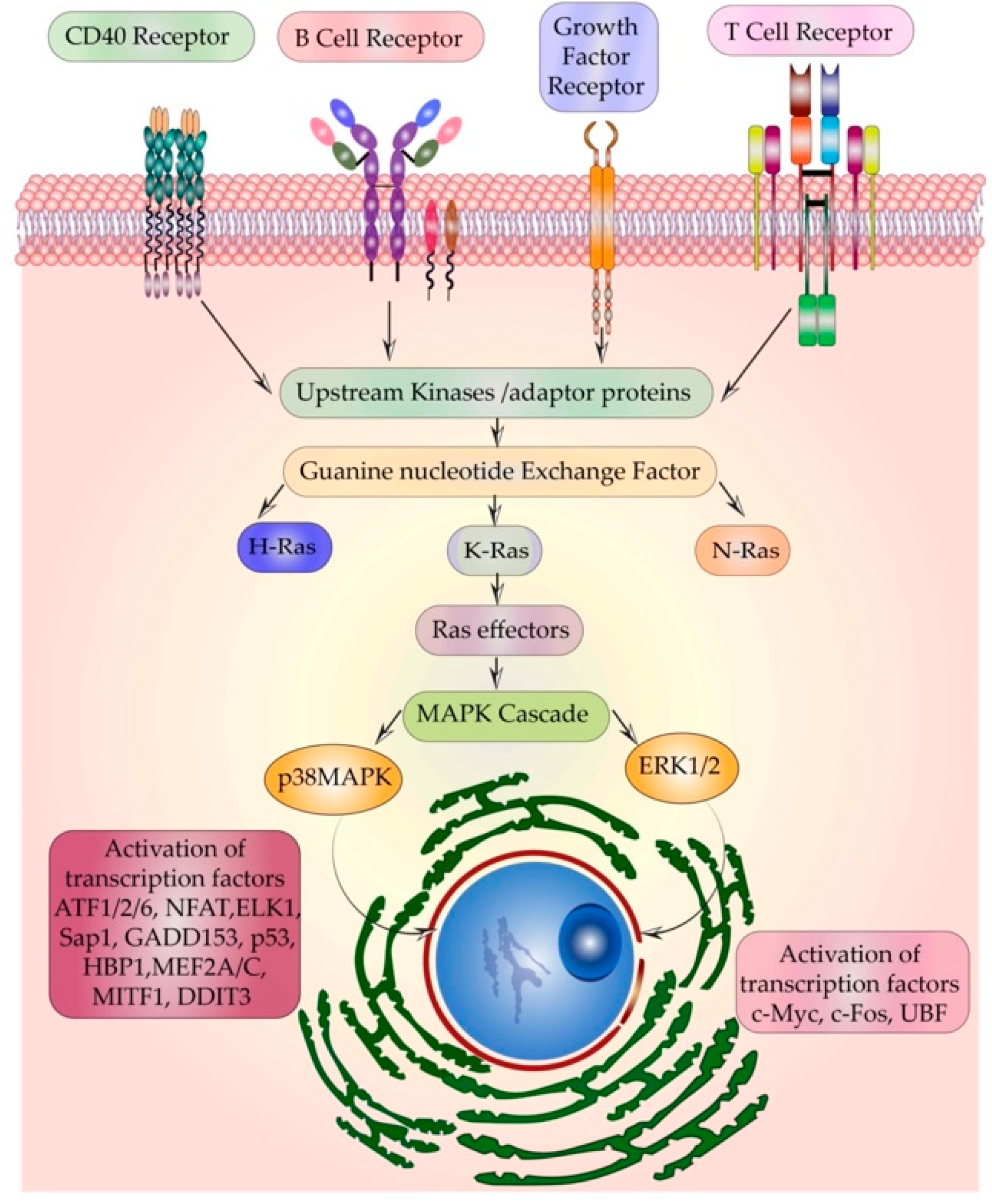
5.1. Multiple Receptors Activating the Same Relay Protein-Ras
5.2. Ras Isoform-Specific Roles in Embryonic Development
5.3. Ras and Infection
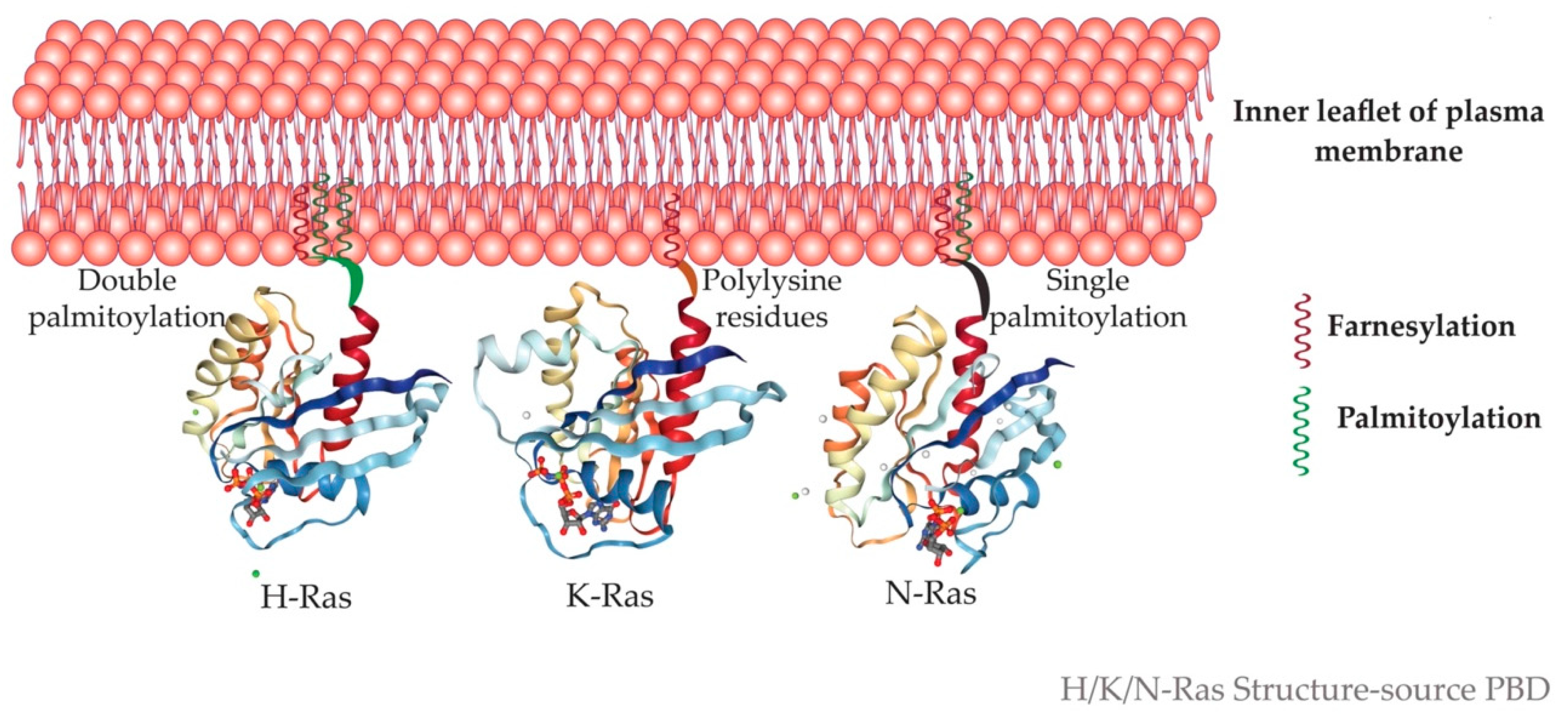
5.4. Isoforms Yet Non-Identical–Subtle Structural Difference in-Ras Isoforms—The G-Domain Isn’t Identical
5.5. Microdomain Localization of Ras Isoforms
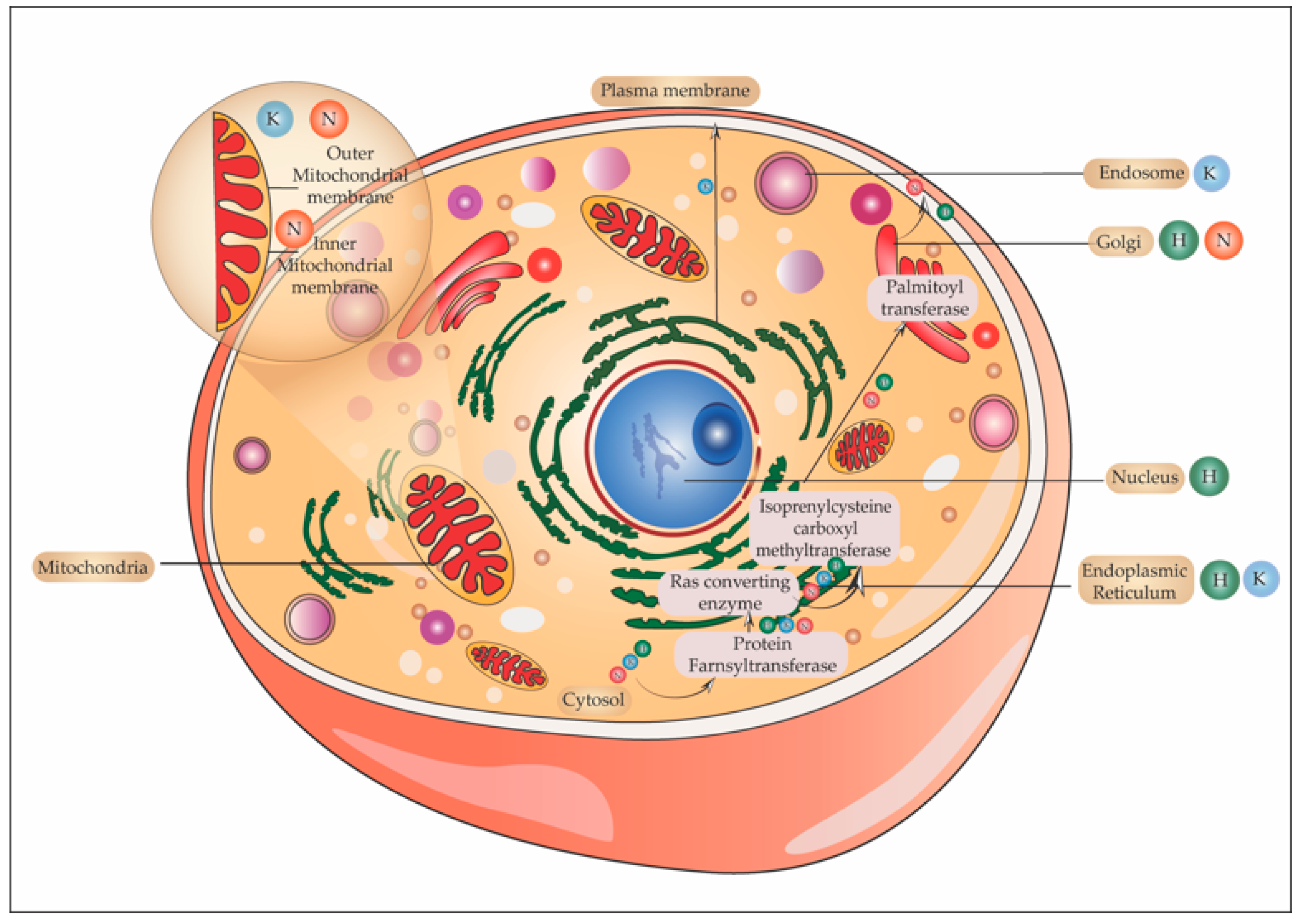
5.6. One Ras and Many Locations
5.6.1. Ras in Golgi
5.6.2. Ras and Endoplasmic Reticulum
5.6.3. Ras in the Endosome
5.6.4. Ras and Mitochondria
5.7. Differential Access and Preference to Activators and Effectors
- Different activation kinetics of Ras isoforms under the same receptor;
- Spatiotemporal segregation of signaling complex, with activators and effectors having different space, time, and thresholds of activation;
- Specific enrichment of substrates for activators or effectors in the microdomains containing specific Ras isoform.
