
The C-Terminal Domain of LRRK2 with the G2019S Substitution Increases Mutant A53T α-Synuclein Toxicity in Dopaminergic Neurons In Vivo

 , ,
, ,  , , add
Show full author list
, , add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
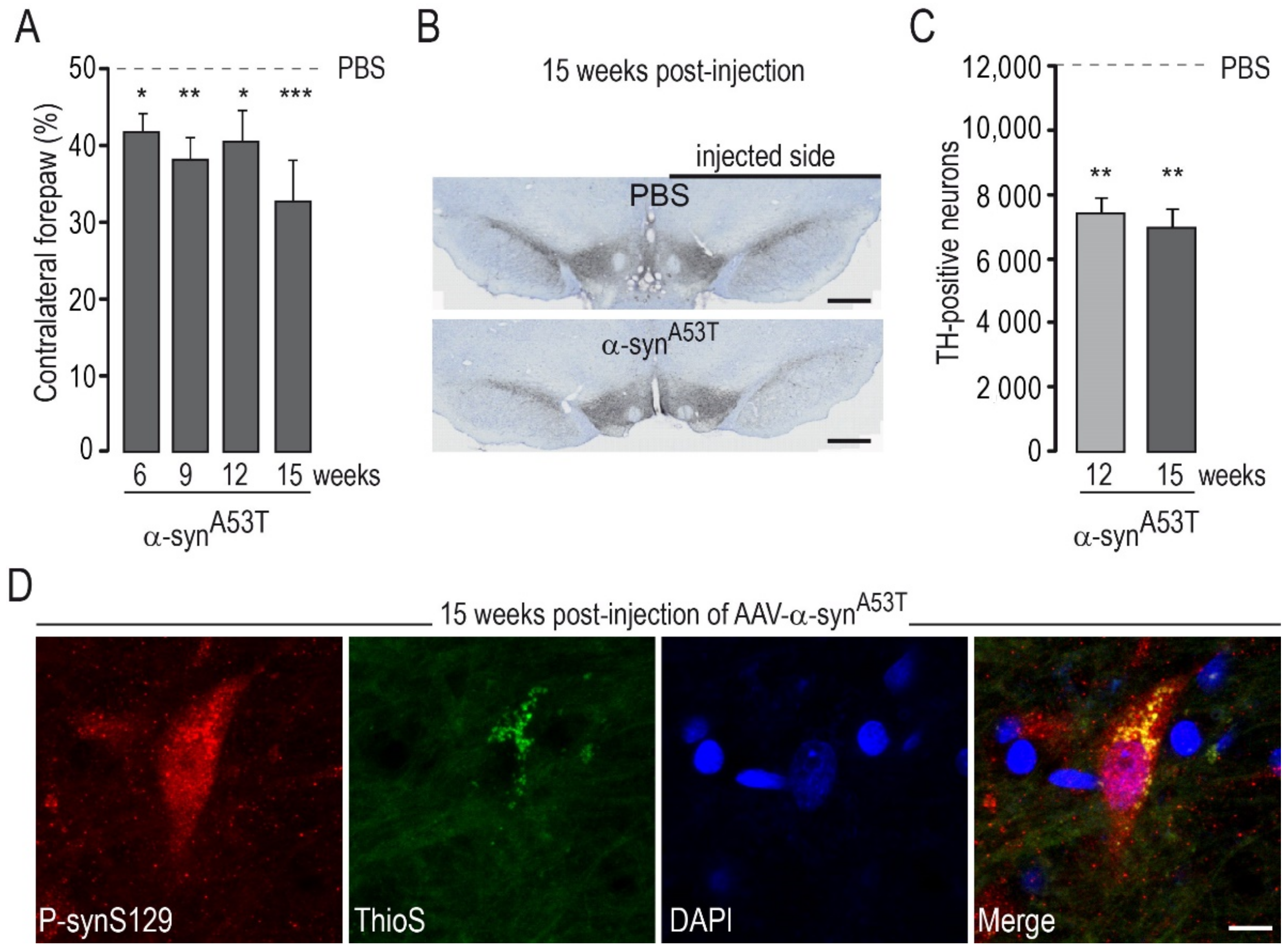
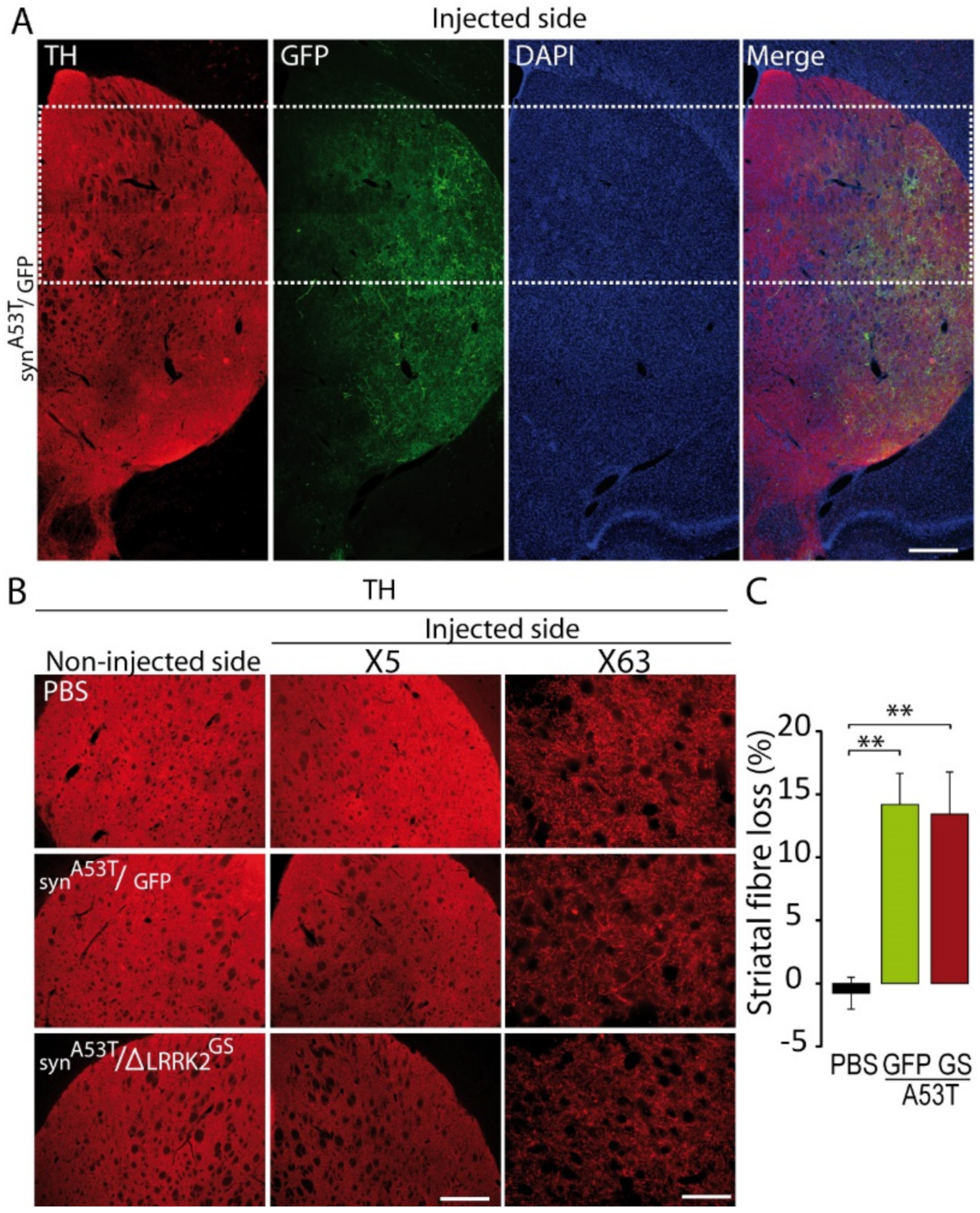
2.1. Determination of the Experimental Conditions to Detect Potential Synergy between AAV-α-SynA53T and AAV-ΔLRRK2G2019S Toxicity
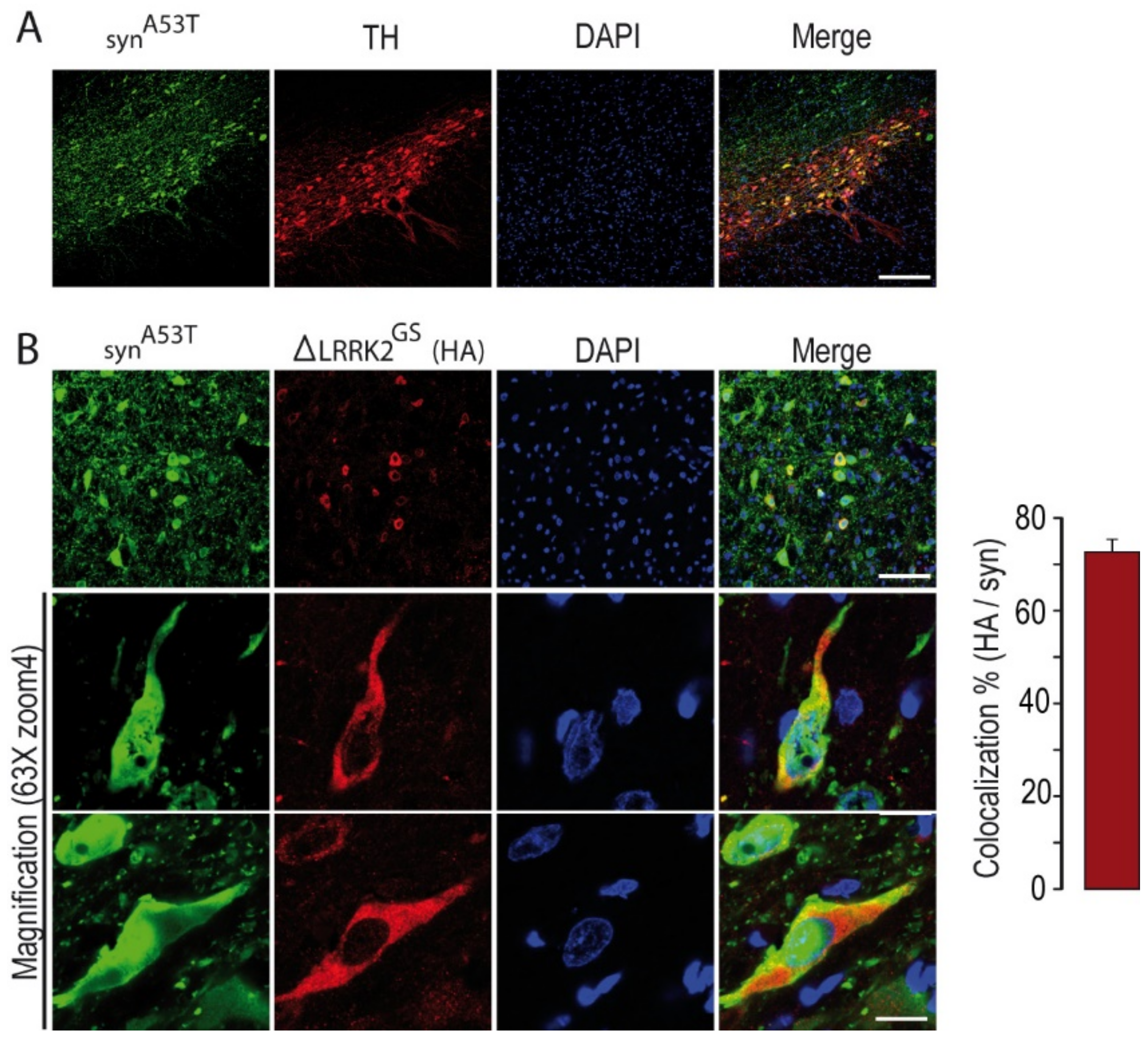
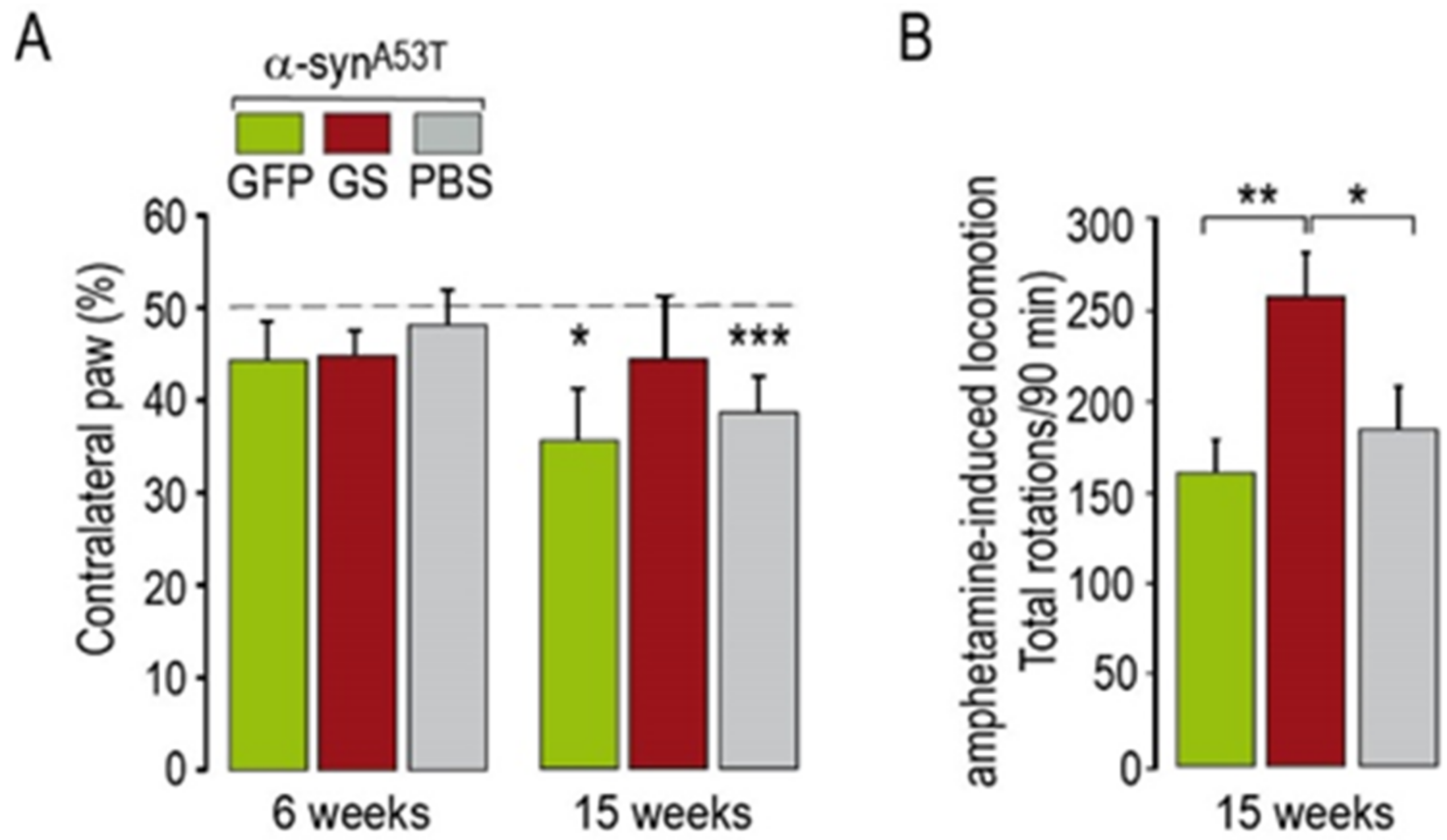
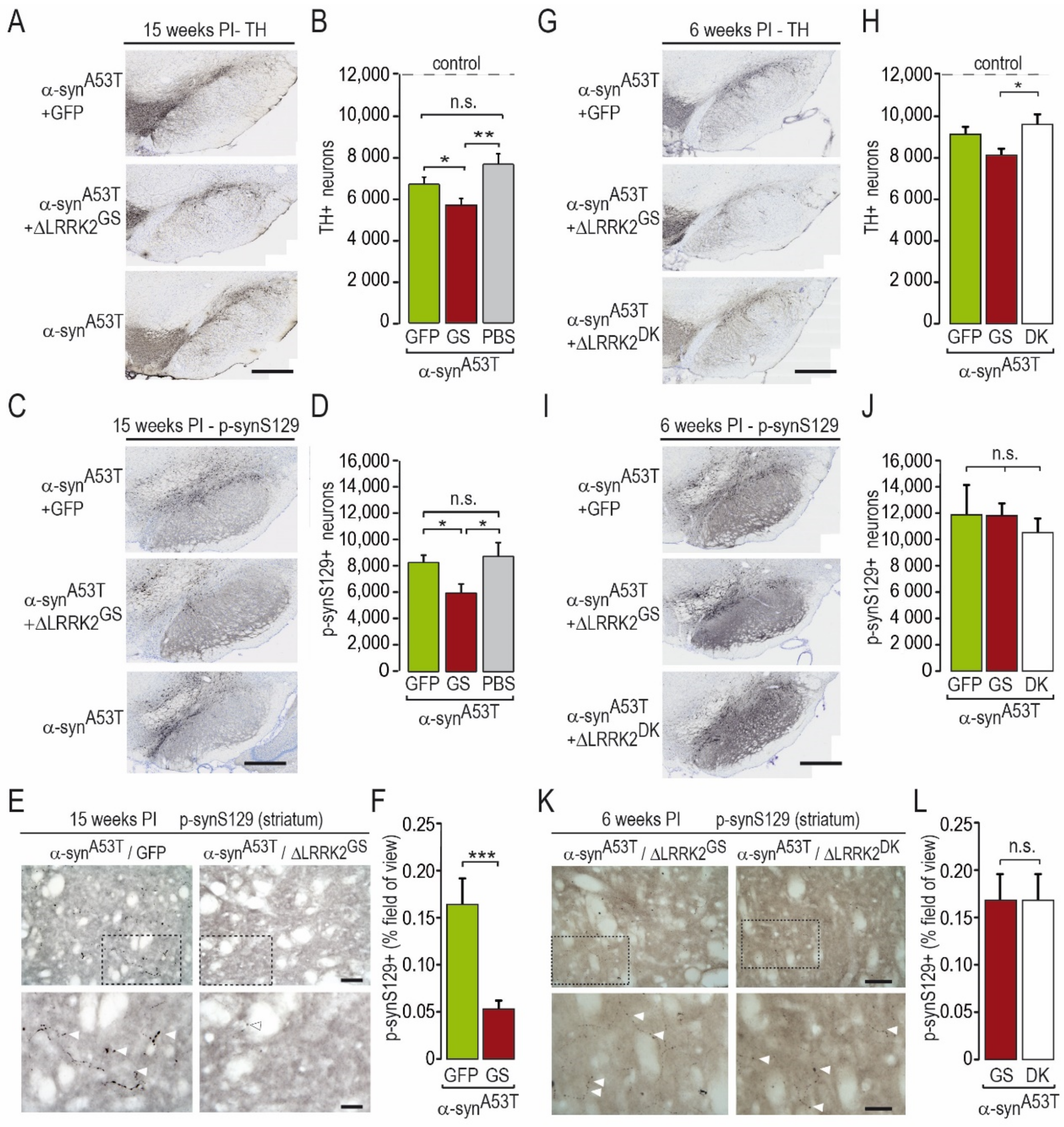
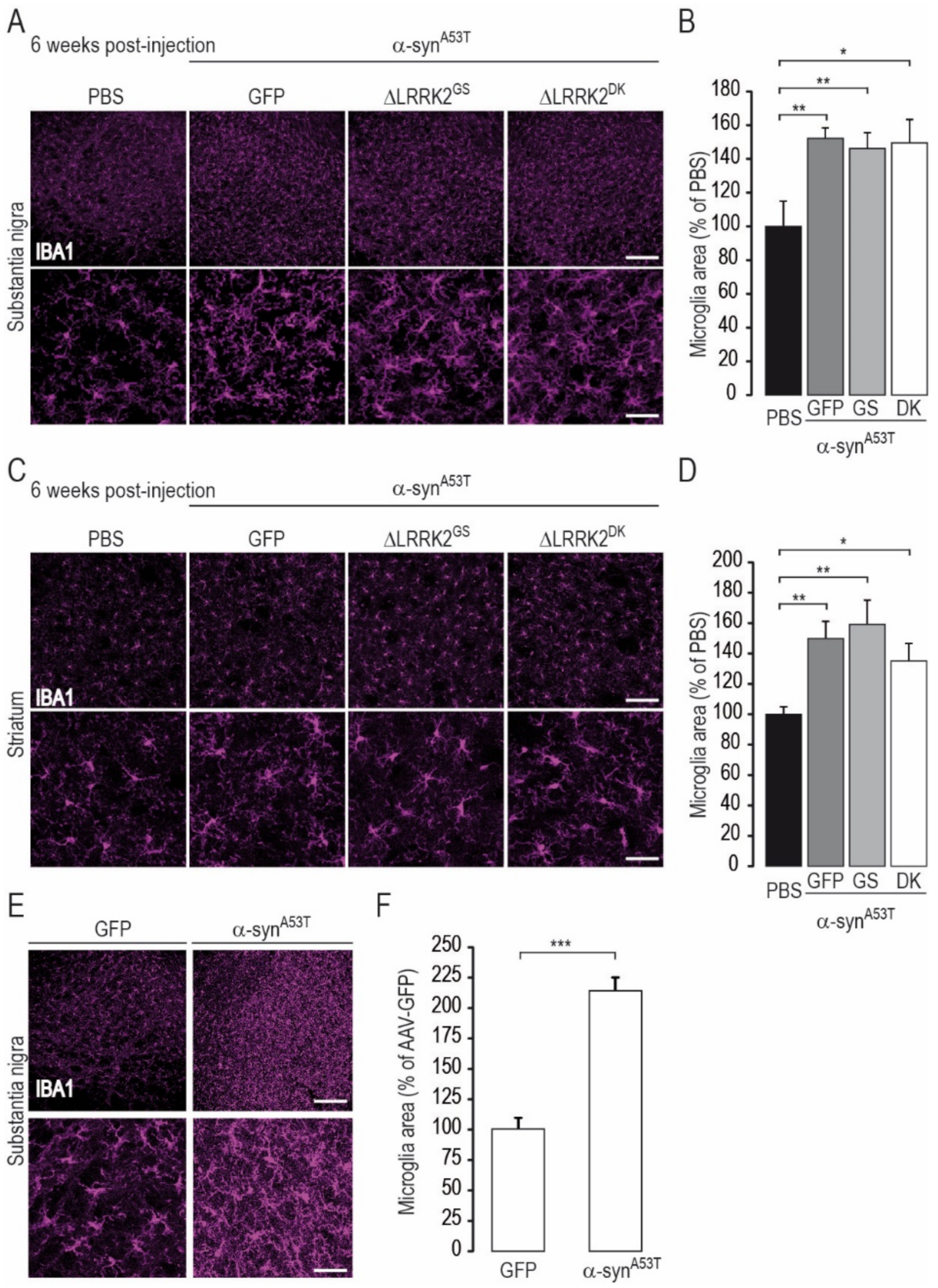
2.2. Effects of Co-Transduction with AAV-α-SynA53T and AAV-∆LRRK2G2019S
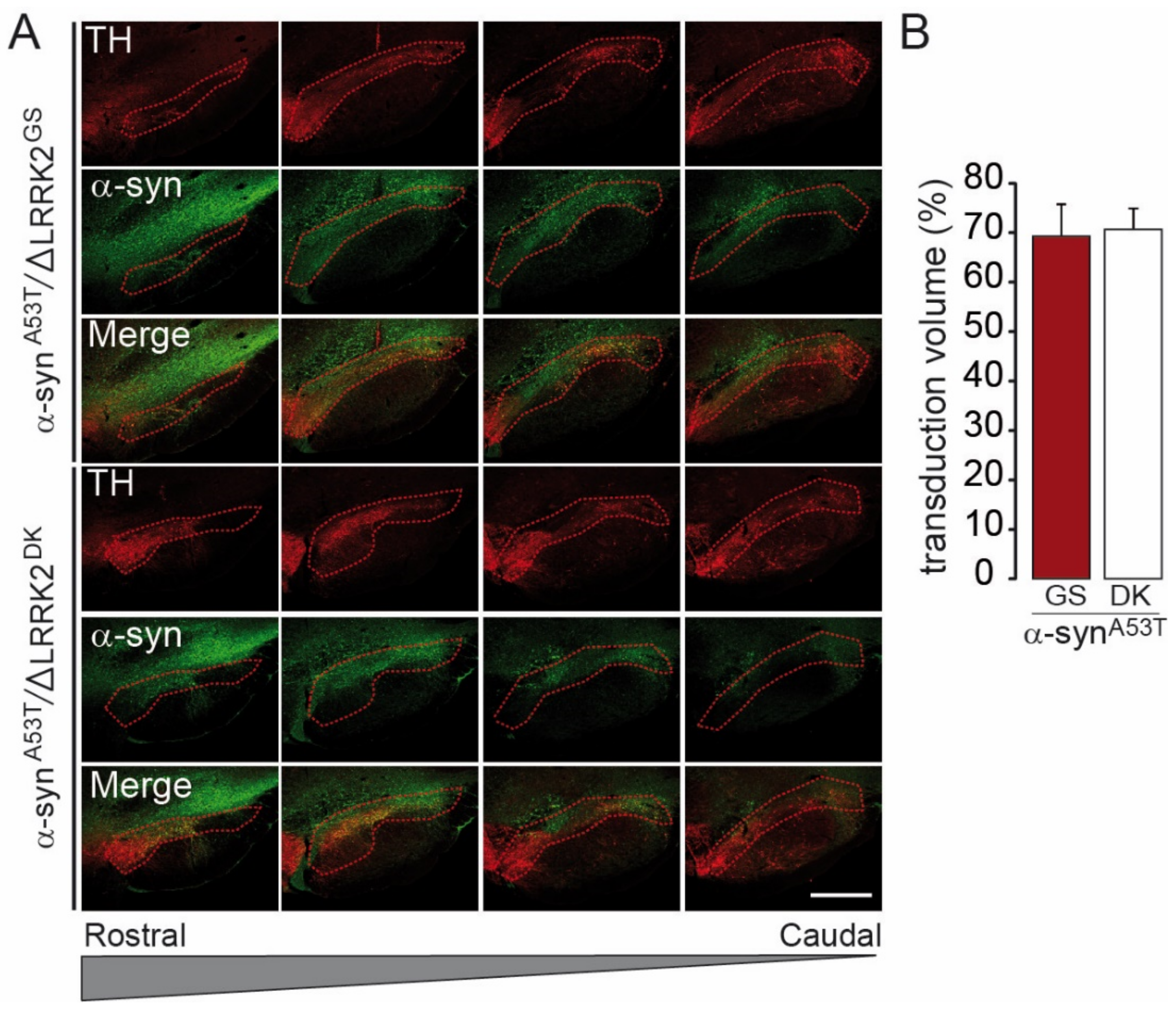
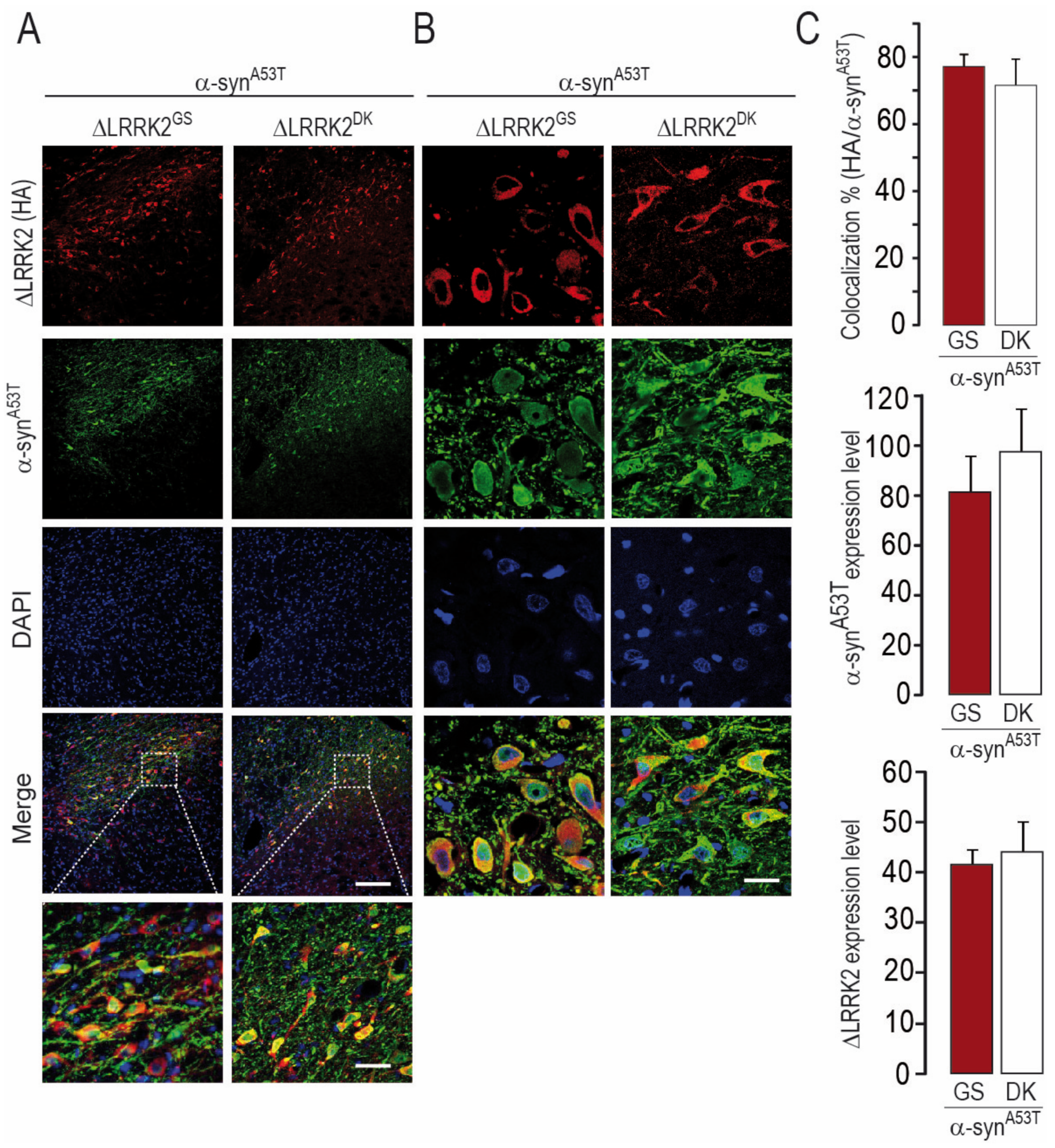
2.3. Differential Effects of AAV-∆LRRK2G2019S and AAV-∆LRRK2DK on AAV-α-SynA53T Toxicity
3. Discussion
4. Materials and Methods
4.1. Adeno-Associated Viral Vectors (AAVs) Construction and Production
4.2. Stereotaxic Injection
4.3. Evaluation of Motor Behavior
4.4. Tissue Processing
4.5. Immunohistological Analysis and Quantification
4.5.1. Immunohistochemistry
4.5.2. Cell Counting
4.5.3. Immunofluorescence
4.5.4. Thioflavin-S Staining
4.5.5. Colocalization
4.5.6. Epifluorescence Intensity Measurement
4.5.7. Microglia Area Measurement
4.5.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ∆LRRK2 | ROC-COR-kinase plus the WD40 domain |
| AAV | adeno-associated virus |
| ANOVA | analysis of variance |
| CamKII | calmodulin-kinase II |
| COR | C-terminus of ROC |
| DA | dopaminergic |
| DK | double-mutant G2019S/D1994A dead kinase |
| GFP | green fluorescent protein |
| GS | G2019S mutation |
| HA | hemagglutinin tag |
| K | kinase domain of LRRK2 |
| LB | lysis buffer (50 mm Tris, pH 8.0, 150 mm NaCl, 1 mm EDTA, 0.5% Triton X-100, 1% NP40, protease inhibitors) |
| LRR | leucin-rich repeats |
| LRRK2 | leucin-rich repeats kinase 2 |
| p-synS129 | α-synuclein phosphorylated at serine 129 |
| PBS | phosphate-buffered saline |
| PBS-T | phosphate-buffered saline with 0.2% Triton X-100 |
| PD | Parkinson’s disease |
| RCK | kinase domain plus the ROC-COR domain |
| ROC | Ras-of-complex protein |
| SNpc | substantia nigra pars compacta |
| SNPs | single-nucleotide polymorphisms |
| TH | tyrosine hydroxylase |
| Vg | viral particle |
| WT | wild-type |
References
