1. Introduction
Tumors of the endocrine system are relatively rare. Nevertheless, thyroid cancers (TCs) are an exception. TCs belong to the five most frequently diagnosed cancers in women, where the highest number of thyroid carcinoma cases is recorded in middle-aged patients [
1]. The most frequently diagnosed TC type is papillary thyroid carcinoma (PTC; up to 80% of all TC cases). The other, less frequent types of TCs are follicular thyroid carcinoma (FTC), anaplastic thyroid carcinoma (ATC), and medullary thyroid cancer (MTC) [
2,
3]. A large fraction of PTCs is accompanied by the presence of certain genetic alternations, such as single nucleotide mutations in the
BRAF gene (especially
BRAF V600E) and
RET gene rearrangements (e.g.,
RET/PTC). Both genes are important regulators of the mitogen-activated protein kinase (MAPK) signaling pathway.
RET encodes for receptor tyrosine kinase and RET/PTC fusion results in ligand-independent activation of the RET kinase. Similarly, single base mutations in the
BRAF gene, which encodes serine-threonine kinase, lead to constitutive activation of MAPK signal transduction [
4]. Mutations in these targets are mutually excluded, and
BRAF mutant alleles are found in most (up to 80%) of all papillary tumors [
5]. Moreover, it has been shown that the presence of the
BRAF V600E allele in PTC is associated with an altered expressional profile of several other genes [
6]. Therefore, such
BRAF V600E-deregulated genes can be considered as potential drivers of malignant transformation and important prognostic or therapeutic targets.
Although the survival rate of the TCs is relatively high, the number of new cases is still growing [
7]. Therefore, identification of new molecular players involved in carcinogenesis and understanding their factual biological functions might be a key factor for effective selection of groups of risk among patients and administration of more personalized treatments.
Here, we present a study on the FERM-domain containing protein 5 (FRMD5), which we found to be predominantly expressed in
BRAF-mutated PTCs. FRMD5 is encoded by the
FRMD5 gene located on chromosome 15 (15q15.3). This 65 kDa protein belongs to the FERM-domain containing protein family, which occupies a unique position in cellular metabolism, performs structural functions, and participates in signal transduction. Although several members of this family have already been linked with tumor progression [
8], the role of FRMD5 in cellular biology and tumorigenesis remains unclear. However, Brazdova et al. (2009) marked FRMD5 as a p53(R273H) target in U251 glioblastoma cells [
9]. Additionally, the role of FRMD5 in p120-catenin-based cell–cell contact and regulation of lung tumor cell progression has been reported [
10]. In other studies, Hu et al. (2014) proposed that FRMD5 may regulate lung cancer cell migration by interacting with integrin β5 cytoplasmic tail and ROCK1 [
11]. Studies performed on the deregulation of the Wnt signaling pathway in colorectal cancer have highlighted FRMD5 as a novel target of the β-catenin/TCF7L2 complex, which plays an important role in tumorigenesis [
12].
In the performed comprehensive analysis, we were particularly interested in studying the relationship between the expressional status of FRMD5 and proliferative, invasiveness, and metastatic potential of PTC cells, as well as the expression of multidrug resistance proteins (MDRs). Our findings supported by NGS and phospho-kinase proteome profiling indicate significant discrepancies in the role of FRMD5 in both wild-type BRAF (BRAF-wt) and BRAF-mutated PTC cells. The data suggest that the mutational status of the cancer-driver BRAF gene might be crucial for the regulation of the expression of FRMD5 and its activity in PTC cells.
3. Discussion
FRMD5 is a still poorly understood FERM-domain protein. Its biological function is unclear. However, relatively high expression of the
FRMD5 gene in various human tissues, including brain, kidney, thyroid, and testis, underlines its importance for biological processes. Former research concerning FRMD3, a close homolog of FRMD5, has emphasized the role of this FERM protein in tumorigenesis [
15]. Likewise, the few studies performed on FRMD5 have also suggested its involvement in carcinogenesis, although its functions in this process still remain indistinct [
9,
10,
11,
12].
Here, we performed an analysis on the role of FRMD5 in thyroid cancer, as we found significant discrepancies in FRMD5 expression between BRAF- and RET-mutated PTCs, as well other types of TCs (TCGA and microarray data analysis of clinical specimens). BRAF and RET tumorigenic genes are the most abundant among TCs and share a common property of signaling via activation of the MEK-ERK kinase pathway. Nevertheless, cells harboring these mutations present unique phenotypic features, signifying the different tumor biology resulting from unequal gene expressional profiles. Based on analyses of FRMD5 expression in human thyroid cancer tissue, it was concluded that the high expression of the FRMD5 gene may be associated with the presence of the BRAF V600E genetic aberration.
Here, we performed an analysis on the role of FRMD5 in thyroid cancer specimens and cell lines, as we found significant discrepancies in FRMD5 expression between BRAF- and RET-mutated PTCs, as well other types of TCs (TCGA and microarray data analysis of clinical specimens). BRAF and RET tumorigenic genes are most abundant among TCs and share a common property of signaling via activation of the MEK-ERK kinase pathway. The nature of imbalance in FRMD5 intracellular yield and expression observed in cells harboring BRAF-wt and BRAF V600E is not clear and might result from overall unequal gene expressional profiles, which determines phenotypic features and unique tumor biology cells harboring these mutations. Based on analyses of FRMD5 expression in human thyroid cancer tissue, it was concluded that high expression of the FRMD5 gene may be associated with the presence of the BRAF V600E genetic aberration.