6. Future Recommendations for Devising Isoform-Specific Targeting
Author Contributions
Funding
Conflicts of Interest
References
- Norden, P.; Kim, D.J.; Barry, D.M.; Cleaver, O.B.; Davis, G.E. Cdc42 and k-Ras Control Endothelial Tubulogenesis through Apical Membrane and Cytoskeletal Polarization: Novel Stimulatory Roles for GTPase Effectors, the Small GTPases, Rac2 and Rap1b, and Inhibitory Influence of Arhgap31 and Rasa1. PLoS ONE 2016, 11, e0147758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Haastert, P.J.; Keizer-Gunnink, I.; Kortholt, A. Coupled excitable Ras and F-actin activation mediates spontaneous pseudopod formation and directed cell movement. Mol. Biol. Cell. 2017, 28, 922–934. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.J. An unidentified virus which causes the rapid production of tumors in mice. Nature 1964, 204, 1104–1105. [Google Scholar] [CrossRef] [PubMed]
- Kirsten, W.H.; Mayer, L.A. Morphologic Responses to a Murine Erythroblastosis Virus2. J. Natl. Cancer Inst. 1967, 39, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Structure of Ras-April 2012, David Goodsell. Available online: https://pdb101.rcsb.org/motm/148 (accessed on 30 May 2021).
- Scolnick, E.M.; Parks, W.P. Harvey sarcoma virus: A second murine type C sarcoma virus with rat genetic information. J. Virol. 1974, 13, 1211–1219. [Google Scholar] [CrossRef] [Green Version]
- Stehelin, D.; Varmus, E.H.; Bishop, J.M.; Vogt, P.K. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nat. Cell Biol. 1976, 260, 170–173. [Google Scholar] [CrossRef]
- Goldfarb, M.P.; Weinberg, R.A. Generation of novel, biologically active Harvey sarcoma viruses via apparent illegitimate recombination. J. Virol. 1981, 38, 136–150. [Google Scholar] [CrossRef] [Green Version]
- Swanstrom, R.; Parker, R.C.; Varmus, H.E.; Bishop, J.M. Transduction of a cellular oncogene: The genesis of Rous sarcoma virus. Proc. Natl. Acad. Sci. USA 1983, 80, 2519–2523. [Google Scholar] [CrossRef] [Green Version]
- Swain, A.; Coffin, J.M. Mechanism of transduction by retroviruses. Science 1992, 255, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Willingham, M.C.; Pastan, I.; Shih, T.Y.; Scolnick, E.M. Localization of the src gene product of the Harvey strain of MSV to plasma membrane of transformed cells by electron microscopic immunocytochemistry. Cell 1980, 19, 1005–1014. [Google Scholar] [CrossRef]
- Ellis, R.W.; Defeo, D.; Shih, T.Y.; Gonda, M.A.; Young, H.A.; Tsuchida, N.; Lowy, D.R.; Scolnick, E.M. The p21 src genes of Harvey and Kirsten sarcoma viruses originate from divergent members of a family of normal vertebrate genes. Nat. Cell Biol. 1981, 292, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, J.B.; Sigal, I.S.; Poe, M.; Scolnick, E.M. Intrinsic GTPase activity distinguishes normal and oncogenic ras p21 molecules. Proc. Natl. Acad. Sci. USA 1984, 81, 5704–5708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buhrman, G.; Holzapfel, G.; Fetics, S.; Mattos, C. Allosteric modulation of Ras positions Q61 for a direct role in catalysis. Proc. Natl. Acad. Sci. USA 2010, 107, 4931–4936. [Google Scholar] [CrossRef] [Green Version]
- Hancock, J.F.; Cadwallader, K.; Paterson, H.; Marshall, C.J. A CAAX or a CAAL motif and a second signal are sufficient for plasma membrane targeting of ras proteins. EMBO J. 1991, 10, 4033–4039. [Google Scholar] [CrossRef]
- Di, Y.; Schroeder, D.C.; Highfield, A.; Readman, J.W.; Jha, A.N. Tissue-specific expression of p53 and ras genes in response to the environmental genotoxicant benzo (α) pyrene in marine mussels. Environ. Sci. Technol. 2011, 45, 8974–8981. [Google Scholar] [CrossRef] [PubMed]
- Leon, J.; Guerrero, I.; Pellicer, A. Differential expression of the ras gene family in mice. Mol. Cell. Biol. 1987, 7, 1535–1540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollock, C.B.; Shirasawa, S.; Sasazuki, T.; Kolch, W.; Dhillon, A.S. Oncogenic K-RAS Is Required to Maintain Changes in Cytoskeletal Organization, Adhesion, and Motility in Colon Cancer Cells. Cancer Res. 2005, 65, 1244–1250. [Google Scholar] [CrossRef] [Green Version]
- Gideon, P.; John, J.; Frech, M.; Lautwein, A.; Clark, R.; Scheffler, E.J.; Wittinghofer, A. Mutational and kinetic analyses of the GTPase-activating protein (GAP)-p21 interaction: The C-terminal domain of GAP is not sufficient for full activity. Mol. Cell. Biol. 1992, 12, 2050–2056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigil, D.; Cherfils, J.; Rossman, K.L.; Der, C.J. Ras superfamily GEFs and GAPs: Validated and tractable targets for cancer therapy? Nat. Rev. Cancer 2010, 10, 842–857. [Google Scholar] [CrossRef] [Green Version]
- Nair, A.; Chakraborty, S.; Banerji, L.A.; Srivastava, A.; Navare, C.; Saha, B. Ras isoforms: Signaling specificities in CD40 pathway. Cell Commun. Signal. 2020, 18, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, L.; Tam, A.; Pomerantz, J.; Wong, M.; Holash, J.; Bardeesy, N.; Shen, Q.; O’Hagan, R.; Pantginis, J.; Zhou, H.; et al. Essential role for oncogenic Ras in tumor maintenance. Nature 1999, 400, 468–472. [Google Scholar] [CrossRef]
- Fisher, G.H.; Wellen, S.L.; Klimstra, D.; Lenczowski, J.M.; Tichelaar, J.W.; Lizak, M.J.; Whitsett, J.A.; Koretsky, A.; Varmus, H.E. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001, 15, 3249–3262. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.D.; Der, C.J. The dark side of Ras: Regulation of apoptosis. Oncogene 2003, 22, 8999–9006. [Google Scholar] [CrossRef]
- Rodenhuis, S. Ras and human tumors. Semin. Cancer Biol. 1992, 3, 241–247. [Google Scholar]
- Al-Kali, A.; Quintás-Cardama, A.; Luthra, R.; Bueso-Ramos, C.; Pierce, S.; Kadia, T.; Borthakur, G.; Estrov, Z.; Jabbour, E.; Faderl, S.; et al. Prognostic impact of RAS mutations in patients with myelodysplastic syndrome. Am. J. Hematol. 2013, 88, 365–369. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic signaling pathways in the cancer genome atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eblen, S.T. Extracellular-Regulated Kinases: Signaling from Ras to ERK Substrates to Control Biological Outcomes. Adv. Cancer Res. 2018, 99–142. [Google Scholar] [CrossRef]
- González-García, A.; Pritchard, C.A.; Paterson, H.F.; Mavria, G.; Stamp, G.; Marshall, C.J. RalGDS is required for tumor formation in a model of skin carcinogenesis. Cancer Cell 2005, 7, 219–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wortzel, I.; Seger, R. The ERK Cascade: Distinct Functions within Various Subcellular Organelles. Genes Cancer 2011, 2, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Catling, A.D.; Schaeffer, H.J.; Reuter, C.W.; Reddy, G.R.; Weber, M.J. A proline-rich sequence unique to MEK1 and MEK2 is required for raf binding and regulates MEK function. Mol. Cell. Biol. 1995, 15, 5214–5225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanoue, T.; Adachi, M.; Moriguchi, T.; Nishida, E. A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat. Cell Biol. 2000, 2, 110–116. [Google Scholar] [CrossRef]
- Jaaro, H.; Rubinfeld, H.; Hanoch, T.; Seger, R. Nuclear translocation of mitogen-activated protein kinase kinase (MEK1) in response to mitogenic stimulation. Proc. Natl. Acad. Sci. USA 1997, 94, 3742–3747. [Google Scholar] [CrossRef] [Green Version]
- Payne, D.M.; Rossomando, A.J.; Martino, P.; Erickson, A.K.; Her, J.H.; Shabanowitz, J.; Hunt, D.F.; Weber, M.J.; Sturgill, T.W. Identification of the regulatory phosphorylation sites in pp42/mitogen-activated protein kinase (MAP kinase). EMBO J. 1991, 10, 885–892. [Google Scholar] [CrossRef]
- Ahn, N.G.; Seger, R.; Bratlien, R.L.; Diltz, C.D.; Tonks, N.K.; Krebs, E.G. Multiple components in an epidermal growth factor-stimulated protein kinase cascade. In vitro activation of a myelin basic protein/microtubule-associated protein 2 kinase. J. Biol. Chem. 1991, 266, 4220–4227. [Google Scholar] [CrossRef]
- Ünal, E.B.; Uhlitz, F.; Blüthgen, N. A compendium of ERK targets. FEBS Lett. 2017, 591, 2607–2615. [Google Scholar] [CrossRef] [Green Version]
- Maik-Rachline, G.; Hacohen-Lev-Ran, A.; Seger, R. Nuclear ERK: Mechanism of Translocation, Substrates, and Role in Cancer. Int. J. Mol. Sci. 2019, 20, 1194. [Google Scholar] [CrossRef] [Green Version]
- Chuderland, D.; Konson, A.; Seger, R. Identification and Characterization of a General Nuclear Translocation Signal in Signaling Proteins. Mol. Cell 2008, 31, 850–861. [Google Scholar] [CrossRef]
- Zehorai, E.; Yao, Z.; Plotnikov, A.; Seger, R. The subcellular localization of MEK and ERK—A novel nuclear translocation signal (NTS) paves a way to the nucleus. Mol. Cell. Endocrinol. 2010, 314, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.-F.; Ng, S.-Y. Functional divergence of the MAP kinase pathway ERK1 and ERK2 activate specific transcription factors. FEBS Lett. 1994, 346, 229–234. [Google Scholar] [CrossRef] [Green Version]