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson Disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Oroz, M.C.; Jahanshahi, M.; Krack, P.; Litvan, I.; Macias, R.; Bezard, E.; Obeso, J.A. Initial Clinical Manifestations of Parkinson’s Disease: Features and Pathophysiological Mechanisms. Lancet Neurol. 2009, 8, 1128–1139. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Braak, E. Pathoanatomy of Parkinson’s Disease. J. Neurol. 2000, 247, II3-II10. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Brice, A. Parkinson’s Disease: From Monogenic Forms to Genetic Susceptibility Factors. Hum. Mol. Genet. 2009, 18, R48–R59. [Google Scholar] [CrossRef]
- Chartier-Harlin, M.C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. Alpha-Synuclein Locus Duplication as a Cause of Familial Parkinson’s Disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Cobb, M.M.; Ravisankar, A.; Skibinski, G.; Finkbeiner, S. IPS Cells in the Study of PD Molecular Pathogenesis. Cell Tissue Res. 2018, 373, 61–77. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. Alpha-Synuclein Locus Triplication Causes Parkinson’s Disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [Green Version]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the α-Synuclein Gene Identified in Families with Parkinson’s Disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [Green Version]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. AlaSOPro Mutation in the Gene Encoding α-Synuclein in Parkinson’s Disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The New Mutation, E46K, of α-Synuclein Causes Parkinson and Lewy Body Dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef]
- Saito, Y.; Kawashima, A.; Ruberu, N.N.; Fujiwara, H.; Koyama, S.; Sawabe, M.; Arai, T.; Nagura, H.; Yamanouchi, H.; Hasegawa, M.; et al. Accumulation of Phosphorylated α-Synuclein in Aging Human Brain. J. Neuropathol. Exp. Neurol. 2003, 62, 644–654. [Google Scholar] [CrossRef] [Green Version]
- Paisán-Ruíz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simón, J.; Van Der Brug, M.; De Munain, A.L.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the Gene Containing Mutations That Cause PARK8-Linked Parkinson’s Disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef] [Green Version]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 Cause Autosomal-Dominant Parkinsonism with Pleomorphic Pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [Green Version]
- Foo, J.N.; Tan, L.C.; Irwan, I.D.; Au, W.-L.; Low, H.Q.; Prakash, K.-M.; Ahmad-Annuar, A.; Bei, J.; Chan, A.Y.; Chen, C.M.; et al. Genome-Wide Association Study of Parkinson’s Disease in East Asians. Hum. Mol. Genet. 2017, 26, 226–232. [Google Scholar] [CrossRef] [Green Version]
- Satake, W.; Nakabayashi, Y.; Mizuta, I.; Hirota, Y.; Ito, C.; Kubo, M.; Kawaguchi, T.; Tsunoda, T.; Watanabe, M.; Takeda, A.; et al. Genome-Wide Association Study Identifies Common Variants at Four Loci as Genetic Risk Factors for Parkinson’s Disease. Nat. Genet. 2009, 41, 1303–1307. [Google Scholar] [CrossRef]