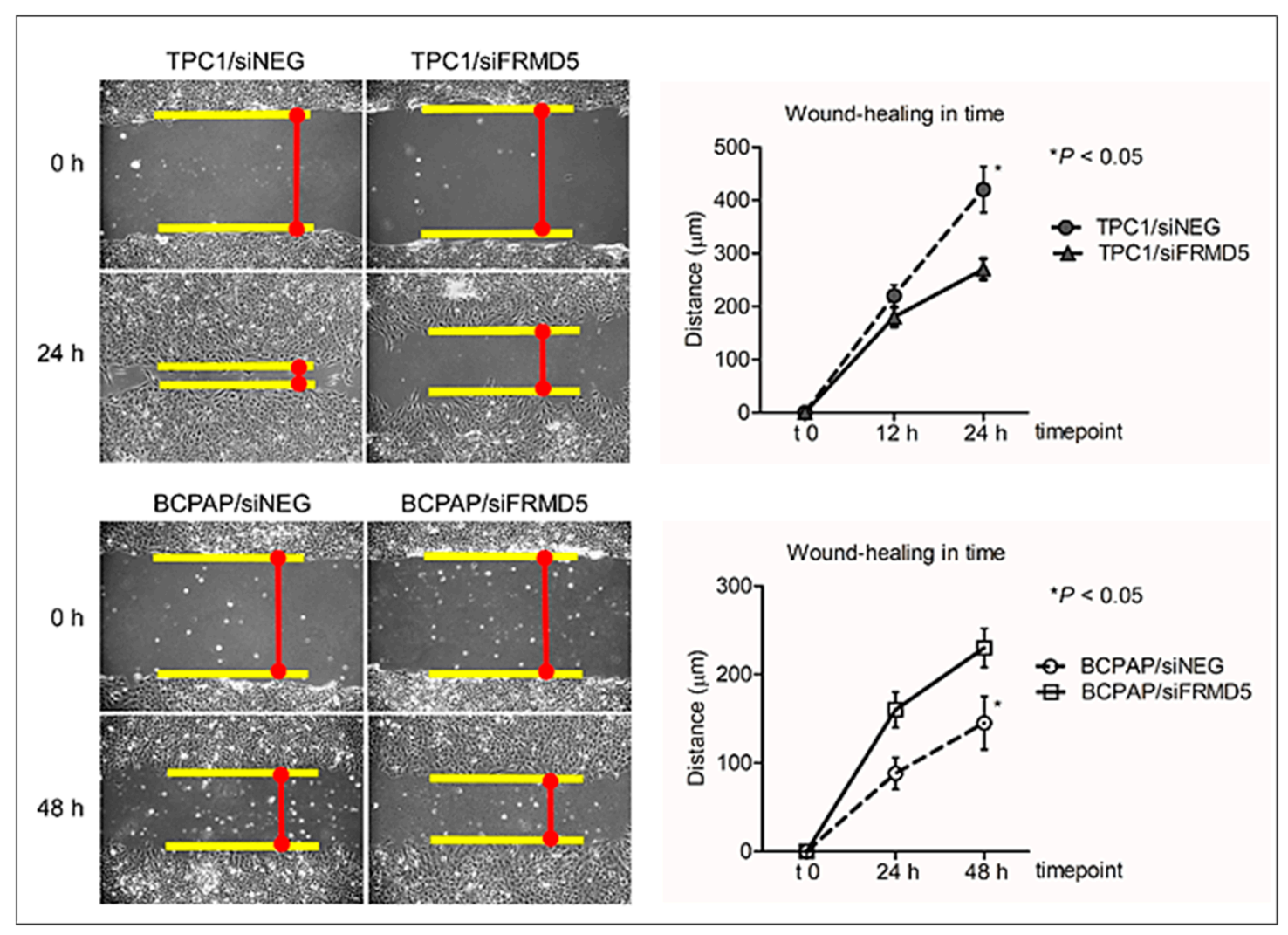
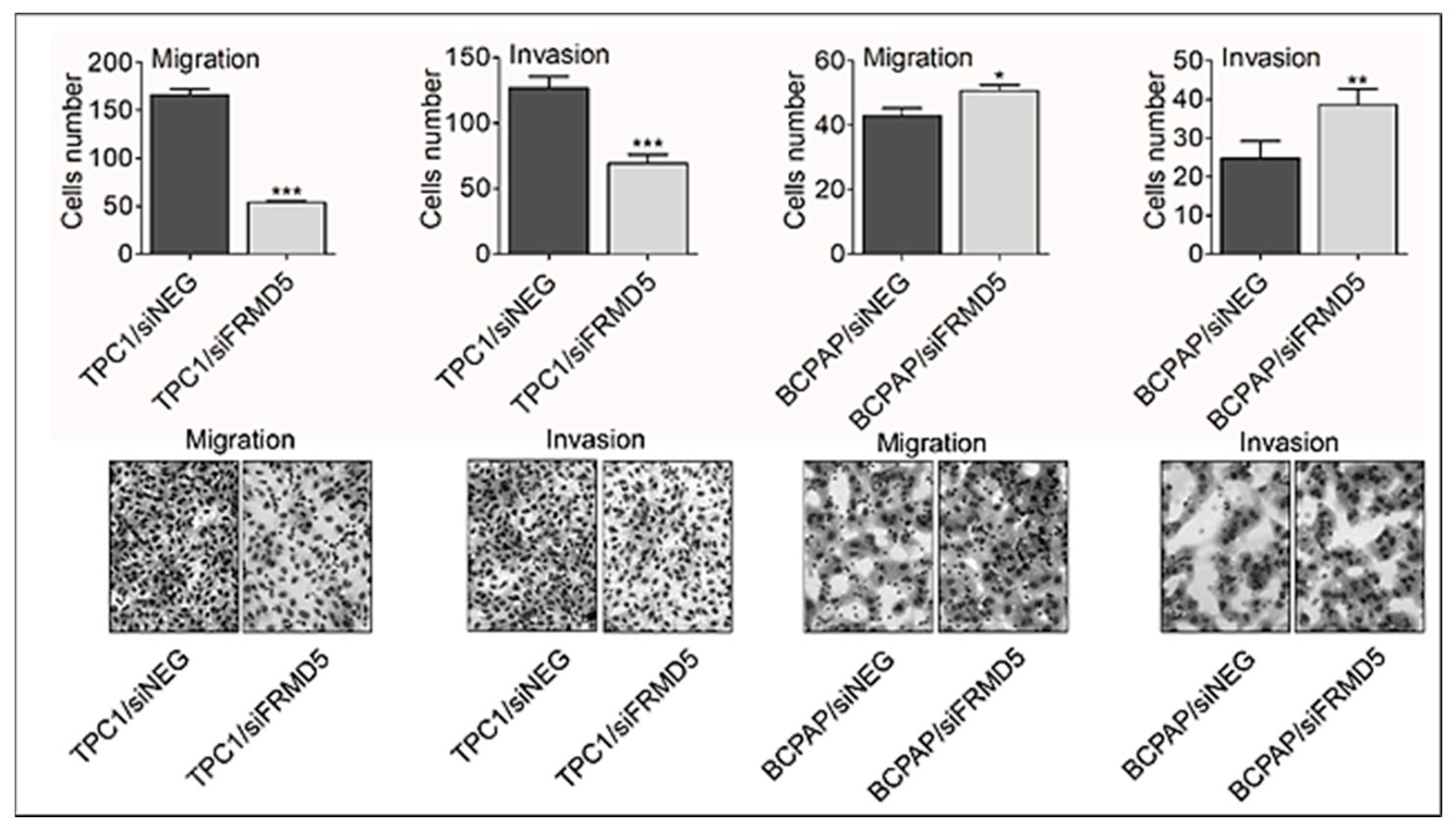
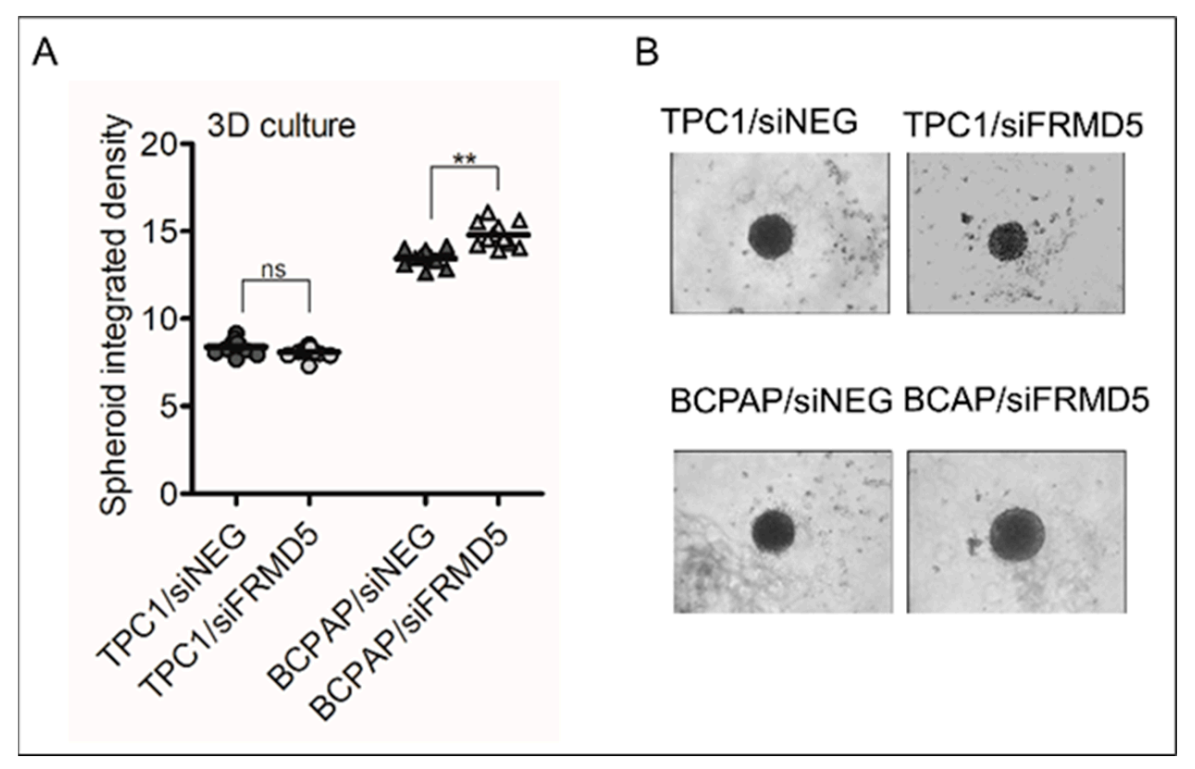
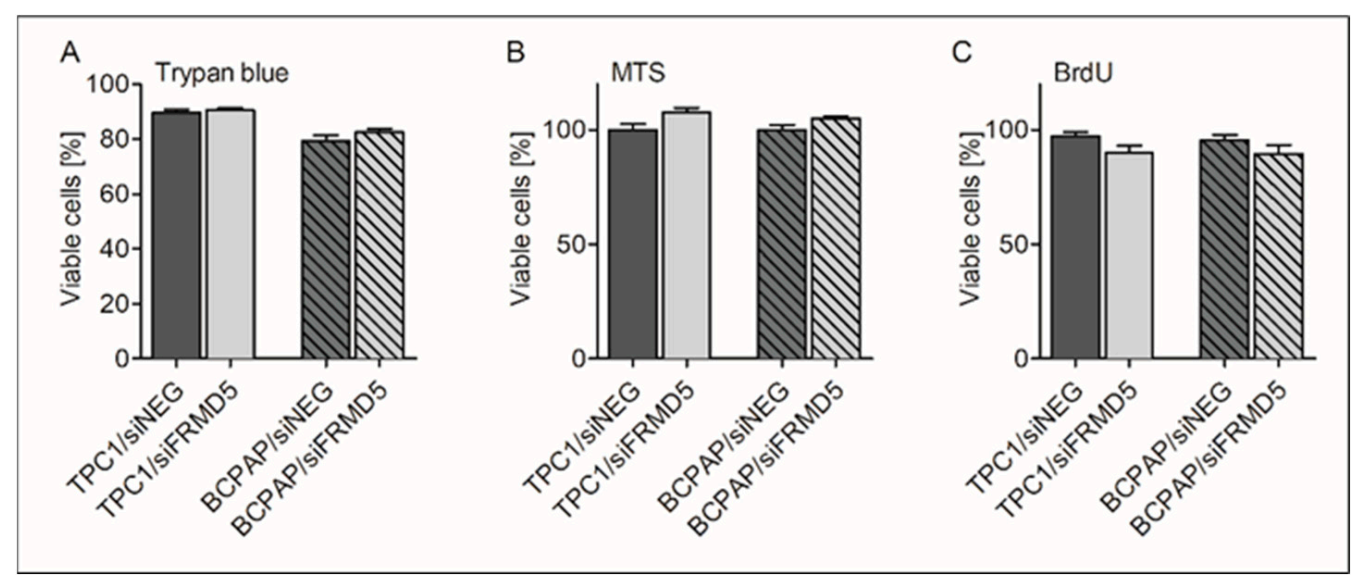
To search for biological processes and define genes associated with FRMD5 expression, we knocked down FRMD5 in PTC-derived, TPC1, and BCPAP cells, harboring RET/PTC and BRAF V600E mutations, respectively. We found that the depletion of FRMD5 significantly increases the migration and invasiveness, as well as promotes spheroid formation of BCPAP cells. In contrast, the depletion of FRMD5 in TPC1 cells strongly suppressed their metastatic potential. The observed diverse impact of FRMD5 knockdown was not associated with alterations in the cells’ viability, proliferation, or apoptosis, as they remained unaffected. Together, these data strongly indicate that the observed disproportions in the motility of FRMD5-deficient cells do not result from altered viability of cells, but rather other processes, such as impaired transduction signal trafficking by major pathways involved in the control of migration processes, cell–cell or cell–matrix adhesion.