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Stefanovsky, V.; Langlois, F.; Gagnon-Kugler, T.; Rothblum, L.I.; Moss, T. Growth Factor Signaling Regulates Elongation of RNA Polymerase I Transcription in Mammals via UBF Phosphorylation and r-Chromatin Remodeling. Mol. Cell 2006, 21, 629–639. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [Green Version]
- McDermott, E.P.; O’Neill, L.A.J. Ras Participates in the Activation of p38 MAPK by Interleukin-1 by Associating with IRAK, IRAK2, TRAF6, and TAK-1. J. Biol. Chem. 2002, 277, 7808–7815. [Google Scholar] [CrossRef] [Green Version]
- Morrison, D.K.; Davis, R.J. Regulation of MAP Kinase Signaling Modules by Scaffold Proteins in Mammals. Annu. Rev. Cell Dev. Biol. 2003, 19, 91–118. [Google Scholar] [CrossRef]
- Hazzalin, C.A.; Cano, E.; Cuenda, A.; Barratt, M.J.; Cohen, P.; Mahadevan, L.C. p38/RK is essential for stress-induced nuclear responses: JNK/SAPKs and c-Jun/ATF-2 phosphorylation are insufficient. Curr. Biol. 1996, 6, 1028–1031. [Google Scholar] [CrossRef] [Green Version]
- del Arco, P.G.; Martínez-Martínez, S.; Maldonado, J.L.; Ortega-Pérez, I.; Redondo, J.M. A role for the p38 MAP kinase pathway in the nuclear shuttling of NFATp. J. Biol. Chem. 2000, 275, 13872–13878. [Google Scholar] [CrossRef] [Green Version]
- Janknecht, R.; Hunter, T. Convergence of MAP kinase pathways on the ternary complex factor Sap-1a. EMBO J. 1997, 16, 1620–1627. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Ron, D. Stress-Induced Phosphorylation and Activation of the Transcription Factor CHOP (GADD153) by p38 MAP Kinase. Science 1996, 272, 1347–1349. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Ma, W.-Y.; Maxiner, A.; Sun, Y.; Dong, Z. p38 Kinase Mediates UV-induced Phosphorylation of p53 Protein at Serine 389. J. Biol. Chem. 1999, 274, 12229–12235. [Google Scholar] [CrossRef] [Green Version]
- Yee, A.S.; Paulson, E.K.; McDevitt, M.A.; Rieger-Christ, K.; Summerhayes, I.; Berasi, S.P.; Kim, J.; Huang, C.-Y.; Zhang, X. The HBP1 transcriptional repressor and the p38 MAP kinase: Unlikely partners in G1 regulation and tumor suppression. Gene 2004, 336, 1–13. [Google Scholar] [CrossRef]
- Zhao, M.; New, L.; Kravchenko, V.V.; Kato, Y.; Gram, H.; Di Padova, F.; Olson, E.N.; Ulevitch, R.J.; Han, J. Regulation of the MEF2 family of transcription factors by p38. Mol. Cell. Biol. 1999, 19, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Pereira, R.C.; Delany, A.M.; Canalis, E. CCAAT/Enhancer Binding Protein Homologous Protein (DDIT3) Induces Osteoblastic Cell Differentiation. Endocrinol. 2004, 145, 1952–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarubin, T.; Jiahuai, H.A. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005, 15, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Mulcahy, L.S.; Smith, M.R.; Stacey, D.W. Requirement for ras proto-oncogene function during serum-stimulated growth of NIH 3T3 cells. Nat. Cell Biol. 1985, 313, 241–243. [Google Scholar] [CrossRef]
- Lowy, D.R.; Willumsen, B.M. Function and regulation of ras. Annu. Rev. Biochem. 1993, 62, 851–891. [Google Scholar] [CrossRef]
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Filmus, J.; Robles, A.; Shi, W.; Wong, M.J.; Colombo, L.L.; Conti, C.J. Induction of cyclin D1 overexpression by activated ras. Oncogene 1994, 9, 3627–3633. [Google Scholar]
- Albanese, C.; Johnson, J.; Watanabe, G.; Eklund, N.; Vu, D.; Arnold, A.; Pestell, R.G. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J. Biol. Chem. 1995, 270, 23589–23597. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.J.; Chao, J.R.; Jiang, M.C.; Ng, S.Y.; Yen, J.J.; Yang-Yen, H.F. Ras transformation results in an elevated level of cyclin D1 and acceleration of G1 progression in NIH 3T3 cells. Mol. Cell. Biol. 1995, 15, 3654–3663. [Google Scholar] [CrossRef] [Green Version]
- Lavoie, J.N.; L’Allemain, G.; Brunet, A.; Müller, R.; Pouysségur, J. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J. Biol. Chem. 1996, 271, 20608–20616. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.S.; Krauss, R.S. Ras induces anchorage-independent growth by subverting multiple adhesion-regulated cell cycle events. Mol. Cell. Biol. 1996, 16, 3370–3380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aktas, H.; Cai, H.; Cooper, G.M. Ras links growth factor signaling to the cell cycle machinery via regulation of cyclin D1 and the Cdk inhibitor p27KIP1. Mol. Cell. Biol. 1997, 17, 3850–3857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawada, M.; Yamagoe, S.; Murakami, Y.; Suzuki, K.; Mizuno, S.; Uehara, Y. Induction of p27Kip1 degradation and anchorage independence by Ras through the MAP kinase signaling pathway. Oncogene 1997, 15, 629–637. [Google Scholar] [CrossRef] [Green Version]
- Serrano, M.; Lin, A.W.; McCurrach, E.M.; Beach, D.; Lowe, S.W. Oncogenic ras Provokes Premature Cell Senescence Associated with Accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.-S.; Lee, E.-J.; Kim, H.-R.C.; Moon, A. p38 kinase is a key signaling molecule for H-Ras-induced cell motility and invasive phenotype in human breast epithelial cells. Cancer Res. 2003, 63, 5454–5461. [Google Scholar]
- De Vos, A.M.; Tong, L.; Milburn, M.V.; Matias, P.M.; Jancarik, J.; Noguchi, S.; Nishimura, S.; Miura, K.; Ohtsuka, E.; Kim, S.H. Three-dimensional structure of an oncogene protein: Catalytic domain of human cH-ras p21. Science 1988, 239, 888–893. [Google Scholar] [CrossRef]
- Pai, E.; Kabsch, W.; Krengel, U.; Holmes, K.C.; John, J.; Wittinghofer, A. Structure of the guanine-nucleotide-binding domain of the Ha-ras oncogene product p21 in the triphosphate conformation. Nat. Cell Biol. 1989, 341, 209–214. [Google Scholar] [CrossRef]
- Zhao, L.; Lobo, S.; Dong, X.; Ault, A.D.; Deschenes, R. Erf4p and Erf2p Form an Endoplasmic Reticulum-associated Complex Involved in the Plasma Membrane Localization of Yeast Ras Proteins. J. Biol. Chem. 2002, 277, 49352–49359. [Google Scholar] [CrossRef] [Green Version]
- Prior, I.A.; Hancock, J.F. Ras trafficking, localization and compartmentalized signalling. Semin. Cell Dev. Biol. 2012, 23, 145–153. [Google Scholar] [CrossRef] [Green Version]
- Leventis, R.; Silvius, J.R. Lipid-Binding Characteristics of the Polybasic Carboxy-Terminal Sequence of K-ras4B. Biochemistry 1998, 37, 7640–7648. [Google Scholar] [CrossRef]
- McLaughlin, S.; Aderem, A. The myristoyl-electrostatic switch: A modulator of reversible protein-membrane interactions. Trends Biochem. Sci. 1995, 20, 272–276. [Google Scholar] [CrossRef]
- Fetics, S.; Young, M.; Buhrman, G.; Mattos, C. Allosteric modulation of H-Ras GTPase. RCSB PDB 2009. [Google Scholar] [CrossRef]
- Tong, Y.; Tempel, W.; Shen, L.; Arrowsmith, C.H.; Edwards, A.M.; Sundstrom, M.; Weigelt, J.; Bochkarev, A.; Park, H. Human K-Ras (Q61H) in complex with a GTP analogue. RCSB PDB 2009. [Google Scholar] [CrossRef]
- Nedyalkova, L.; Tong, Y.; Tempel, W.; Shen, L.; Loppnau, P.; Arrowsmith, C.H.; Edwards, A.M.; Bountra, C.; Weigelt, J.; Bochkarev, A.; et al. Crystal structure of the human NRAS GTPase bound with GDP. RCSB PDB 2008. [Google Scholar] [CrossRef]
- Rose, A.S.; Bradley, A.R.; Valasatava, Y.; Duarte, J.M.; Prlić, A.; Rose, P.W. NGL viewer: Web-based molecular graphics for large complexes. Bioinformatics 2018, 34, 3755–3758. [Google Scholar] [CrossRef] [Green Version]
- Baum, C.; Kirschmeier, P. Preclinical and clinical evaluation of farnesyltransferase inhibitors. Curr. Oncol. Rep. 2003, 5, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Whyte, D.B.; Kirschmeier, P.; Hockenberry, T.N.; Nunez-Oliva, I.; James, L.; Catino, J.J.; Bishop, W.R.; Pai, J.-K. K- and N-Ras Are Geranylgeranylated in Cells Treated with Farnesyl Protein Transferase Inhibitors. J. Biol. Chem. 1997, 272, 14459–14464. [Google Scholar] [CrossRef] [Green Version]
- Liu, V.; Sjogren, A.-K.M.; Karlsson, C.; Ibrahim, M.X.; Andersson, K.; Olofsson, F.J.; Wahlstrom, A.M.; Dalin, M.; Yu, H.; Chen, Z.; et al. Targeting the protein prenyltransferases efficiently reduces tumor development in mice with K-RAS-induced lung cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 6471–6476. [Google Scholar] [CrossRef] [Green Version]
- Lobell, R.B.; Omer, A.C.; Abrams, M.T.; Bhimnathwala, H.G.; Brucker, M.J.; Buser, A.C.; Davide, J.P.; Desolms, S.J.; Dinsmore, C.J.; Ellis-Hutchings, M.S.; et al. Evaluation of farnesyl:protein transferase and geranylgeranyl: Protein transferase inhibitor combinations in preclinical models. Cancer Res. 2001, 61, 8758–8768. [Google Scholar]
- Sinensky, M.; Beck, L.A.; Leonard, S.; Evans, R. Differential inhibitory effects of lovastatin on protein isoprenylation and sterol synthesis. J. Biol. Chem. 1990, 265, 19937–19941. [Google Scholar] [CrossRef]