- Gilks, W.P.; Abou-Sleiman, P.M.; Gandhi, S.; Jain, S.; Singleton, A.; Lees, A.J.; Shaw, K.; Bhatia, K.P.; Bonifati, V.; Quinn, N.P.; et al. A Common LRRK2 Mutation in Idiopathic Parkinson’s Disease. Lancet 2005, 365, 415–416. [Google Scholar] [CrossRef]
- Healy, D.G.; Falchi, M.; O’Sullivan, S.S.; Bonifati, V.; Durr, A.; Bressman, S.; Brice, A.; Aasly, J.; Zabetian, C.P.; Goldwurm, S.; et al. Phenotype, Genotype, and Worldwide Genetic Penetrance of LRRK2-Associated Parkinson’s Disease: A Case-Control Study. Lancet Neurol. 2008, 7, 583–590. [Google Scholar] [CrossRef] [Green Version]
- Biskup, S.; West, A.B. Zeroing in on LRRK2-Linked Pathogenic Mechanisms in Parkinson’s Disease. Biochimica et Biophysica Acta (BBA) Mol. Basis Dis. 2009, 1792, 625–633. [Google Scholar] [CrossRef] [Green Version]
- Yahalom, G.; Orlev, Y.; Cohen, O.S.; Kozlova, E.; Friedman, E.; Inzelberg, R.; Hassin-Baer, S. Motor Progression of Parkinson’s Disease with the Leucine-Rich Repeat Kinase 2 G2019S Mutation: Disease Progression in PD with G2019S Mutation. Mov. Disord. 2014, 29, 1057–1060. [Google Scholar] [CrossRef] [PubMed]
- Cresto, N.; Gardier, C.; Gubinelli, F.; Gaillard, M.-C.; Liot, G.; West, A.B.; Brouillet, E. The Unlikely Partnership Between LRRK2 and α-Synuclein in Parkinson’s Disease. Eur. J. Neurosci. 2018, 49, 339–363. [Google Scholar] [CrossRef]
- Ben Romdhan, S.; Farhat, N.; Nasri, A.; Lesage, S.; Hdiji, O.; Ben Djebara, M.; Landoulsi, Z.; Stevanin, G.; Brice, A.; Damak, M.; et al. LRRK2 G2019S Parkinson’s Disease with More Benign Phenotype than Idiopathic. Acta Neurol. Scand. 2018, 138, 425–431. [Google Scholar] [CrossRef]
- Sayad, M.; Zouambia, M.; Chaouch, M.; Ferrat, F.; Nebbal, M.; Bendini, M.; Lesage, S.; Brice, A.; Brahim Errahmani, M.; Asselah, B. Greater Improvement in LRRK2 G2019S Patients Undergoing Subthalamic Nucleus Deep Brain Stimulation Compared to Non-Mutation Carriers. BMC Neurosci. 2016, 17, 6. [Google Scholar] [CrossRef] [Green Version]
- Kalia, L.V.; Lang, A.E.; Hazrati, L.-N.; Fujioka, S.; Wszolek, Z.K.; Dickson, D.W.; Ross, O.A.; Van Deerlin, V.M.; Trojanowski, J.Q.; Hurtig, H.I.; et al. Clinical Correlations With Lewy Body Pathology in LRRK2-Related Parkinson Disease. JAMA Neurol. 2015, 72, 100–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.D.; Shin, J.-H.; VanKampen, J.; Petrucelli, L.; West, A.B.; Ko, H.S.; Lee, Y.; Maguire-Zeiss, K.A.; Bowers, W.J.; Federoff, H.J.; et al. Inhibitors of Leucine Rich Repeat Kinase 2 (LRRK2) Protect Against LRRK2-Models of Parkinson’s Disease. Nat. Med. 2010, 16, 998–1000. [Google Scholar] [CrossRef] [Green Version]