Partly, the effect of FRMD5 on the metastatic potential of PTC cells stays in accordance with previously published studies on glioblastoma, lung, and colorectal cancer (mentioned in the Introduction), suggesting its pro-migratory potential. Nevertheless, our findings also imply its antimetastatic character in
BRAF-mutated cells, indicating that the expressional status and functions of FRMD5 may not be dependent on the type of cancer tissue but rather on genetic alternations in cancer cells. Interestingly, such an anti-migratory gene expression-dependent effect was previously reported for podoplanin, a membrane glycoprotein, which is also predominantly expressed in mutant
BRAF V600E PTC cells [
16].
Altered motility of FRMD5-deficient TPC1 and BCPAP cells was also associated with increased or decreased (respectively) adhesion to selected ECM proteins. Therefore, discrepancies in adherence of siFRMD5-treated TPC1 and BCPAP cells may at least partially explain the detected opposite migratory patterns.
Another novel finding concerned the ability of FRMD5 to modulate multidrug resistance genes. Intriguingly, depletion of FRMD5 in both cell lines resulted in the upregulation of key ABC transporter genes (P-gp and MRP6). This, in turn, led to increased survival of the cells treated with the chemotherapeutic agent (DOX). We cannot exclude that FRMD5 affects the expression of MDR genes indirectly. We consider that the mechanism of MDR regulation is rather independent of the BRAF mutational status, as a decrease in DOX sensitivity was observed in both tested cell lines. As induction of drug resistance by FRMD5 is unclear, the mechanism and critical molecular players involved in this phenomenon must be further elucidated.
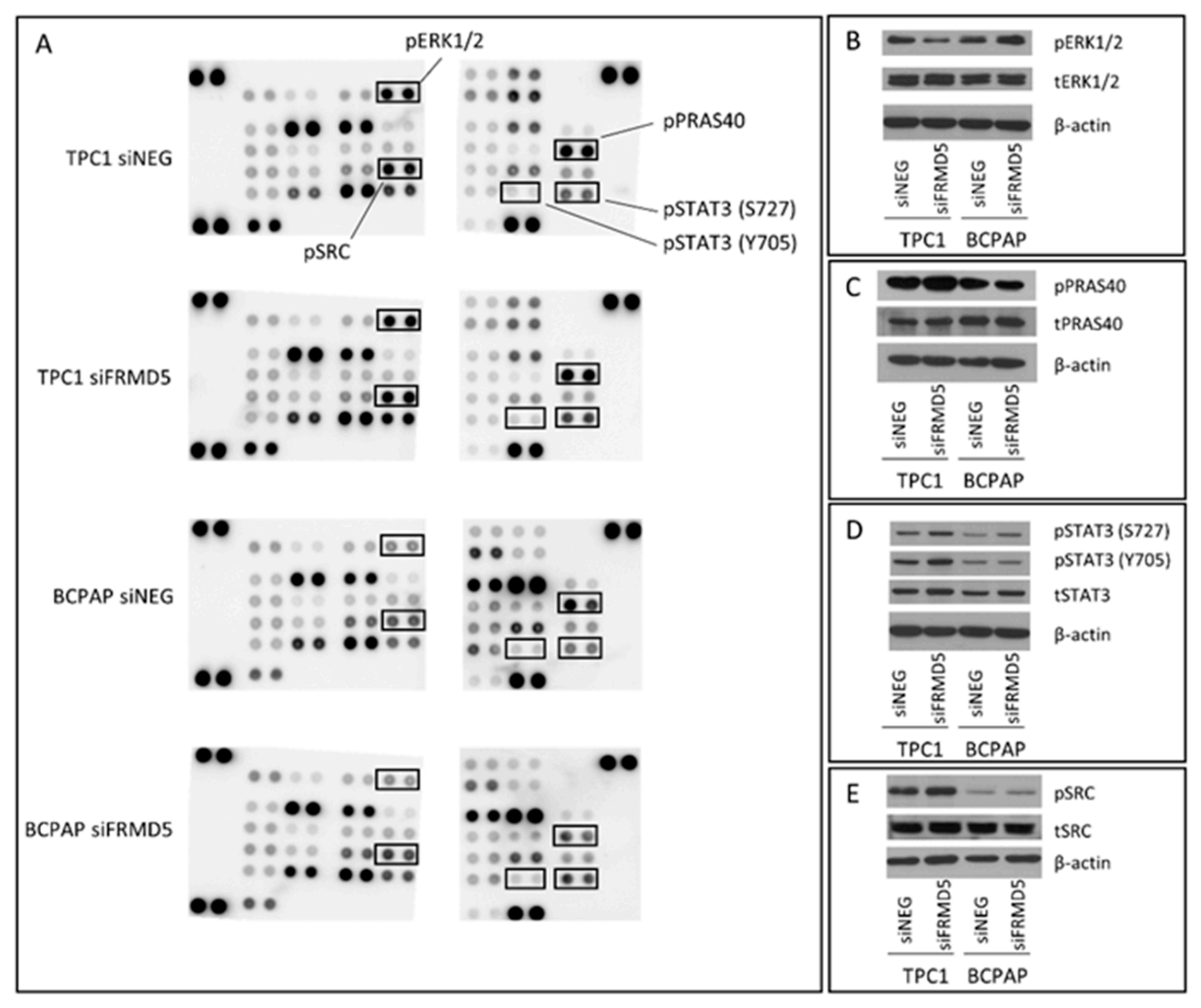
Phospho-kinase array and RNA-Seq analyses were completed to identify signaling pathways and genes associated with the observed FRMD5 phenotype. The study on the profile of phospho-kinases revealed that lack of FRMD5 may significantly disturb the activity of various signaling pathways. The activation status of several kinases, including SRC and two STAT family kinases (3 and 5), was upregulated in both cell lines, while other kinases were deregulated uniquely in only one of the cell lines. For example, two STAT (1 and 6) and extracellular signal-regulated kinases 1/2 (ERK1/2) proteins were upregulated only in BCPAP cells, while c-JUN was repressed in TPC1
FRMD5-deficient cells. Upregulation of STAT family proteins is especially intriguing, as they are partners of JAK, another FERM-domain protein, and play a significant role in tumorigenic processes. Disrupted JAK-STAT signaling affects cellular growth and transformation, preventing apoptosis or tumor formation [
17]. Therefore, it can be considered that an interaction between the FRMD5 protein and the JAK-STAT axis may exist.