- Cho, K.-J.; Hill, M.M.; Chigurupati, S.; Du, G.; Parton, R.G.; Hancock, J.F. Therapeutic Levels of the Hydroxmethylglutaryl-Coenzyme A Reductase Inhibitor Lovastatin Activate Ras Signaling via Phospholipase D2. Mol. Cell. Biol. 2011, 31, 1110–1120. [Google Scholar] [CrossRef] [Green Version]
- Bergo, M.O.; Ambroziak, P.; Gregory, C.; George, A.; Otto, J.C.; Kim, E.; Nagase, H.; Casey, P.J.; Balmain, A.; Young, S.G. Absence of the CAAX Endoprotease Rce1: Effects on Cell Growth and Transformation. Mol. Cell. Biol. 2002, 22, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Wahlstrom, A.M.; Cutts, B.A.; Karlsson, C.; Andersson, K.M.E.; Liu, M.; Sjogren, A.-K.M.; Swolin, B.; Young, S.G.; Bergo, M.O. Rce1 deficiency accelerates the development of K-RAS–induced myeloproliferative disease. Blood 2006, 109, 763–768. [Google Scholar] [CrossRef] [Green Version]
- Wahlstrom, A.M.; Cutts, B.A.; Liu, V.; Lindskog, A.; Karlsson, C.; Sjogren, A.-K.M.; Andersson, K.; Young, S.; Bergo, M.O. Inactivating Icmt ameliorates K-RAS–induced myeloproliferative disease. Blood 2008, 112, 1357–1365. [Google Scholar] [CrossRef] [Green Version]
- Court, H.; Amoyel, M.; Hackman, M.; Lee, K.E.; Xu, R.; Miller, G.; Bar-Sagi, D.; Bach, E.A.; Bergã, M.O.; Philips, M.R. Isoprenylcysteine carboxylmethyltransferase deficiency exacerbates KRAS-driven pancreatic neoplasia via Notch suppression. J. Clin. Investig. 2013, 123, 4681–4694. [Google Scholar] [CrossRef] [Green Version]
- Bergo, M.O.; Lieu, H.D.; Gavino, B.J.; Ambroziak, P.; Otto, J.C.; Casey, P.J.; Walker, Q.M.; Young, S.G. On the physiological importance of endoproteolysis of CAAX proteins: Heart-specific RCE1 knockout mice develop a lethal cardiomyopathy. J. Biol. Chem. 2004, 279, 4729–4736. [Google Scholar] [CrossRef] [Green Version]
- Christiansen, J.R.; Kolandaivelu, S.; Bergo, M.O.; Ramamurthy, V. RAS-converting enzyme 1-mediated endoproteolysis is required for trafficking of rod phosphodiesterase 6 to photoreceptor outer segments. Proc. Natl. Acad. Sci. USA 2011, 108, 8862–8866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Jiao, B.; Zhang, R.; Zhao, H.; Zhang, C.; Wu, M.; Li, D.; Zhao, X.; Qiu, Q.; Li, J.; et al. Palmitoylacyltransferase Zdhhc9 inactivation mitigates leukemogenic potential of oncogenic Nras. Leukemia 2015, 30, 1225–1228. [Google Scholar] [CrossRef]
- Dekker, F.; Rocks, O.; Vartak, N.; Menninger, S.; Hedberg, C.; Balamurugan, R.; Wetzel, S.; Renner, S.; Gerauer, M.; Schölermann, B.; et al. Small-molecule inhibition of APT1 affects Ras localization and signaling. Nat. Chem. Biol. 2010, 6, 449–456. [Google Scholar] [CrossRef]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [Green Version]
- Patgiri, A.; Yadav, K.K.; Arora, P.S.; Bar-Sagi, D. An orthosteric inhibitor of the Ras-Sos interaction. Nat. Chem. Biol. 2011, 7, 585–587. [Google Scholar] [CrossRef] [Green Version]
- Roberts, E.A.; Araki, T.; Swanson, K.D.; Montgomery, K.T.; Schiripo, A.T.; Joshi, A.V.; Li, L.; Yassin, Y.; Tamburino, A.M.; Neel, B.G.; et al. Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat. Genet. 2006, 39, 70–74. [Google Scholar] [CrossRef]
- Rojas, J.M.; Oliva, J.L.; Santos, E. Mammalian Son of Sevenless Guanine Nucleotide Exchange Factors: Old Concepts and New Perspectives. Genes Cancer 2011, 2, 298–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteban, L.M.; Fernández-Medarde, A.; López, E.; Yienger, K.; Guerrero, C.; Ward, J.M.; Tessarollo, L.; Santos, E. Ras-guanine nucleotide exchange factor sos2 is dispensable for mouse growth and development. Mol. Cell. Biol. 2000, 20, 6410. [Google Scholar] [CrossRef]
- Baltanás, F.C.; Pérez-Andrés, M.; Ginel-Picardo, A.; Díaz, D.; Jimeno, D.; Liceras-Boillos, P.; Kortum, R.L.; Samelson, L.E.; Orfao, A.; Santos, E. Functional Redundancy of Sos1 and Sos2 for Lymphopoiesis and Organismal Homeostasis and Survival. Mol. Cell. Biol. 2013, 33, 4562–4578. [Google Scholar] [CrossRef] [Green Version]
- Lepri, F.; De Luca, A.; Stella, L.; Rossi, C.; Baldassarre, G.; Pantaleoni, F.; Cordeddu, V.; Williams, B.J.; Dentici, M.L.; Caputo, V.; et al. SOS1 mutations in Noonan syndrome: Molecular spectrum, structural insights on pathogenic effects, and genotype-phenotype correlations. Hum. Mutat. 2011, 32, 760–772. [Google Scholar] [CrossRef] [Green Version]
- Beneteau, C.; Cavé, H.; Moncla, A.; Dorison, N.; Munnich, A.; Verloes, A.; Leheup, B. SOS1 and PTPN11 mutations in five cases of Noonan syndrome with multiple giant cell lesions. Eur. J. Hum. Genet. 2009, 17, 1216–1221. [Google Scholar] [CrossRef]
- Sondermann, H.; Nagar, B.; Bar-Sagi, D.; Kuriyan, J. Computational docking and solution X-ray scattering predict a membrane-interacting role for the histone domain of the Ras activator son of sevenless. Proc. Natl. Acad. Sci USA 2005, 102, 16632–16637. [Google Scholar] [CrossRef] [Green Version]
- Cordeddu, V.; Yin, J.C.; Gunnarsson, C.; Virtanen, C.; Drunat, S.; Lepri, F.; De Luca, A.; Rossi, C.; Ciolfi, A.; Pugh, T.J.; et al. Activating Mutations Affecting the Dbl Homology Domain of SOS2 Cause Noonan Syndrome. Hum. Mutat. 2015, 36, 1080–1087. [Google Scholar] [CrossRef] [Green Version]
- Hart, T.C.; Zhang, Y.; Gorry, M.C.; Hart, P.S.; Cooper, M.; Marazita, M.L.; Marks, J.M.; Cortelli, J.R.; Pallos, D. A Mutation in the SOS1 Gene Causes Hereditary Gingival Fibromatosis Type 1. Am. J. Hum. Genet. 2002, 70, 943–954. [Google Scholar] [CrossRef] [Green Version]
- Owens, M.; Kivuva, E.; Quinn, A.; Brennan, P.; Caswell, R.; Allen, H.L.; Vaidya, B.; Ellard, S. SOS1frameshift mutations cause pure mucosal neuroma syndrome, a clinical phenotype distinct from multiple endocrine neoplasia type 2B. Clin. Endocrinol. 2015, 84, 715–719. [Google Scholar] [CrossRef]
- Gupta, S.; Ramjaun, A.R.; Haiko, P.; Wang, Y.; Warne, P.H.; Nicke, B.; Nye, E.; Stamp, G.; Alitalo, K.; Downward, J. Binding of Ras to Phosphoinositide 3-Kinase p110α Is Required for Ras- Driven Tumorigenesis in Mice. Cell 2007, 129, 957–968. [Google Scholar] [CrossRef] [Green Version]
- Gulbins, E.; Brenner, B.; Schlottmann, K.; Koppenhoefer, U.; Linderkamp, O.; Coggeshall, K.M.; Lang, F. Activation of the Ras signaling pathway by the CD40 receptor. J. Immunol. 1996, 157, 2844–2850. [Google Scholar] [PubMed]
- Downward, J.; Graves, J.D.; Warne, P.H.; Rayter, S.; Cantrell, D.A. Stimulation of p21ras upon T-cell activation. Nat. Cell Biol. 1990, 346, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Swan, K.A.; Alberola-Ila, J.; Gross, J.A.; Appleby, M.W.; Forbush, K.A.; Thomas, J.F.; Perlmutter, R.M. Involvement of p21ras distinguishes positive and negative selection in thymocytes. EMBO J. 1995, 14, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Ebinu, J.O.; Stang, S.L.; Teixeira, C.; Bottorff, D.A.; Hooton, J.; Blumberg, P.M.; Barry, M.; Bleakley, R.C.; Ostergaard, H.L.; Stone, J.C. RasGRP links T-cell receptor signaling to Ras. Blood J. Am. Soc. Hematol. 2000, 95, 3199–3203. [Google Scholar]
- Crabtree, G. Contingent genetic regulatory events in T lymphocyte activation. Science 1989, 243, 355–361. [Google Scholar] [CrossRef]
- Jacob, A.; Cooney, D.; Pradhan, M.; Coggeshall, K.M. Convergence of Signaling Pathways on the Activation of ERK in B Cells. J. Biol. Chem. 2002, 277, 23420–23426. [Google Scholar] [CrossRef] [Green Version]
- Roose, J.P.; Mollenauer, M.; Ho, M.; Kurosaki, T.; Weiss, A. Unusual Interplay of Two Types of Ras Activators, RasGRP and SOS, Establishes Sensitive and Robust Ras Activation in Lymphocytes. Mol. Cell. Biol. 2007, 27, 2732–2745. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; Kim, H.G.; Kim, E.; Han, S.Y.; Aziz, N.; Yi, Y.-S.; Kim, S.; Lee, Y.; Yoo, B.C.; Han, J.-W.; et al. Isoprenylcysteine Carboxyl Methyltransferase and Its Substrate Ras Are Critical Players Regulating TLR-Mediated Inflammatory Responses. Cells 2020, 9, 1216. [Google Scholar] [CrossRef]
- Xu, H.; An, H.; Yu, Y.; Zhang, M.; Qi, R.; Cao, X. Ras Participates in CpG Oligodeoxynucleotide Signaling through Association with Toll-like Receptor 9 and Promotion of Interleukin-1 Receptor-associated Kinase/Tumor Necrosis Factor Receptor-associated Factor 6 Complex Formation in Macrophages. J. Biol. Chem. 2003, 278, 36334–36340. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, S.; Srivastava, A.; Jha, M.K.; Nair, A.; Pandey, S.P.; Srivastava, N.; Kumari, S.; Singh, S.; Krishnasastry, M.V.; Saha, B. Inhibition of CD40-Induced N-Ras Activation Reduces Leishmania major Infection. J. Immunol. 2015, 194, 3852–3860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koera, K.; Nakamura, K.; Nakao, K.; Miyoshi, J.; Toyoshima, K.; Hatta, T.; Otani, H.; Aiba, A.; Katsuki, M. K-Ras is essential for the development of the mouse embryo. Oncogene 1997, 15, 1151–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, K.; Ichise, H.; Nakao, K.; Hatta, T.; Otani, H.; Sakagami, H.; Kondo, H.; Katsuki, M. Partial functional overlap of the three ras genes in mouse embryonic development. Oncogene 2007, 27, 2961–2968. [Google Scholar] [CrossRef] [Green Version]