- West, A.B.; Moore, D.J.; Choi, C.; Andrabi, S.A.; Li, X.; Dikeman, D.; Biskup, S.; Zhang, Z.; Lim, K.-L.; Dawson, V.L.; et al. Parkinson’s Disease-Associated Mutations in LRRK2 Link Enhanced GTP-Binding and Kinase Activities to Neuronal Toxicity. Hum. Mol. Genet. 2007, 16, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Jeong, G.R.; Lee, B.D. Pathological Functions of LRRK2 in Parkinson’s Disease. Cells 2020, 9, 2565. [Google Scholar] [CrossRef]
- Seol, W.; Nam, D.; Son, I. Rab GTPases as Physiological Substrates of LRRK2 Kinase. Exp. Neurobiol. 2019, 28, 134–145. [Google Scholar] [CrossRef]
- Steger, M.; Tonelli, F.; Ito, G.; Davies, P.; Trost, M.; Vetter, M.; Wachter, S.; Lorentzen, E.; Duddy, G.; Wilson, S.; et al. Phosphoproteomics Reveals That Parkinson’s Disease Kinase LRRK2 Regulates a Subset of Rab GTPases. eLife 2016, 5, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Steger, M.; Diez, F.; Dhekne, H.S.; Lis, P.; Nirujogi, R.S.; Karayel, O.; Tonelli, F.; Martinez, T.N.; Lorentzen, E.; Pfeffer, S.R.; et al. Systematic Proteomic Analysis of LRRK2-Mediated Rab GTPase Phosphorylation Establishes a Connection to Ciliogenesis. eLife 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Di Maio, R.; Hoffman, E.K.; Rocha, E.M.; Keeney, M.T.; Sanders, L.H.; De Miranda, B.R.; Zharikov, A.; Van Laar, A.; Stepan, A.F.; Lanz, T.A.; et al. LRRK2 Activation in Idiopathic Parkinson’s Disease. Sci. Transl. Med. 2018, 10, eaar5429. [Google Scholar] [CrossRef] [Green Version]
- Cresto, N.; Gaillard, M.-C.; Gardier, C.; Gubinelli, F.; Diguet, E.; Bellet, D.; Legroux, L.; Mitja, J.; Auregan, G.; Guillermier, M.; et al. The C-Terminal Domain of LRRK2 with the G2019S Mutation Is Sufficient to Produce Neurodegeneration of Dopaminergic Neurons in Vivo. Neurobiol. Dis. 2020, 134, 104614. [Google Scholar] [CrossRef]
- O’Hara, D.M.; Pawar, G.; Kalia, S.K.; Kalia, L.V. LRRK2 and α-Synuclein: Distinct or Synergistic Players in Parkinson’s Disease? Front. Neurosci. 2020, 14, 577. [Google Scholar] [CrossRef] [PubMed]
- Daher, J.P.L.; Volpicelli-Daley, L.A.; Blackburn, J.P.; Moehle, M.S.; West, A.B. Abrogation of α-Synuclein–Mediated Dopaminergic Neurodegeneration in LRRK2-Deficient Rats. Proc. Natl. Acad. Sci. USA 2014, 111, 9289–9294. [Google Scholar] [CrossRef] [Green Version]