Furthermore, a detailed analysis of ERK1/2 in FERM-depleted cells showed their increased activation in BCPAP and decreased phosphorylation in TPC1 cell lines. As ERK1/2 is one of the major regulators of the mitogen-activated protein kinase (MAPK) pathway, their deregulation may partially explain the observed variances in metastases of
FRMD5-silenced TPC1 and BCPAP cells. In addition, other phosphokinases, AKT and cAMP response element-binding protein (CREB), were upregulated in TPC1 and downregulated in BCPAP
FRMD5-depleted cells. AKT is a critical regulator of the PI3K/AKT/mTOR signaling pathway [
18], while CREB is considered a key mediator in carcinogenesis, including invasion and metastasis [
19]. As apoptosis was not significantly affected by
FRMD5 silencing in both cell lines, an alternative role of the PI3K/AKT/mTOR pathway must be considered.
The role of altered adhesion, hampering migration of siFRMD5-treated TPC1 cells, may be supported by the activation of the SRC kinase in FRMD5-depleted cells. Surprisingly, β-catenin, another important protein involved in the regulation of cell–cell adhesion and previously marked as one of the potential FRMD5 partners, was not affected in FRMD5-silenced cells.
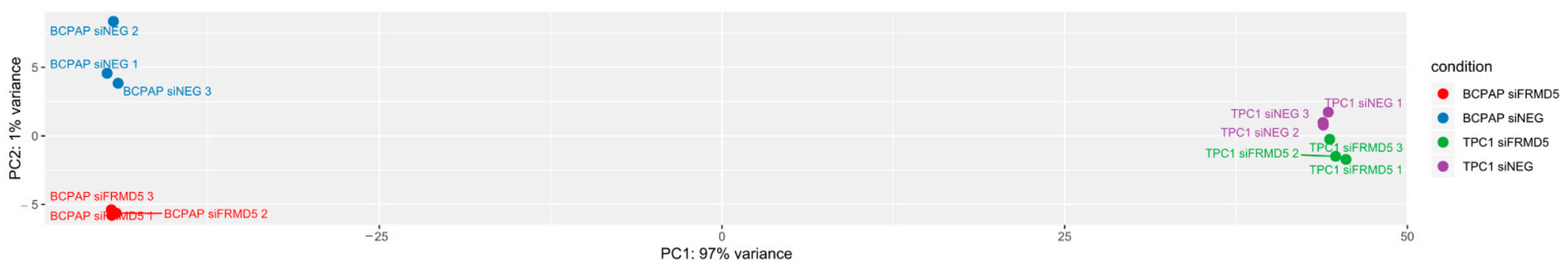
The analysis using the RNA-Seq technique revealed hundreds of genes, which were up- or downregulated in FRMD5-suppressed TPC1 and BCPAP cells. Examination of the GO term enrichment of the differentially expressed genes in both studied cell lines revealed distinct differences. In siFRMD5-treated TPC1 cells, none of the GO terms of the upregulated genes were linked with cell migration. In contrast, a similar analysis of GO terms performed for siFRMD5-treated BCPAP cells revealed significant upregulation of genes associated with extracellular matrix organization and migration. GO terms representing downregulated genes in TPC1/siFRMD5 cells were associated with the suppression of the cells’ motility. In contrast, gene ontology enrichment analysis of the downregulated genes in BCPAP/siFRMD5 cells showed changes in processes related to cilium organization and movement, microtubule- and cytoskeleton-dependent transport.
Additional analysis of alternatively expressed genes in siFRMD5-treated cells showed significant upregulation of matrix metalloproteinases (MMPs)-encoding genes (e.g., MMP1 and MMP9) in BCPAP cells. MMPs are critical enzymes targeting extracellular matrix components, including collagen, elastin, fibronectin, and laminin proteins. MMP-driven degradation of ECM results in the remodeling of cells and promotes their detachment. Furthermore, MMPs affect cell adhesion receptors, such as E-cadherin or integrins, and therefore directly disrupt cell–cell adhesion [
20]. As MMPs are the most significant enzymes in the degradation of the extracellular matrix, their observed upregulation may explain both observed increased motility and decreased adhesion of BCPAP/siFRMD5 cells. Overall, RNA-Seq and GO term profiles stay in accordance with the observed phenotype of FRMD5-deficient PTC-derived cells, which showed significant changes in migratory properties.
In conclusion, we have provided a novel insight into the role of FRMD5 in cancer cell biology. Moreover, it is the first trial reporting a genome-wide analysis of gene expression in FRMD5-deficient cells. Our findings indicate that the biological properties of FRMD5 are strongly dependent on the mutational status of cancer cells. It has been shown that lack of FRMD5 promotes the cells’ migration, likely via activation of the MAPK pathway of BRAF-mutated BCPAP cells but decreases motility of PTC-derived BRAF-wt TPC1 cells. It has also been presented that FRMD5 depletion affects the expression of various other contributors of metastases, mainly by regulating cellular matrix organization, cell–cell, and cell–matrix adhesion.
4. Materials and Methods
4.1. Retrieval of TCGA and GTEx Data
A visualization website GEPIA (Gene Expression Profiling Interactive Analysis), available at
http://gepia2.cancer-pku.cn/#analysis (accessed on 24 May 2021), was used to analyze
FRMD5 expression in cancer and normal thyroid tissues in the Genotype Tissue Expression (GTEx) and the Cancer Genome Atlas (TCGA) projects data. The expression analyses were performed using the following terms: Gene,
FRMD5; Dataset (cancer name), THCA; log 2-fold change cutoff, 1;
p-value cut-off, 0.05; differential methods, ANOVA.
4.2. Microarray Data Analysis
FRMD5 gene expression analysis was examined using previously published microarray data [
6,
14]. The dataset contained 54 samples of PTC, including 30
BRAF-wt and 24
BRAF V600E. The quality of the data was checked using dedicated software—Affymetrix Expression Console. Analysis was performed on gene and exome levels according to Affymetrix’s whitepaper. All data passed the quality test. Further analysis was performed in R Studio. Raw data were pre-processed; background subtraction, normalization, and summarization were performed with the fRMA algorithm implemented in the fRMA package. Arguments specifying methods for each step were selected as follows: for background correction, it was RMA, for normalization—quantile method, and for summarization: median polish. Due to the occurrence of batch effect between two series of sample processing, batch effect removal was necessary. It was performed with COMBAT (Empirical Bayes method), implemented in SVA package. Afterward, control probe sets were removed, and only those described as “main” in Affymetrix documentation were used for further analysis. Afterward, the
p-value and false discovery rate (FDR) were calculated to detect the significance of expression deregulation in the
FRMD5 gene. The boxplots with the expression of
FRMD5 in both subsets were created using the ggplot2 package.