- Plowman, S.J.; Williamson, D.J.; O’Sullivan, M.J.; Doig, J.; Ritchie, A.-M.; Harrison, D.J.; Melton, D.W.; Arends, M.J.; Hooper, M.L.; Patek, C.E. While K-ras Is Essential for Mouse Development, Expression of the K-ras 4A Splice Variant Is Dispensable. Mol. Cell. Biol. 2003, 23, 9245–9250. [Google Scholar] [CrossRef] [Green Version]
- Fuentes-Mateos, R.; Jimeno, D.; Gómez, C.; Calzada, N.; Fernández-Medarde, A.; Santos, E. Concomitant deletion of HRAS and NRAS leads to pulmonary immaturity, respiratory failure and neonatal death in mice. Cell Death Dis. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Jha, M.K.; Sarode, A.Y.; Saha, B. Ras isoforms selectively regulate antigen-specific immune response. Cytokine 2020, 126, 154914. [Google Scholar] [CrossRef]
- Ballester, R.; Furth, M.E.; Rosen, O.M. Phorbol ester-and protein kinase C-mediated phosphorylation of the cellular Kirste N-Ras gene product. J. Biol. Chem. 1987, 262, 2688–2695. [Google Scholar] [CrossRef]
- Parker, J.A.; Mattos, C. The K-Ras, N-Ras, and H-Ras Isoforms: Unique Conformational Preferences and Implications for Targeting Oncogenic Mutants. Cold Spring Harb. Perspect. Med. 2017, 8, a031427. [Google Scholar] [CrossRef]
- Parker, J.A.; Mattos, C. The Ras–Membrane Interface: Isoform-Specific Differences in the Catalytic Domain. Mol. Cancer Res. 2015, 13, 595–603. [Google Scholar] [CrossRef] [Green Version]
- Buhrman, G.; O′connor, C.; Zerbe, B.; Kearney, B.M.; Napoleon, R.; Kovrigina, E.A.; Vajda, S.; Kozakov, D.; Kovrigin, E.L.; Mattos, C. Analysis of Binding Site Hot Spots on the Surface of Ras GTPase. J. Mol. Biol. 2011, 413, 773–789. [Google Scholar] [CrossRef] [Green Version]
- Kearney, B.M.; Johnson, C.W.; Roberts, D.M.; Swartz, P.; Mattos, C. DRoP: A Water Analysis Program Identifies Ras-GTP-Specific Pathway of Communication between Membrane-Interacting Regions and the Active Site. J. Mol. Biol. 2014, 426, 611–629. [Google Scholar] [CrossRef]
- Johnson, C.W.; Reid, D.; Parker, J.A.; Salter, S.; Knihtila, R.; Kuzmic, P.; Mattos, C. The small GTPases K-Ras, N-Ras, and H-Ras have distinct biochemical properties determined by allosteric effects. J. Biol. Chem. 2017, 292, 12981–12993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, C.; Kovrigin, E.L. Global Conformational Dynamics in Ras. Biochemistry 2008, 47, 10244–10246. [Google Scholar] [CrossRef]
- Pereyra, M.; Méndez, E. The protein surface as a thermodynamic frontier: A fractal approach. Appl. Thermodyn. Biol. Mater. Sci. 2011, 243–258, Chapter 9. [Google Scholar] [CrossRef] [Green Version]
- Rotblat, B.; Prior, I.; Muncke, C.; Parton, R.; Kloog, Y.; Henis, Y.I.; Hancock, J.F. Three Separable Domains Regulate GTP-Dependent Association of H-ras with the Plasma Membrane. Mol. Cell. Biol. 2004, 24, 6799–6810. [Google Scholar] [CrossRef] [Green Version]
- Plowman, S.; Hancock, J. Ras signaling from plasma membrane and endomembrane microdomains. Biochim. Biophys. Acta (BBA) Bioenerg. 2005, 1746, 274–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plowman, S.J.; Muncke, C.; Parton, R.; Hancock, J.F. H-ras, K-ras, and inner plasma membrane raft proteins operate in nanoclusters with differential dependence on the actin cytoskeleton. Proc. Natl. Acad. Sci. USA 2005, 102, 15500–15505. [Google Scholar] [CrossRef] [Green Version]
- Hancock, J.F.; Parton, R. Ras plasma membrane signalling platforms. Biochem. J. 2005, 389, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunsveld, L.; Waldmann, H.; Huster, D. Membrane binding of lipidated Ras peptides and proteins—The structural point of view. Biochim. Biophys. Acta (BBA) Biomembr. 2009, 1788, 273–288. [Google Scholar] [CrossRef] [Green Version]
- Welman, A.; Burger, M.M.; Hagmann, J. Structure and function of the C-terminal hypervariable region of K-Ras4B in plasma membrane targetting and transformation. Oncogene 2000, 19, 4582–4591. [Google Scholar] [CrossRef] [Green Version]
- Elad-Sfadia, G.; Haklai, R.; Balan, E.; Kloog, Y. Galectin-3 Augments K-Ras Activation and Triggers a Ras Signal That Attenuates ERK but Not Phosphoinositide 3-Kinase Activity. J. Biol. Chem. 2004, 279, 34922–34930. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.; Mu, H.; Jean-Francois, F.; Lakshman, B.; Sarkar-Banerjee, S.; Zhuang, Y.; Zeng, Y.; Gao, W.; Zaske, A.M.; Nissley, D.V.; et al. Membrane curvature sensing of the lipid-anchored K-Ras small GTPase. Life Sci. Alliance 2019, 2, e201900343. [Google Scholar] [CrossRef]
- Zhu, X.; Assoian, R.K. Integrin-dependent activation of MAP kinase: A link to shape-dependent cell proliferation. Mol. Biol. Cell 1995, 6, 273–282. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, P.M.; Ghosh-Choudhury, N.; Moyer, M.L.; Mott, E.G.; Thomas, A.C.; Foster, A.B.; Greenberg, N.M.; Kreisberg, I.J. Role of RhoA activation in the growth and morphology of a murine prostate tumor cell line. Oncogene 1999, 18, 4120–4130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augsten, M.; Pusch, R.; Biskup, C.; Rennert, K.; Wittig, U.; Beyer, K.; Blume, A.; Wetzker, R.; Friedrich, K.; Rubio, I. Live-cell imaging of endogenous Ras-GTP illustrates predominant Ras activation at the plasma membrane. EMBO Rep. 2006, 7, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Mitin, N.Y.; Ramocki, M.B.; Zullo, A.J.; Der, C.J.; Konieczny, S.F.; Taparowsky, E.J. Identification and Characterization of Rain, a Novel Ras-interacting Protein with a Unique Subcellular Localization. J. Biol. Chem. 2004, 279, 22353–22361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bivona, T.G.; de Castro, I.P.; Ahearn, I.; Grana, T.M.; Chiu, V.K.; Lockyer, P.J.; Cullen, P.; Pellicer, A.; Cox, A.D.; Philips, M.R. Phospholipase Cγ activates Ras on the Golgi apparatus by means of RasGRP1. Nat. Cell Biol. 2003, 424, 694–698. [Google Scholar] [CrossRef] [PubMed]
- Arozarena, I.; Matallanas, D.; Berciano, M.T.; Sanz-Moreno, V.; Calvo, F.; MuñozM, T.; Egea, G.; Lafarga, M.; Crespo, P. Activation of H-Ras in the Endoplasmic Reticulum by the RasGRF Family Guanine Nucleotide Exchange Factors. Mol. Cell. Biol. 2004, 24, 1516–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, V.K.; Bivona, T.; Hach, A.; Sajous, J.B.; Silletti, J.; Wiener, H.; Ii, R.L.J.; Cox, A.D.; Philips, M.R. Ras signalling on the endoplasmic reticulum and the Golgi. Nat. Cell Biol. 2002, 4, 343–350. [Google Scholar] [CrossRef]
- Rocks, O.; Peyker, A.; Kahms, M.; Verveer, P.J.; Koerner, C.; Lumbierres, M.; Kuhlmann, J.; Waldmann, H.; Wittinghofer, A.; Bastiaens, P.I.H. An Acylation Cycle Regulates Localization and Activity of Palmitoylated Ras Isoforms. Science 2005, 307, 1746–1752. [Google Scholar] [CrossRef]
- Torii, S.; Kusakabe, M.; Yamamoto, T.; Maekawa, M.; Nishida, E. Sef Is a Spatial Regulator for Ras/MAP Kinase Signaling. Dev. Cell 2004, 7, 33–44. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.F.; Ma, Z.; Liu, Z.; Terada, L.S. Nox4-derived H2O2 mediates endoplasmic reticulum signaling through local Ras ac-tivation. Mol. Cell. Biol. 2010, 30, 3553–3568. [Google Scholar] [CrossRef] [Green Version]
- Di Guglielmo, G.M.; Baass, P.C.; Ou, W.J.; Posner, B.I.; Bergeron, J.J. Compartmentalization of SHC, GRB2 and mSOS, and hy-perphosphorylation of Raf-1 by EGF but not insulin in liver parenchyma. EMBO J. 1994, 13, 4269–4277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pol, A.; Calvo, M.; Enrich, C. Isolated endosomes from quiescent rat liver contain the signal transduction machinery: Differential distribution of activated Raf-1 and Mek in the endocytic compartment. FEBS Lett. 1998, 441, 34–38. [Google Scholar] [CrossRef] [Green Version]
- Tsutsumi, K.; Fujioka, Y.; Tsuda, M.; Kawaguchi, H.; Ohba, Y. Visualization of Ras-PI3K interaction in the endosome using BiFC. Cell. Signal. 2009, 21, 1672–1679. [Google Scholar] [CrossRef]
- Lu, A.; Tebar, F.; Alvarez-Moya, B.; Lopez, A.L.; Calvo, M.; Enrich, C.; Agell, N.; Nakamura, T.; Matsuda, M.; Bachs, O. A clathrin-dependent pathway leads to KRas signaling on late endosomes en route to lysosomes. J. Cell Biol. 2009, 184, 863–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrade, A.W.; Silva, A.M.; Alves, V.S.; Salgado, A.P.C.; Melo, M.B.; Andrade, H.M.; Dall’Orto, F.V.; Garcia, A.S.; Silveira, T.N.; Gazzinelli, R.T. Early endosome localization and activity of RasGEF1b, a toll-like receptor-inducible Ras guanine-nucleotide exchange factor. Genes Immun. 2010, 11, 447–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bivona, T.G.; Quatela, S.E.; Bodemann, B.O.; Ahearn, I.; Soskis, M.J.; Mor, A.; Miura, J.; Wiener, H.H.; Wright, L.; Saba, S.G.; et al. PKC Regulates a Farnesyl-Electrostatic Switch on K-Ras that Promotes its Association with Bcl-Xl on Mitochondria and Induces Apoptosis. Mol. Cell 2006, 21, 481–493. [Google Scholar] [CrossRef]