- Daher, J.P.L.; Abdelmotilib, H.A.; Hu, X.; Volpicelli-Daley, L.A.; Moehle, M.S.; Fraser, K.B.; Needle, E.; Chen, Y.; Steyn, S.J.; Galatsis, P.; et al. Leucine-Rich Repeat Kinase 2 (LRRK2) Pharmacological Inhibition Abates α-Synuclein Gene-Induced Neurodegeneration. J. Biol. Chem. 2015, 290, 19433–19444. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Parisiadou, L.; Gu, X.-L.; Wang, L.; Shim, H.; Sun, L.; Xie, C.; Long, C.-X.; Yang, W.-J.; Ding, J.; et al. Leucine-Rich Repeat Kinase 2 Regulates the Progression of Neuropathology Induced by Parkinson’s-Disease-Related Mutant Alpha-Synuclein. Neuron 2009, 64, 807–827. [Google Scholar] [CrossRef] [Green Version]
- Skibinski, G.; Nakamura, K.; Cookson, M.R.; Finkbeiner, S. Mutant LRRK2 Toxicity in Neurons Depends on LRRK2 Levels and Synuclein But Not Kinase Activity or Inclusion Bodies. J. Neurosci. 2014, 34, 418–433. [Google Scholar] [CrossRef] [PubMed]
- West, A.B. Achieving Neuroprotection with LRRK2 Kinase Inhibitors in Parkinson Disease. Exp. Neurol. 2017, 298, 236–245. [Google Scholar] [CrossRef]
- Wallings, R.; Manzoni, C.; Bandopadhyay, R. Cellular Processes Associated with LRRK2 Function and Dysfunction. FEBS J. 2015, 282, 2806–2826. [Google Scholar] [CrossRef]
- Russo, I.; Bubacco, L.; Greggio, E. LRRK2 and Neuroinflammation: Partners in Crime in Parkinson’s Disease? J. Neuroinflammation 2014, 11, 52. [Google Scholar] [CrossRef] [Green Version]
- Russo, I.; Kaganovich, A.; Ding, J.; Landeck, N.; Mamais, A.; Varanita, T.; Biosa, A.; Tessari, I.; Bubacco, L.; Greggio, E.; et al. Transcriptome Analysis of LRRK2 Knock-out Microglia Cells Reveals Alterations of Inflammatory- and Oxidative Stress-Related Pathways upon Treatment with α-Synuclein Fibrils. Neurobiol. Dis. 2019, 129, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Ciron, C.; Lengacher, S.; Dusonchet, J.; Aebischer, P.; Schneider, B.L. Sustained Expression of PGC-1a in the Rat Nigrostriatal System Selectively Impairs Dopaminergic Function. Hum. Mol. Genet. 2012, 21, 1861–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsika, E.; Glauser, L.; Moser, R.; Fiser, A.; Daniel, G.; Sheerin, U.-M.; Lees, A.; Troncoso, J.C.; Lewis, P.A.; Bandopadhyay, R.; et al. Parkinson’s Disease-Linked Mutations in VPS35 Induce Dopaminergic Neurodegeneration. Hum. Mol. Genet. 2014, 23, 4621–4638. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Z.; Zhang, S.; Bustos, D.; Kleinheinz, T.; Le Pichon, C.E.; Dominguez, S.L.; Solanoy, H.O.; Drummond, J.; Zhang, X.; Ding, X.; et al. Ser1292 Autophosphorylation Is an Indicator of LRRK2 Kinase Activity and Contributes to the Cellular Effects of PD Mutations. Sci. Trans. Med. 2012, 4, 164ra161-164ra161. [Google Scholar] [CrossRef] [PubMed]