4.3. Cell Culture
The experiments were performed on cell lines derived from human papillary thyroid cancer, BCPAP bearing the
BRAF V600E mutation, and TPC1 bearing
RET/PTC rearrangement. BCPAP cells were purchased from the German Collection of Microorganisms. TPC1 cells were kindly provided by Prof. M. Santoro (The University of Naples Federico II, Naples, Italy) and were authenticated by short tandem repeat (STR) analysis at the American Type Culture Collection (ATCC), as already described [
21]. Cells were propagated in Roswell Park Memorial Institute (RPMI) 1640 medium (Cat No. SH30027.FS, HyClone, Cytiva, Marlborough, MA, USA) supplemented with 10% fetal bovine serum (FBS; Cat No. SV30160.03, HyClone, Cytiva). Cells were cultured in a humidified incubator at 37 °C with 5% CO
2.
4.4. Silencing of FRMD5 by Small Interfering RNA (siRNA)
The
FRMD5 gene was silenced by transfection with siRNA specifically targeting human FRMD5 (siFRMD5; siRNA ID: s39794, Silencer Select, ThermoFisher Scientific, Inc., Rockford, IL, USA) or MISSION siRNA Universal Negative Control (siNEG; Cat No. SIC001, Sigma-Aldrich, Steinheim, Germany), and Lipofectamine 2000 (Cat No. 11668019, ThermoFisher Scientific, Inc.) in Opti-MEM I Reduced Serum Medium (Cat No. 11058021, ThermoFisher Scientific, Inc.), as previously described [
22]. The efficiency of
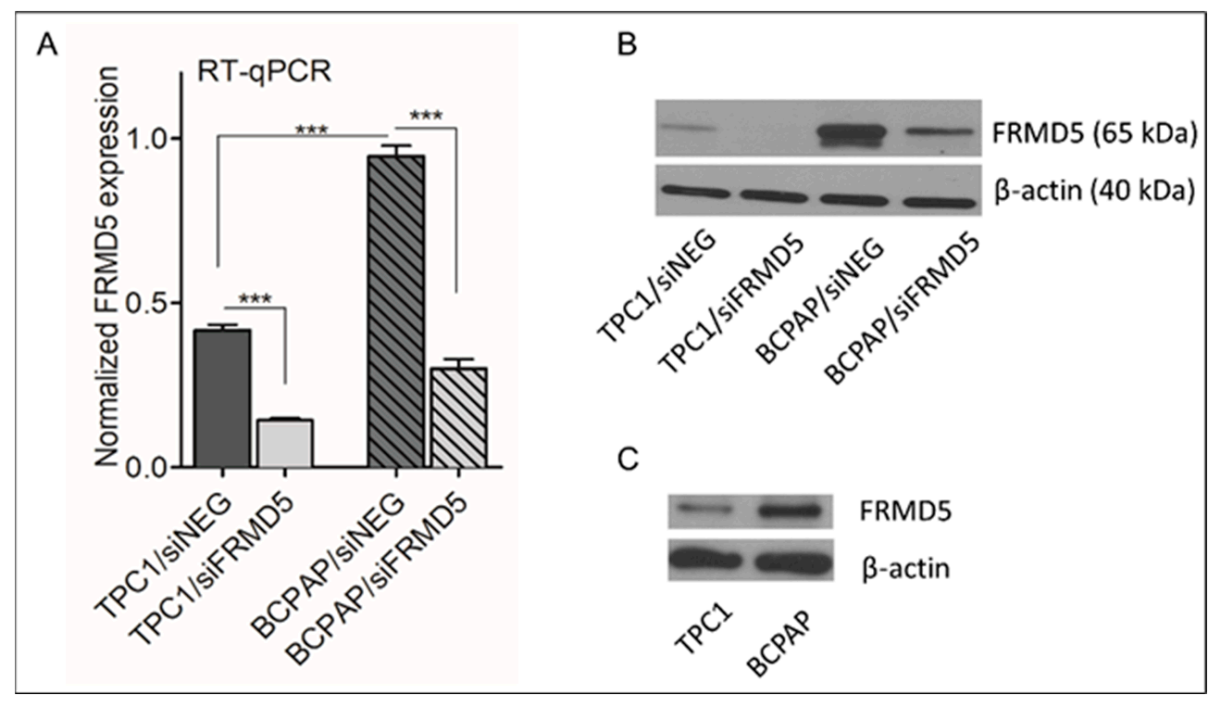
FRMD5-knockdown was verified by RT-qPCR and Western blotting.
4.5. Total RNA Extraction, Reverse Transcription, and RT-qPCR
Forty-eight hours post-transfection, total RNA was isolated from cells using GeneMATRIX Universal RNA Purification Kit (Cat No. E3598, EURx, Gdansk, Poland) with the step of the on-column DNase I (Cat No. 1009-100, A&A Biotechnology, Gdynia, Poland) digestion according to the producer’s instructions. Five hundred nanograms of total RNA was reverse transcribed using a High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor (Cat No. 4374966, ThermoFisher Scientific, Inc.), following the recommended protocol. The expression of the genes was quantified by RT-qPCR using a 5x HOT FIREPol EvaGreen qPCR Mix Plus (Cat No. 08-25-00020, Solis BioDyne, Tartu, Estonia), 6-times diluted cDNA, and 0.5 µM of specific oligonucleotide primers (listed in
Table 2). RT-qPCR was performed in the CFX96 Detection System (Bio-Rad, Hercules, CA, USA) with one cycle of 15 min at 95 °C, followed by 40 cycles of 15 s at 95 °C, 20 s at 58 °C, and 20 s at 72 °C. The expression of target genes was calculated using the 2
−ΔΔCt method and normalized to the
18S rRNA reference gene.