- Wolfman, J.C.; Planchon, S.M.; Liao, J.; Wolfman, A. Structural and functional consequences of cN-Ras constitutively associ-ated with intact mitochondria. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2006, 1763, 1108–1124. [Google Scholar] [CrossRef] [Green Version]
- Rebollo, A.; Pérez-Sala, D.; Martínez-A, C. Bcl-2 differentially targets K-, N-, and H-Ras to mitochondria in IL-2 supple-mented or deprived cells: Implications in prevention of apoptosis. Oncogene 1999, 18, 4930–4939. [Google Scholar] [CrossRef] [Green Version]
- Contente, S.; Yeh, T.-J.A.; Friedman, R.M. H-Ras Localizes to Cell Nuclei and Varies with the Cell Cycle. Genes Cancer 2011, 2, 166–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Roy, S.; Apolloni, A.; Lane, A.; Hancock, J.F. Ras Isoforms Vary in Their Ability to Activate Raf-1 and Phosphoinositide 3-Kinase. J. Biol. Chem. 1998, 273, 24052–24056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollag, G.; McCormick, F. Differential regulation of rasGAP and neurofibromatosis gene product activities. Nat. Cell Biol. 1991, 351, 576–579. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liang, H.; Rodkey, T.; Ariotti, N.; Parton, R.G.; Hancock, J.F. Signal Integration by Lipid-Mediated Spatial Cross Talk between Ras Nanoclusters. Mol. Cell. Biol. 2013, 34, 862–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Du, G.; Skowronek, K.; Frohman, M.A.; Bar-Sagi, D. Phospholipase D2-generated phosphatidic acid couples EGFR stimulation to Ras activation by Sos. Nat. Cell Biol. 2007, 9, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [Green Version]
- Resat, H.; Straatsma, T.P.; Dixon, D.A.; Miller, J.H. The arginine finger of RasGAP helps Gln-61 align the nucleophilic water in GAP-stimulated hydrolysis of GTP. Proc. Natl. Acad. Sci. USA 2001, 98, 6033–6038. [Google Scholar] [CrossRef] [Green Version]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Ihle, N.T.; Byers, L.A.; Kim, E.S.; Saintigny, P.; Lee, J.J.; Blumenschein, G.R.; Tsao, A.; Liu, S.; Larsen, J.E.; Wang, J.; et al. Effect of KRAS Oncogene Substitutions on Protein Behavior: Implications for Signaling and Clinical Outcome. J. Natl. Cancer Inst. 2012, 104, 228–239. [Google Scholar] [CrossRef]
- Smith, M.J.; Ikura, M. Integrated RAS signaling defined by parallel NMR detection of effectors and regulators. Nat. Chem. Biol. 2014, 10, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nat. Cell Biol. 2013, 503, 548–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patricelli, M.P.; Janes, M.R.; Li, L.-S.; Hansen, R.; Peters, U.; Kessler, L.V.; Chen, Y.; Kucharski, J.M.; Feng, J.; Ely, T.; et al. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov. 2016, 6, 316–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janes, M.R.; Zhang, J.; Li, L.-S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [Green Version]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS (G12C) inhibitor AMG 510 drives anti-tumor immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Khwaja, A.; Rodriguez-Viciana, P.; Wennström, S.; Warne, P.H.; Downward, J. Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. EMBO J. 1997, 16, 2783–2793. [Google Scholar] [CrossRef]
- Niederst, M.J.; Engelman, J.A. Bypass Mechanisms of Resistance to Receptor Tyrosine Kinase Inhibition in Lung Cancer. Sci. Signal. 2013, 6, re6. [Google Scholar] [CrossRef] [Green Version]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef]
- Sakamoto, K.; Kamada, Y.; Sameshima, T.; Yaguchi, M.; Niida, A.; Sasaki, S.; Miwa, M.; Ohkubo, S.; Sakamoto, J.-I.; Kamaura, M.; et al. K-Ras(G12D)-selective inhibitory peptides generated by random peptide T7 phage display technology. Biochem. Biophys. Res. Commun. 2017, 484, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Zhang, Y.; Wang, G.; Mwangi, P.M.; Cai, H.; Li, R. Recombinant KRAS G12D Protein Vaccines Elicit Significant An-ti-Tumor Effects in Mouse CT26 Tumor Models. Front. Oncol. 2020, 10, 1326. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Robbins, P.F.; Lu, Y.-C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cafri, G.; Gartner, J.J.; Zaks, T.; Hopson, K.; Levin, N.; Paria, B.C.; Parkhurst, M.R.; Yossef, R.; Lowery, F.J.; Jafferji, M.S.; et al. mRNA vaccine–induced neoantigen-specific T cell immunity in patients with gastrointestinal cancer. J. Clin. Investig. 2020, 130, 5976–5988. [Google Scholar] [CrossRef]
- Smith, M.J.; Neel, B.G.; Ikura, M. NMR-based functional profiling of RASopathies and oncogenic RAS mutations. Proc. Natl. Acad. Sci. USA 2013, 110, 4574–4579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, K.W.; Tsai, S.T.; Hattori, T.; Fedele, C.; Koide, A.; Yang, C.; Hou, X.; Zhang, Y.; Neel, B.G.; O’Bryan, J.P.; et al. Selective and noncovalent targeting of RAS mutants for inhibition and degradation. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Skora, A.D.; Douglass, J.; Hwang, M.S.; Tam, A.J.; Blosser, R.L.; Gabelli, S.; Cao, J.; Diaz, L.A.; Papadopoulos, N.; Kinzler, K.W.; et al. Generation of MANAbodies specific to HLA-restricted epitopes encoded by somatically mutated genes. Proc. Natl. Acad. Sci. USA 2015, 112, 9967–9972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Wei, X.; Jin, S.; Wu, Y.; Zhao, W.; Xu, Y.; Pan, L.; Zhou, Z.; Chen, S. TCR-mimic antibody-drug conjugates targeting intracellular tumor-specific mutant antigen KRAS G12V mutation. Asian J. Pharm. Sci. 2020, 15, 777–785. [Google Scholar] [CrossRef]
- Li, S.; Liu, S.; Deng, J.; Akbay, E.A.; Hai, J.; Ambrogio, C.; Zhang, L.; Zhou, F.; Jenkins, R.W.; Adeegbe, D.O.; et al. Assessing ther-apeutic efficacy of MEK inhibition in a KRASG12C-driven mouse model of lung cancer. Clin. Cancer Res. 2018, 24, 4854–4864. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Liang, S.-Q.; Schmid, R.A.; Peng, R.-W. New Horizons in KRAS-Mutant Lung Cancer: Dawn After Darkness. Front. Oncol. 2019, 9, 953. [Google Scholar] [CrossRef] [Green Version]
- Haigis, K.M. KRAS Alleles: The Devil Is in the Detail. Trends Cancer 2017, 3, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Arbour, K.C.; Jordan, E.; Kim, H.; Dienstag, J.; Yu, H.A.; Sanchez-Vega, F.; Lito, P.; Berger, M.; Solit, D.B.; Hellmann, M.; et al. Effects of Co-occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non–Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 334–340. [Google Scholar] [CrossRef] [Green Version]
- Ucar, G.; Ergun, Y.; Esen, S.A.; Acikgoz, Y.; Dirikoc, M.; Esen, I.; Bal, Ö.; Uncu, D. Prognostic and predictive value of KRAS mutation number in metastatic colorectal cancer. Medicine 2020, 99, e22407. [Google Scholar] [CrossRef] [PubMed]
- Ottaiano, A.; Normanno, N.; Facchini, S.; Cassata, A.; Nappi, A.; Romano, C.; Silvestro, L.; De Stefano, A.; Rachiglio, A.M.; Roma, C.; et al. Study of Ras Mutations’ Prognostic Value in Metastatic Colorectal Cancer: STORIA Analysis. Cancers 2020, 12, 1919. [Google Scholar] [CrossRef]
- Modest, D.P.; Ricard, I.; Heinemann, V.; Hegewisch-Becker, S.; Schmiegel, W.; Porschen, R.; Stintzing, S.; Graeven, U.; Arnold, D.; Von Weikersthal, L.F.; et al. Outcome according to KRAS-, NRAS-and BRAF-mutation as well as KRAS mutation variants: Pooled analysis of five randomized trials in metastatic colorectal cancer by the AIO colorectal cancer study group. Ann. Oncol. 2016, 27, 1746–1753. [Google Scholar] [CrossRef]
- Chuang, S.C.; Huang, C.W.; Chen, Y.T.; Ma, C.J.; Tsai, H.L.; Chang, T.K.; Su, W.C.; Hsu, W.H.; Kuo, C.H.; Wang, J.Y. Effect of KRAS and NRAS mutations on the prognosis of patients with synchronous metastatic colorectal cancer presenting with liver-only and lung-only metastases. Oncol. Lett. 2020, 20, 2119–2130. [Google Scholar] [CrossRef]
- Tosi, F.; Magni, E.; Amatu, A.; Mauri, G.; Bencardino, K.; Truini, M.; Veronese, S.; De Carlis, L.; Ferrari, G.; Nichelatti, M.; et al. Effect of KRAS and BRAF mutations on survival of metastatic colorectal cancer after liver resection: A systematic review and meta-analysis. Clin. Col. Cancer 2017, 16, e153–e163. [Google Scholar] [CrossRef]
- Ottaiano, A.; Circelli, L.; Lombardi, A.; Scala, S.; Martucci, N.; Galon, J.; Buonanno, M.; Scognamiglio, G.; Botti, G.; Hermitte, F.; et al. Genetic trajectory and immune microenvironment of lung-specific oligometastatic colorectal cancer. Cell Death Dis. 2020, 11, 1. [Google Scholar]