- Gombash, S.E.; Manfredsson, F.P.; Kemp, C.J.; Kuhn, N.C.; Fleming, S.M.; Egan, A.E.; Grant, L.M.; Ciucci, M.R.; MacKeigan, J.P.; Sortwell, C.E. Morphological and Behavioral Impact of AAV2/5-Mediated Overexpression of Human Wildtype Alpha-Synuclein in the Rat Nigrostriatal System. PLoS ONE 2013, 8, e81426. [Google Scholar] [CrossRef] [Green Version]
- Hoenen, C.; Gustin, A.; Birck, C.; Kirchmeyer, M.; Beaume, N.; Felten, P.; Grandbarbe, L.; Heuschling, P.; Heurtaux, T. Alpha-Synuclein Proteins Promote Pro-Inflammatory Cascades in Microglia: Stronger Effects of the A53T Mutant. PLoS ONE 2016, 11, e0162717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greggio, E.; Jain, S.; Kingsbury, A.; Bandopadhyay, R.; Lewis, P.; Kaganovich, A.; van der Brug, M.P.; Beilina, A.; Blackinton, J.; Thomas, K.J.; et al. Kinase Activity Is Required for the Toxic Effects of Mutant LRRK2/Dardarin. Neurobiol. Dis. 2006, 23, 329–341. [Google Scholar] [CrossRef]
- Smith, W.W.; Pei, Z.; Jiang, H.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Kinase Activity of Mutant LRRK2 Mediates Neuronal Toxicity. Nat. Neurosci. 2006, 9, 1231–1233. [Google Scholar] [CrossRef]
- Brooks, S.P.; Dunnett, S.B. Tests to Assess Motor Phenotype in Mice: A User’s Guide. Nat. Rev. Neurosci. 2009, 10, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Björklund, A.; Dunnett, S.B. The Amphetamine Induced Rotation Test: A Re-Assessment of Its Use as a Tool to Monitor Motor Impairment and Functional Recovery in Rodent Models of Parkinson’s Disease. J. Parkinsons Dis. 2019, 9, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Sossi, V.; de la Fuente-Fernández, R.; Nandhagopal, R.; Schulzer, M.; McKenzie, J.; Ruth, T.J.; Aasly, J.O.; Farrer, M.J.; Wszolek, Z.K.; Stoessl, J.A. Dopamine Turnover Increases in Asymptomatic LRRK2 Mutations Carriers. Mov. Disord. 2010, 25, 2717–2723. [Google Scholar] [CrossRef]
- Melrose, H.L.; Dächsel, J.C.; Behrouz, B.; Lincoln, S.J.; Yue, M.; Hinkle, K.M.; Kent, C.; Korvatska, E.; Taylor, J.P.; Witten, L.; et al. Impaired Dopaminergic Neurotransmission and Microtubule-Associated Protein Tau Alterations in Human LRRK2 Transgenic Mice. Neurobiol. Dis. 2010, 40, 503–517. [Google Scholar] [CrossRef] [Green Version]
- Ramonet, D.; Daher, J.P.L.; Lin, B.M.; Stafa, K.; Kim, J.; Banerjee, R.; Westerlund, M.; Pletnikova, O.; Glauser, L.; Yang, L.; et al. Dopaminergic Neuronal Loss, Reduced Neurite Complexity and Autophagic Abnormalities in Transgenic Mice Expressing G2019S Mutant LRRK2. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Neifert, S.; Karuppagounder, S.S.; Stankowski, J.N.; Lee, B.D.; Grima, J.C.; Chen, G.; Ko, H.S.; Lee, Y.; Swing, D.; et al. Overexpression of Parkinson’s Disease-Associated Mutation LRRK2 G2019S in Mouse Forebrain Induces Behavioral Deficits and α-Synuclein Pathology. eNeuro 2017, 4, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Daher, J.P.L.; Pletnikova, O.; Biskup, S.; Musso, A.; Gellhaar, S.; Galter, D.; Troncoso, J.C.; Lee, M.K.; Dawson, T.M.; Dawson, V.L.; et al. Neurodegenerative Phenotypes in an A53T α-Synuclein Transgenic Mouse Model Are Independent of LRRK2. Hum. Mol. Genet. 2012, 21, 2420–2431. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Neifert, S.; Karuppagounder, S.S.; Liu, Q.; Stankowski, J.N.; Lee, B.D.; Ko, H.S.; Lee, Y.; Grima, J.C.; Mao, X.; et al. Robust Kinase- and Age-Dependent Dopaminergic and Norepinephrine Neurodegeneration in LRRK2 G2019S Transgenic Mice. Proc. Natl. Acad. Sci. USA 2018, 115, 201712648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galvan, L.; Francelle, L.; Gaillard, M.-C.; de Longprez, L.; Carrillo-de Sauvage, M.-A.; Liot, G.; Cambon, K.; Stimmer, L.; Luccantoni, S.; Flament, J.; et al. The Striatal Kinase DCLK3 Produces Neuroprotection against Mutant Huntingtin. Brain 2018, 141, 1434–1454. [Google Scholar] [CrossRef] [PubMed]
- Francelle, L.; Galvan, L.; Gaillard, M.-C.; Guillermier, M.; Houitte, D.; Bonvento, G.; Petit, F.; Jan, C.; Dufour, N.; Hantraye, P.; et al. Loss of the Thyroid Hormone-Binding Protein Crym Renders Striatal Neurons More Vulnerable to Mutant Huntingtin in Huntington’s Disease. Hum. Mol. Genet. 2015, 24, 1563–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damiano, M.; Diguet, E.; Malgorn, C.; D’Aurelio, M.; Galvan, L.; Petit, F.; Benhaim, L.; Guillermier, M.; Houitte, D.; Dufour, N.; et al. A Role of Mitochondrial Complex II Defects in Genetic Models of Huntington’s Disease Expressing N-Terminal Fragments of Mutant Huntingtin. Hum. Mol. Genet. 2013, 22, 3869–3882. [Google Scholar] [CrossRef] [Green Version]
- Galvan, L.; Lepejová, N.; Gaillard, M.-C.; Malgorn, C.; Guillermier, M.; Houitte, D.; Bonvento, G.; Petit, F.; Dufour, N.; Héry, P.; et al. Capucin Does Not Modify the Toxicity of a Mutant Huntingtin Fragment in Vivo. Neurobiol. Aging 2012, 33, 1845.e5–6. [Google Scholar] [CrossRef]
- Francelle, L.; Galvan, L.; Gaillard, M.-C.; Petit, F.; Bernay, B.; Guillermier, M.; Bonvento, G.; Dufour, N.; Elalouf, J.-M.; Hantraye, P.; et al. Striatal Long Noncoding RNA Abhd11os Is Neuroprotective against an N-Terminal Fragment of Mutant Huntingtin in Vivo. Neurobiol. Aging 2015, 36, 1601-e7. [Google Scholar] [CrossRef]