4.6. RNA-Seq Library Preparation and Sequencing
The integrity of the extracted RNA was evaluated using the 2100 Bioanalyzer instrument (Agilent Technologies, Inc., St. Clara, CA, USA) and the Agilent RNA 6000 Nano Kit (Cat No. 5067-1511, Agilent Technologies, Inc.). Samples with RIN (RNA integrity number) values of 8 or greater were taken to construct a cDNA library. The Ion AmpliSeq Transcriptome Human Gene Expression Panel (Cat No. A31446, ThermoFisher Scientific, Inc.) was applied for the preparation of the library, following the manufacturer’s protocol [
23]. In brief, cDNA libraries were prepared with the Ion AmpliSeq Transcriptome Human Gene Expression Panel (ThermoFisher Scientific, Inc.) according to the manufacturer’s protocol. RNA samples were reverse-transcribed, and cDNA libraries were generated and quantified on the Bioanalyzer 2100 with a High Sensitivity DNA Kit (Agilent Technologies, Inc.). Eight barcoded library templates (50 pM) were mixed and loaded onto Ion PI chips with Ion Chef Instrument and sequenced on an Ion Proton Sequencer (ThermoFisher Scientific, Inc.) with the Ion PI Hi-Q Chef Kit, according to the manufacturer’s instructions. Signal processing and base calling was conducted using the Ion Torrent Suite v5.10 (
https://github.com/iontorrent/TS, accessed on 17 January 2020). The read-outs were mapped to the hg19 AmpliSeq Transcriptome v1 genome with tmap v5.10. Read counts per gene were obtained with HTSeq-count [
24] v0.6, using default parameters. Normalization and differential gene expression estimations were performed using DESeq2 [
25] v1.18.1, using default parameters and options. A gene was considered differentially expressed when the adjusted
p-value was less than 0.05. Functional analysis of differentially expressed genes was conducted with the Cytoscape [
26] (v3.7.2) plugin ClueGo [
27] (v2.5.7), on a database consisting of Biological Process (BP) and Molecular Function (MF) categories in Gene Ontology [
28] (GO, version 8 May 2020). Benjamini–Hochberg
p-value adjustment was used, and terms with adjusted
p-value less than 0.05 were considered significant. GO enrichment visualization was prepared with GOplot [
29] R package v1.0.2, using a reduction of similar terms (90% for BCPAP and 70% for TPC1).
4.7. Total Protein Isolation and Western Blotting
Seventy-two hours after transfection, the cells were washed 3 times with chilled phosphate-buffered saline (PBS; pH 7.3) and lysed with RIPA Lysis and Extraction Buffer (Cat No. 89900, ThermoFisher Scientific, Inc.) supplemented with Pierce Phosphate Inhibitor Cocktail (Cat No. A32957, ThermoFisher Scientific, Inc.), Complete Protease Inhibitor Cocktail (Cat No. 4693116001, Roche, Basel, Switzerland), and Viscolase (Cat No. 1010-100, A&A Biotechnology) on ice for 30 min. Protein extraction from tissue samples was performed as described previously [
30]. After one freeze-thaw cycle, the total protein concentration in the cell lysate was determined using the BCA Protein Assay Kit (Cat No. 23225, ThermoFisher Scientific, Inc.). The membrane was further processed as already described, with some minor modifications [
31]. Twenty to thirty micrograms of the total protein lysate was resolved in 9% SDS-PAGE under reducing conditions and subsequently electro-transferred to a PVDF membrane (Cat No. IPVH00010, Merck Millipore, Tullagreen, Ireland). After one hour blocking in 5% skimmed milk in Tris-buffered saline (TBS) supplemented with 0.1% Tween 20 (TBST) at room temperature (RT) and extensive washing in TBST, the membrane was probed overnight at 4 °C with a primary antibody diluted in 5% skimmed milk-TBST or 5% BSA (Cat No. 7906, Sigma-Aldrich)-TBST (primary antibodies and blocking agents used in the study are listed in
Table 3). After extensive washing, the membrane was incubated for 1 h at RT with secondary antibodies (1:5000 in 1% skimmed milk-TBST): goat anti-rabbit immunoglobulins/horseradish peroxidase (HRP; Cat No. P0448, Dako, Carpinteria, CA, USA), or goat anti-mouse immunoglobulins/HRP (Cat No. 115-035-146, Jackson ImmunoResearch Lab, Inc., West Grove, PA, USA). Then, the membrane was intensively washed, and signals from reactive bands were visualized using the SuperSignal West Dura Extended Duration Substrate (Cat No. 34076, ThermoFisher Scientific, Inc.) or SuperSignal West Pico PLUS Chemiluminescent Substrate (Cat No. 34577, ThermoFisher Scientific, Inc.).
4.8. Phospho-Kinase Proteome Profiling
The assay was performed using the Human Phospho-Kinase Array Kit (Cat No. ARY003C, R&D Systems), according to the manufacturer’s instructions. Seventy-two hours post-transfection, the cells were rinsed three times with chilled phosphate-buffered saline (PBS; pH 7.3) and lysed with Lysis Buffer 6 supplemented with Complete Protease Inhibitor Cocktail (Roche), Pierce Phosphate Inhibitor Cocktail (ThermoFisher Scientific, Inc.), and nuclease (Viscolase; A&A Biotechnology) on ice for 30 min. After a single freeze–thaw cycle, the total protein concentration in the cell lysate was determined using a BCA Protein Assay Kit (ThermoFisher Scientific, Inc.). The array membranes were blocked in Array Buffer 1 for 1 h and incubated overnight with 600 µg of total protein lysate in Array Buffer 1 at 4 °C. Then, they were probed with a Detection Antibody Cocktail for 2 h, followed by 30-min incubation with Streptavidin-HRP solution. After each step, the membranes were washed three times with Wash Buffer. The chemiluminescent signal was developed using the Chemi Reagent Mix and captured on the Mini HD9 acquisition system (Uvitec Ltd., Cambridge, UK). The results were confirmed by Western blot analysis.
4.9. Cell Migration and Matrigel Invasion Assays
The cell migration and invasion assays were determined using 8-μm pore non-coated (Cat No. 353097, Corning, Inc., Corning, New York, NY, USA) and Matrigel-coated (Cat No. 354480, Corning) inserts, respectively, as already described [
16,
22]. Briefly, 48-h after transfection, the harvested cells (2 × 10
5) were added to the upper chambers in a serum-free medium and cultured for a further 24 h. Complete medium in the lower well was used as a chemoattractant. Cells that migrated or invaded through the membranes underwent fixation and staining using a RAL Diff-Quik kit (Cat No. 720555-0000, RAL Diagnostics, Martillac, France) and the cells were counted using Olympus BX41 microscope (Olympus Corporation, Tokyo, Japan) with a 40× objective lens.
4.10. In Vitro Wound Healing Motility Assay
A wound-healing assay was performed as already described [
22]. Briefly, siRNA-treated cells were seeded on a 6-well plate and cultured until the formation of a nearly confluent monolayer. Then, a scratch wound was created using a sterile 200 μL pipette tip. Any cellular debris was removed by washing with Dulbecco’s phosphate-buffered saline without calcium and magnesium (D-PBS; Cat No. SH30028.FS, HyClone, Cytiva), and then growth medium was added. Images of the scratched areas were captured between 0 and 48 h using a light microscope (10× lens; AxioObserver D1, Carl Zeiss AG, Oberkochen, Germany) equipped with AxioVision LE software (Carl Zeiss AG). The cell migration distance was calculated by measuring the wound width, dividing it by two, and subtracting this value from the initial half-wound width. For the 10× lens, 1 pixel is equal to 1.026 µm.