- Ottaiano, A.; Nasti, G.; Santorsola, M.; Altieri, V.; Di Fruscio, G.; Circelli, L.; Luce, A.; Cossu, A.M.; Scognamiglio, G.; Perri, F.; et al. KRAS mutational regression is associated with oligo-metastatic status and good prognosis in metastatic colorectal cancer. Front. Oncol. 2021, 11, 970. [Google Scholar] [CrossRef]
- Ottaiano, A.; Caraglia, M.; Di Mauro, A.; Botti, G.; Lombardi, A.; Galon, J.; Luce, A.; D’amore, L.; Perri, F.; Santorsola, M.; et al. Evolution of Mutational Landscape and Tumor Immune-Microenvironment in Liver Oligo-Metastatic Colorectal Cancer. Cancers 2020, 12, 3073. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Hong, M.; Shin, M.K.; Kim, B.C.; Shin, H.S.; Yu, E.; Hong, S.M.; Kim, J.; Chun, S.M.; Kim, T.I.; et al. KRAS and PIK3CA mutations in colorectal adenocarcinomas correlate with aggressive histological features and behavior. Hum. Pathol. 2017, 65, 21–30. [Google Scholar] [CrossRef]
- Aktories, K. Bacterial protein toxins that modify host regulatory GTPases. Nat. Rev. Genet. 2011, 9, 487–498. [Google Scholar] [CrossRef]
- Koh, E.; Cho, H.-S. NleB/SseKs ortholog effectors as a general bacterial monoglycosyltransferase for eukaryotic proteins. Curr. Opin. Struct. Biol. 2021, 68, 215–223. [Google Scholar] [CrossRef]
- Popoff, M.R.; Geny, B. Rho/Ras-GTPase-dependent and -independent activity of clostridial glucosylating toxins. J. Med. Microbiol. 2011, 60, 1057–1069. [Google Scholar] [CrossRef] [Green Version]
- Dreger, S.C.; Schulz, F.; Huelsenbeck, J.; Gerhard, R.; Hofmann, F.; Just, I.; Genth, H. Killing of Rat Basophilic Leukemia Cells by Lethal Toxin from Clostridium sordellii: Critical Role of Phosphatidylinositide 3′-OH Kinase/Akt Signaling. Biochemistry 2009, 48, 1785–1792. [Google Scholar] [CrossRef]
- Schorch, B.; Heni, H.; Zahaf, N.-I.; Brummer, T.; Mione, M.; Schmidt, G.; Papatheodorou, P.; Aktories, K. Targeting oncogenic Ras by the Clostridium perfringens toxin TpeL. Oncotarget 2018, 9, 16489–16500. [Google Scholar] [CrossRef] [Green Version]
- Ganesan, A.K.; Frank, D.W.; Misra, R.P.; Schmidt, G.; Barbieri, J.T. Pseudomonas aeruginosa Exoenzyme S ADP-ribosylates Ras at Multiple Sites. J. Biol. Chem. 1998, 273, 7332–7337. [Google Scholar] [CrossRef] [Green Version]
- Antic, I.; Biancucci, M.; Zhu, Y.; Gius, D.R.; Satchell, K.J.F. Site-specific processing of Ras and Rap1 Switch I by a MARTX toxin effector domain. Nat. Commun. 2015, 6, 7396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavin, H.E.; Satchell, K.J.F. MARTX toxins as effector delivery platforms. Pathog. Dis. 2015, 73, ftv092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahaf, N.-I.; Schmidt, G. Bacterial Toxins for Cancer Therapy. Toxins 2017, 9, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-S.; Jun, S.-Y.; Kim, Y.-S. Critical Issues in the Development of Immunotoxins for Anticancer Therapy. J. Pharm. Sci. 2020, 109, 104–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, X.; Feig, L.A. Basis for Signaling Specificity Difference between Sos and Ras-GRF Guanine Nucleotide Exchange Factors. J. Biol. Chem. 2001, 276, 47248–47256. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Primary Tissue Type | (Mutated) Total Samples Tested | % of Point Mutations | (Mutated) Total Samples Tested | % of Point Mutations | (Mutated) Total Samples Tested | % of Point Mutations |
|---|---|---|---|---|---|---|---|
| H-Ras | H-Ras | K-Ras | K-Ras | N-Ras | N-Ras | ||
| 1 | Adrenal gland | (45)1303 | 3.45 | (1)1331 | 0.88 | (13)1276 | 1.02 |
| 2 | Autonomic ganglia | (6)1597 | 0.38 | (1)1572 | 0.06 | (11)1673 | 0.66 |
| 3 | Biliary tract | (9)1728 | 0.52 | (958)5367 | 19.49 | (58)2133 | 2.72 |
| 4 | Bone | (8)1060 | 0.75 | (14)1184 | 1.27 | (13)1209 | 1.08 |
| 5 | Breast | (54)8192 | 0.66 | (150)11297 | 1.42 | (33)7763 | 0.43 |
| 6 | Central nervous system | (11)4150 | 0.27 | (47)4881 | 1.21 | (54)4800 | 1.13 |
| 7 | Cervix | (28)1249 | 2.24 | (151)2569 | 6.03 | (10)1191 | 0.84 |
| 8 | Endometrium | (14)2384 | 0.59 | (720)4663 | 15.89 | (53)1637 | 3.24 |
| 9 | Eye | 340 | − | (7)476 | 1.47 | (40)888 | 4.5 |
| 10 | Fallopian tube | 3 | − | 9 | − | 6 | − |
| 11 | Female genital tract (site indeterminate) | 20 | − | (1)24 | 8.33 | 22 | − |
| 12 | Gastrointestinal tract (site indeterminate) | 1 | − | (70)1083 | 6.46 | 477 | − |
| 13 | Genital tract | (2)285 | 0.7 | (36)576 | 6.42 | (12)565 | 2.12 |
| 14 | Haematopoietic and lymphoid | (24)12623 | 0.19 | (1124)24788 | 4.91 | (2517)28829 | 8.73 |
| 15 | Kidney | (7)31127 | 0.22 | (41)4277 | 1.01 | (12)3345 | 0.36 |
| 16 | Large intestine | (56)5989 | 0.94 | (25902)80015 | 33.4 | (617)16211 | 3.81 |
| 17 | Liver | (7)2902 | 0.24 | (112(3604 | 3.3 | (34)3164 | 1.07 |
| 18 | Lung | (45)7450 | 0.6 | (6419)43284 | 16.08 | (145)16486 | 0.88 |
| 19 | Mediastinum | 1 | − | 1 | − | 1 | − |
| 20 | Meninges | (12)283 | 4.24 | (4)317 | 1.26 | (23)359 | 6.41 |
| 21 | NS | (17)1251 | 1.36 | (103)1808 | 6.19 | (458)2674 | 17.13 |
| 22 | Oesophagus | (9)2668 | 0.34 | (71)3203 | 2.28 | (5)2142 | 0.23 |
| 23 | Ovary | (4)1677 | 0.24 | (899)6913 | 13.57 | (31)1916 | 1.62 |
| 24 | Pancreas | (3)3135 | 0.1 | (6767)12289 | 56.38 | (26)3513 | 0.74 |
| 25 | Paratesticular tissues | * | * | 1 | − | * | * |
| 26 | Parathyroid | (2)151 | 1.32 | 136 | − | 135 | − |
| 27 | Penis | (25)209 | 11.6 | (13)433 | 3.23 | (3)128 | 2.34 |
| 28 | Pericardium | − | 3 | − | 2 | − | |
| 29 | Perineum | − | 2 | − | 2 | − | |
| 30 | Peritoneum | − | (166)371 | 45.28 | (1)149 | 0.67 | |
| 31 | Pituitary | (12)364 | 3.3 | 389 | 390 | ||
| 32 | Placenta | − | 14 | − | 6 | − | |
| 33 | Pleura | − | (7)739 | 0.95 | (4)581 | 0.69 | |
| 34 | Prostate | (61)3978 | 1.53 | (136)4827 | 2.86 | (30)3953 | 0.76 |
| 35 | Salivary gland | (93)783 | 11.88 | (12)701 | 1.71 | (5)573 | 0.87 |
| 36 | Skin | (648)6529 | 9.92 | (177)6286 | 3.07 | (2408)15500 | 15.54 |
| 37 | Small intestine | (1)316 | 0.32 | (256)1232 | 21.27 | (2)420 | 0.48 |
| 38 | Soft tissue | (74)2767 | 2.67 | (118)4577 | 2.67 | (70)3171 | 2.21 |
| 39 | Stomach | (37)3399 | 1.09 | (389)6472 | 6.13 | (28)2597 | 1.08 |
| 40 | Testis | (5)675 | 0.74 | (61)1199 | 6.51 | (26)1041 | 2.5 |
| 41 | Thymus | (9)393 | 2.29 | (9)652 | 1.38 | (4)469 | 0.85 |
| 42 | Thyroid | (451)10503 | 4.29 | (239)12387 | 2.11 | (933)11422 | 8.17 |
| 43 | Upper aerodigestive tract | (228)3911 | 5.83 | (95)5146 | 2.06 | (51)3449 | 1.48 |
| 44 | Urinary tract | (321)3727 | 8.61 | (155)2803 | 5.71 | (43)2514 | 1.71 |
| 45 | Vagina | − | 5 | − | 4 | − | |
| 46 | Vulva | (20)175 | 11.43 | (2)199 | 1.01 | 175 | − |
| S. No. | Subtype of NS | Mutated Gene |
|---|---|---|
| 1 | NS1 | PTPN11 |
| 2 | NS2 | LZTR1 |
| 3 | NS3 | KRAS |
| 4 | NS4 | SOS1 |
| 5 | NS5 | RAF1 |
| 6 | NS6 | NRAS |
| 7 | NS7 | BRAF |
| 8 | NS8 | RIT1 |
| 9 | NS9 | SOS2 |
| 10 | NS10 | LZTR1 |
| 11 | NS11 | MRAS |
| 12 | NS12 | RRAS2 |
| 13 | NS13 | MAPK1 |
| S. No. | Category | Condition | Intervention/Treatment | Clinical trial status |
|---|---|---|---|---|
| 1 | H-Ras mutation | HRAS Gene Mutation HNSCC | Drug: Tipifarnib Device: H-Ras Detection Assay | Phase 2 |
| 2 | H-Ras mutation | Thyroid Cancer Squamous Cell Carcinoma Head and Neck Cancer (HNSCC) H-Ras Mutant Tumor Other Squamous Cell Carcinoma (SCC) With H-Ras Mutant Tumor | Drug: Tipifarnib | Phase 2 |
| 3 | H-Ras mutation | Recurrent Adrenal Gland Pheochromocytoma Recurrent Ectomesenchymoma Recurrent Ependymoma Recurrent Ewing Sarcoma Recurrent Hepatoblastoma Recurrent Kidney Wilms Tumor Recurrent Langerhans Cell Histiocytosis Recurrent Malignant Germ Cell Tumor Recurrent Malignant Glioma Recurrent Medulloblastoma Recurrent Melanoma Recurrent Neuroblastoma Recurrent Non-Hodgkin Lymphoma Recurrent Osteosarcoma Recurrent Peripheral Primitive Neuroectodermal Tumor Recurrent Rhabdoid Tumor Recurrent Rhabdoid Tumor of the Kidney Recurrent Rhabdomyosarcoma Recurrent Soft Tissue Sarcoma Recurrent Thyroid Gland Carcinoma Recurrent WHO Grade II Glioma Refractory Adrenal Gland Pheochromocytoma Refractory Ependymoma Refractory Ewing Sarcoma Refractory Hepatoblastoma Refractory Langerhans Cell Histiocytosis Refractory Malignant Germ Cell Tumor Refractory Malignant Glioma Refractory Medulloblastoma Refractory Melanoma Refractory Neuroblastoma Refractory Non-Hodgkin Lymphoma Refractory Osteosarcoma Refractory Peripheral Primitive Neuroectodermal Tumor Refractory Rhabdoid Tumor Refractory Rhabdoid Tumor of the Kidney Refractory Rhabdomyosarcoma Refractory Soft Tissue Sarcoma Refractory Thyroid Gland Carcinoma Refractory WHO Grade II Glioma | Drug: Tipifarnib | Phase 2 |
| 4 | H-Ras mutation | Non-Small Cell Lung Cancer | Drug: Tipifarnib | Phase 2 |
| 5 | H-Ras mutation | Urothelial Carcinoma | Drug: Tipifarnib | Phase 2 |