4.11. Cell Adhesion Assay
The assay was performed using a colorimetric ECM Adhesion Array Kit (Cat No. ECM 540, Merck Millipore), following the manufacturer’s instructions. Seventy-two hours after FRMD5 silencing, cells were detached with HyQTase Cell Detachment Solution (Cat No. SV30030.01, HyClone, Cytiva) and quenched with Dulbecco’s Modified Eagle Medium (DMEM; Cat No. 10-013-CV, Corning, Inc.) supplemented with 5% BSA. Then, 100 µL of cell suspension (1 × 106/mL in Assay Buffer) was added to each well. After a two-hour incubation, the plate was washed with Assay Buffer and stained with Cell Stain Solution for 5 min. The excess stain was removed by washing with deionized water, and the plate was air-dried. After the dye solubilization with Extraction Buffer for 10 min, the absorbance was determined using Synergy 2 Multi-Mode Microplate Reader (BioTek Instruments, Inc., Winooski, VT, USA) at 560 nm.
4.12. Cell Proliferation Assay
The assay was performed using BrdU-based colorimetric Cell Proliferation ELISA (Cat No. 11647229001, Roche) following the manufacturer’s protocol. Five thousand siRNA-transfected cells in a final volume of 100 µL were seeded in 96-well plates (8 replicates). Forty-eight hours later, 10 µL of BrdU labeling solution was added to each well, and the incubation was continued for an extra 7 h. The culture medium was removed, cells were fixed, and DNA was denatured by adding FixDenat. Then, cells were incubated with a BrdU-specific antibody for 90 min, and unbound antibody conjugates were removed in three washing cycles. After 15-min of incubation with tetramethylbenzidine (TMB) substrate, the absorbance was determined using the Synergy 2 Multi-Mode Microplate Reader (BioTek Instruments, Inc.) at 370 nm with a reference wavelength of 492 nm and finally expressed as the percentage of control (siNEG-treated cells).
4.13. Cell Viability Assay (MTS-Based Assay)
The assay was performed using the MTS CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay (MTS; Cat No. G3581, Promega Corporation, Madison, WI, USA) according to the manufacturer’s protocol. Five thousand siRNA-transfected cells in a final volume of 100 µL were seeded in 96-well plates (12 replicates). After 48 h, 20 µL of MTS reagent was added to each well, and the incubation was continued for an additional 3 h. In some experiments, 48 h post-transfection, the culture medium was supplemented with doxorubicin (Cat No. T1020, TargetMol, Boston, MA, USA), at a final concentration of 10 µM and MTS-based cell viability assay was performed 24 h later. The absorbance readings were recorded at 490 nm with a reference wavelength of 650 nm using the Synergy 2 Multi-Mode Microplate Reader (BioTek Instruments, Inc.). The data are expressed as the percent of control (siNEG-treated cells).
4.14. Cell Viability Assay (Trypan Blue-Based Assay)
Seventy-two hours after transfection, the trypan blue exclusion assay was performed, as described elsewhere [
32,
33]. Briefly, all cells (both attached and unattached) were harvested, centrifuged, resuspended in D-PBS (HyClone, Cytiva), and stained with trypan blue dye (NanoEnTek, Inc., Seoul, Korea) at a final concentration of 0.2%. The number of total and viable cells was determined using an automated cell counter (EVE; NanoEnTek, Inc.). Results are expressed as the percent of viable cells.
4.15. Cell Viability Assay (Annexin V/Propidium Iodide-Based Flow Cytometry)
The viability of siRNA-treated cells (for 72 h) using the FITC Annexin V Apoptosis Detection Kit I (Cat No. 556547, BD Biosciences, San Diego, CA, USA) was performed, as suggested by the manufacturer. Briefly, all cells were mixed, washed with D-PBS (HyClone, Cytiva), resuspended in Binding Buffer at a final concentration of 106 cells/mL. Afterward, 100 µL (1 × 105) of cell suspension was probed with 5 µL of FITC-conjugated Annexin V and 5 µL of propidium iodide for 15 min protected from light. Then, 400 µL of Binding Buffer was added to each tube, and cells were analyzed by flow cytometry using a BD Accuri C6 Plus flow cytometer and dedicated BD Biosciences software (v.1.0.23.1; BD Biosciences).
4.16. Confocal Microscopy
The transfected cells (1 × 10
5) were seeded in 6-well plates containing uncoated cover glasses in a final volume of 2 mL. Forty-eight hours later, the culture medium was supplemented with doxorubicin (Cat No. T1020, TargetMol) at a final concentration of 10 µM, and the incubation was continued for a further 17 h. Next, the cells were fixed with 4% paraformaldehyde (Cat. No. 6148, Sigma-Aldrich) in PBS, as already described [
31], followed by permeabilization with 0.25% Triton X-100 (Cat No. X-100, Sigma-Aldrich) in deionized water for 3 min. After washing with PBS, the cells were blocked with 2% BSA (Cat No. 7906, Sigma-Aldrich) in TBST for 1 h and stained with phalloidin conjugated with FITC (2 µg/mL in PBS; Cat No. P5282-.1MG, Sigma-Aldrich) and 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI; 0.4 µg/mL in deionized water; Cat No. P5282-.1MG, Sigma-Aldrich) for 30 min and 2 min, respectively. After a final wash with PBS and mounting using Fluorescence Mounting Medium (Cat No. S3023, Dako), the cells were examined using the Zeiss LSM800 confocal unit supplied with a plan-apochromatic 63x/1.4 oil DIC M27 lens (Carl Zeiss AG), as previously described [
31].
4.17. Spheroid Formation Assay
Forty-eight hours after transfection, 1 × 10
4 of 0.05% trypsin-EDTA (HyClone, Cytiva)-detached cells in a final volume of 100 µL were seeded in Costar ultra-low attachment 96-well round-bottom plates (Cat No. 7007, Corning, Inc.) in complete medium (12 replicates). Spheroid formation was monitored and visualized using AxioObserver D1 microscope (10× lens; Carl Zeiss AG) and AxioVision LE software (Carl Zeiss AG) as previously described [
32]. The area of spheroids was calculated using ImageJ (NIH) software. The results are presented as the percentage of spheroid area compared to controls (siNEG-treated cells).
4.18. Statistical Analysis
All experiments were performed at least three times. Quantitative data are expressed as mean ± standard deviation (SD). The data were analyzed using GraphPad Prism 6.0 for Windows (GraphPad, Inc., San Diego, CA, USA). For statistical purposes, nonparametric Mann–Whitney U test and one-way ANOVA followed by Bonferroni posthoc comparative test or t-student test were used. Statistical significance was considered at p-values < 0.05.


 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}