| 6 | H-Ras mutation | Colorectal Cancer | Drug: ISIS 2503 | Phase 2 |
| 7 | H-Ras mutation | Pancreatic Cancer | Drug: ISIS 2503 | Phase 2 |
| 8 | H-Ras mutation | Cancer Malignancy Neoplasia Neoplasm Neoplasm Metastasis Colon Cancer Colonic Neoplasms Colon Cancer Liver Metastasis Metastatic Cancer Metastatic Melanoma Metastatic Colon Cancer Metastatic Lung Cancer Non-Small Cell Lung Cancer Metastatic Pancreatic Cancer Pancreas Cancer Pancreas Adenocarcinoma Pancreas Neoplasm Metastatic Nonsmall Cell Lung Cancer Metastatic Pancreatic Cancer | Drug: ASN007: ascending doses Drug: ASN007 RD | Phase 1 |
| 1 | K-RasG12C | Neoplasms Advanced Solid Tumors Non-small Cell Lung Cancer Colorectal Cancer | Drug: JNJ-74699157 | Phase 1 |
| 2 | K-RasG12C | Advanced Cancer Metastatic Cancer Malignant Neoplastic Disease | Drug: MRTX849 Drug: TNO155 | Phase ½ |
| 3 | K-RasG12C | Advanced Non-Small Cell Lung Cancer Metastatic Cancer | Drug: MRTX849 in Combination with Pembrolizumab | Phase 2 |
| 4 | K-RasG12C | Advanced Cancer Metastatic Cancer Malignant Neoplastic Disease | Drug: MRTX849 Drug: Pembrolizumab Drug: Cetuximab Drug: Afatinib | Phase 1/2 |
| 5 | K-RasG12C | Metastatic Non-Small Cell Lung Cancer Advanced Non-Small Cell Lung Cancer | Drug: MRTX849 Drug: Docetaxel | Phase 3 |
| 6 | K-RasG12C | Advanced Colorectal Cancer Metastatic Colorectal Cancer | Drug: MRTX849 Biological: Cetuximab Drug: mFOLFOX6 Regimen Drug: FOLFIRI Regimen | Phase 3 |
| 7 | K-RasG12C | Non-Small Cell Lung Cancer Colorectal Cancer Advanced Solid Tumors | Drug: GDC-6036 Drug: Atezolizumab Drug: Cetuximab Drug: Bevacizumab Drug: Erlotinib | Phase 1 |
| 8 | K-RasG12C | Lung Adenocarcinoma Lung Non-Small Cell Carcinoma Recurrent Lung Non-Squamous Non-Small Cell Carcinoma Stage IV Lung Cancer AJCC v8 Stage IVA Lung Cancer AJCC v8 Stage IVB Lung Cancer AJCC v8 | Drug: Sotorasib | Phase 2 |
| 9 | K-RasG12C | KRAS G12C Mutant Solid Tumors Carcinoma, Non-Small-Cell Lung Carcinoma, Colorectal Cancer of Lung Cancer of the Lung Lung Cancer Neoplasms, Lung Neoplasms, Pulmonary Pulmonary Cancer Pulmonary Neoplasms | Drug: JDQ443 Drug: TNO155 Biological: spartalizumab | |
| 10 | K-RasG12C | Advanced/Metastatic Solid Tumors With KRAS p.G12C Mutation | Drug: AMG 510 | Phase 1 |
| 11 | K-RasG12C | Advanced Solid Tumors Kirsten Rat Sarcoma (KRAS) pG12C Mutation | Drug: Sotorasib Drug: PD1 inhibitor Drug: MEK inhibitor Drug: SHP2 allosteric inhibitor Drug: Pan-ErbB tyrosine kinase inhibitor Drug: PD-L1 inhibitor Drug: EGFR inhibitor Drug: Chemotherapeutic regimen Drug: PD-1 inhibitor Drug: mTOR inhibitor Drug: CDK inhibitor Drug: VEGF inhibitor | Phase 1 |
| 12 | K-RasG12C | KRAS p.G12C Mutant Advanced Solid Tumors | Drug: AMG 510 Drug: Anti PD-1/L1 Drug: Midazolam | Phase 1/2 |
| 13 | K-RasG12C | Non-Small-cell Lung Cancer Locally Advanced Unresectable NSCLC Locally Advanced Metastatic NSCLC | Drug: AMG 510 | |
| 14 | K-RasG12C | KRAS p, G12c Mutated /Advanced Metastatic NSCLC | Drug: AMG 510 Drug: Docetaxel | Phase 3 |
| 15 | K-RasG12C | Solid Tumor, Adult NSCLC CRC | Drug: D-1553 Drug: Other | Phase 1/2 |
| 16 | K-RasG12C | Advanced EGFR mutant Non-Small cell Lung Cancer (NSCLC), KRAS G12-mutant NSCLC, Esophageal Squamous Cell Cancer (SCC), Head/Neck SCC, Melanoma | Drug: TNO155 Drug: TNO155 in combination with EGF816 (nazartinib) | Phase 1 |
| 17 | K-RasG12C/ K-RasG12D | Advanced or Metastatic Solid Tumors | Drug: TAS0612 | Phase 1 |
| 18 | K-RasG12C | Tumor, Solid | Drug: BBP-398 (Formerly Known as IACS-15509) | Phase 1 |
| 19 | K-RasG12C | Carcinoma, Non-Small Cell Lung | Drug: carboplatin Drug: paclitaxel Drug: Bevacizumab Drug: Pemetrexed Drug: cisplatin | Phase 3 |
| 20 | K-RasG12C | KRAS Gene Mutation Recurrent Lung Non-Small Cell Carcinoma Stage IV Lung Non-Small Cell Cancer AJCC v7 | Drug: Docetaxel Other: Laboratory Biomarker Analysis Drug: Trametinib | Phase 2 |
| 21 | K-RasG12C | Advanced Solid Tumor Non-Small Cell Lung Cancer Colorectal Cancer | Drug: LY3499446 Drug: Abemaciclib Drug: Cetuximab Drug: Erlotinib Drug: Docetaxel | Phase 1/2 |
| 1 | K-RasG12D | Non-Small Cell Lung Cancer | Drug: Bortezomib Drug: Acyclovir | Phase 2 |
| 2 | K-RasG12D | Minimal Residual Disease KRAS G12D KRAS G12R NRAS G12D NRAS G12R Pancreatic Ductal Adenocarcinoma Colorectal Cancer Non-small Cell Lung Cancer Ovarian Cancer Cholangiocarcinoma Bile Duct Cancer Gallbladder Carcinoma | Drug: ELI-002 (Dose Escalation) Drug: ELI-002 (at the RP2D) Other: Observation | Phase 1/2 |
| 3 | K-RASG12D | TNBC—Triple-Negative Breast Cancer Head and Neck Squamous Cell Carcinoma Squamous Cell Carcinoma of Anal Canal Uveal Melanoma Glioblastoma Colorectal Cancer Chordoma Squamous Cell Carcinoma of the Lung KRAS G12D KRAS G13D EGFR Amplification Epithelial Ovarian Cancer Hepatocellular Carcinoma Anaplastic Thyroid Cancer Pancreas Cancer | Drug: BCA101 Drug: Pembrolizumab | Phase 1 |
| 4 | K-RasG12D | Advanced or Metastatic Solid Tumors | Drug: TAS0612 | Phase 1 |
| 1 | K-RasG12V | Pancreatic Cancer Pancreatic Neoplasms Pancreatic Ductal Adenocarcinoma Advanced Cancer | Drug: Cyclophosphamide Drug: Fludarabine Biological: Mutant KRAS G12V-specific TCR transduced autologous T cells Drug: Anti-PD-1 monoclonal antibody | Phase 1/2 |
| 2 | K-RasG12V | Non-Small Cell Lung Cancer KRAS Activating Mutation | Drug: VS-6766 Drug: VS-6766 and Defactinib | Phase 2 |
| 1 | K-RasG13D | TNBC—Triple-Negative Breast Cancer Head and Neck Squamous Cell Carcinoma Squamous Cell Carcinoma of Anal Canal Uveal Melanoma Glioblastoma Colorectal Cancer Chordoma Squamous Cell Carcinoma of the Lung KRAS G12D KRAS G13D EGFR Amplification Epithelial Ovarian Cancer Hepatocellular Carcinoma Anaplastic Thyroid Cancer Pancreas Cancer | Drug: BCA101 Drug: Pembrolizumab | Phase 1 |
| 1 | N-Ras mutation | KRAS Gene Mutation Metastatic Colorectal Carcinoma NRAS Gene Mutation Stage III Colorectal Cancer AJCC v8 Stage IIIA Colorectal Neuroendocrine Tumor AJCC v8 Stage IIIB Colorectal Cancer AJCC v8 Stage IIIC Colorectal Cancer AJCC v8 Stage IV Colorectal Cancer AJCC v8 Stage IVA Colorectal Cancer AJCC v8 Stage IVB Colorectal Cancer AJCC v8 Stage IVC Colorectal Cancer AJCC v8 Unresectable Carcinoma | Drug: Binimetinib Drug: Palbociclib Drug: Trifluridine and Tipiracil Hydrochloride | Phase 2 |
| 2 | N-Ras mutation | BRAF V600E Negative KRAS Gene Mutation Negative Locally Advanced Unresectable Colorectal Adenocarcinoma Metastatic Colorectal Adenocarcinoma NRAS Gene Mutation Negative Stage III Colorectal Cancer AJCC v8 Stage IIIA Colorectal Cancer AJCC v8 Stage IIIB Colorectal Cancer AJCC v8 Stage IIIC Colorectal Cancer AJCC v8 Stage IV Colorectal Cancer AJCC v8 Stage IVA Colorectal Cancer AJCC v8 Stage IVB Colorectal Cancer AJCC v8 Stage IVC Colorectal Cancer AJCC v8 | Biological: Cetuximab Drug: Irinotecan Biological: Panitumumab Drug: Regorafenib | Phase 2 |
| 3 | N-Ras mutation | Solid Tumor | Drug: ARQ 736 | Phase 1 |
| 4 | N-Ras mutation | Cancer Lung Cancer Metastatic Immunotherapy | Drug: PDR001 | Phase 2 |
| 5 | N-Ras mutation | Metastatic or Unresectable Cutaneous Melanoma | Drug: MEK162 Drug: Dacarbazine | Phase 3 |
| 6 | N-Ras mutation | BRAF or NRAS Mutant Metastatic Melanoma | Drug: MEK162 | Phase 2 |
| 7 | N-Ras mutation | Advanced Lymphoma Advanced Malignant Solid Neoplasm Hematopoietic and Lymphoid Cell Neoplasm Refractory Lymphoma Refractory Malignant Solid Neoplasm Refractory Plasma Cell Myeloma | Drug: Binimetinib | Phase 2 |
| 8 | N-Ras mutation | Solid Tumor | Drug: HM95573 | Phase 1 |
| 9 | N-Ras mutation | Lung Cancer, Non-Small Cell | Drug: GSK1120212 Drug: docetaxel | Phase 2 |
| 10 | N-Ras mutation | Metastatic Malignant Solid Neoplasm Refractory Malignant Solid Neoplasm Unresectable Malignant Solid Neoplasm | Biological: Navitoclax Drug: Trametinib | Phase 1/2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nair, A.; Kubatzky, K.F.; Saha, B. Ras Isoforms from Lab Benches to Lives—What Are We Missing and How Far Are We? Int. J. Mol. Sci. 2021, 22, 6508. https://doi.org/10.3390/ijms22126508
Nair A, Kubatzky KF, Saha B. Ras Isoforms from Lab Benches to Lives—What Are We Missing and How Far Are We? International Journal of Molecular Sciences. 2021; 22(12):6508. https://doi.org/10.3390/ijms22126508
Chicago/Turabian StyleNair, Arathi, Katharina F. Kubatzky, and Bhaskar Saha. 2021. "Ras Isoforms from Lab Benches to Lives—What Are We Missing and How Far Are We?" International Journal of Molecular Sciences 22, no. 12: 6508. https://doi.org/10.3390/ijms22126508
APA StyleNair, A., Kubatzky, K. F., & Saha, B. (2021). Ras Isoforms from Lab Benches to Lives—What Are We Missing and How Far Are We? International Journal of Molecular Sciences, 22(12), 6508. https://doi.org/10.3390/ijms22126508