- Deng, J.; Lewis, P.A.; Greggio, E.; Sluch, E.; Beilina, A.; Cookson, M.R. Structure of the ROC Domain from the Parkinson’s Disease-Associated Leucine-Rich Repeat Kinase 2 Reveals a Dimeric GTPase. Proc. Natl. Acad. Sci. USA 2008, 105, 1499–1504. [Google Scholar] [CrossRef] [Green Version]
- Gloeckner, C.J.; Kinkl, N.; Schumacher, A.; Braun, R.J.; O’Neill, E.; Meitinger, T.; Kolch, W.; Prokisch, H.; Ueffing, M. The Parkinson Disease Causing LRRK2 Mutation I2020T Is Associated with Increased Kinase Activity. Hum. Mol. Genet. 2006, 15, 223–232. [Google Scholar] [CrossRef]
- Greggio, E.; Zambrano, I.; Kaganovich, A.; Beilina, A.; Taymans, J.-M.; Daniëls, V.; Lewis, P.; Jain, S.; Ding, J.; Syed, A.; et al. The Parkinson Disease-Associated Leucine-Rich Repeat Kinase 2 (LRRK2) Is a Dimer That Undergoes Intramolecular Autophosphorylation. J. Biol. Chem. 2008, 283, 16906–16914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, G.; Iwatsubo, T. Re-Examination of the Dimerization State of Leucine-Rich Repeat Kinase 2: Predominance of the Monomeric Form. Biochem. J. 2012, 441, 987–998. [Google Scholar] [CrossRef]
- Kett, L.R.; Boassa, D.; Ho, C.C.-Y.; Rideout, H.J.; Hu, J.; Terada, M.; Ellisman, M.; Dauer, W.T. LRRK2 Parkinson Disease Mutations Enhance Its Microtubule Association. Hum. Mol. Genet. 2012, 21, 890–899. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, F.; Yabata, T.; Ohta, E.; Maekawa, T.; Shimada, N.; Suzuki, M.; Maruyama, H.; Ichikawa, T.; Obata, F. LRRK2 Phosphorylates Tubulin-Associated Tau but Not the Free Molecule: LRRK2-Mediated Regulation of the Tau-Tubulin Association and Neurite Outgrowth. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Deniston, C.K.; Salogiannis, J.; Mathea, S.; Snead, D.M.; Lahiri, I.; Matyszewski, M.; Donosa, O.; Watanabe, R.; Böhning, J.; Shiau, A.K.; et al. Structure of LRRK2 in Parkinson’s Disease and Model for Microtubule Interaction. Nature 2020. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Buschauer, R.; Böhning, J.; Audagnotto, M.; Lasker, K.; Lu, T.-W.; Boassa, D.; Taylor, S.; Villa, E. The In Situ Structure of Parkinson’s Disease-Linked LRRK2. Cell 2020, 182, 1508–1518.e16. [Google Scholar] [CrossRef] [PubMed]
- Volpicelli-Daley, L.A.; Abdelmotilib, H.; Liu, Z.; Stoyka, L.; Daher, J.P.L.; Milnerwood, A.J.; Unni, V.K.; Hirst, W.D.; Yue, Z.; Zhao, H.T.; et al. G2019S-LRRK2 Expression Augments α-Synuclein Sequestration into Inclusions in Neurons. J. Neurosci. 2016, 36, 7415–7427. [Google Scholar] [CrossRef]
- Bieri, G.; Brahic, M.; Bousset, L.; Couthouis, J.; Kramer, N.J.; Ma, R.; Nakayama, L.; Monbureau, M.; Defensor, E.; Schüle, B.; et al. LRRK2 Modifies α-Syn Pathology and Spread in Mouse Models and Human Neurons. Acta Neuropathol. 2019, 137, 961–980. [Google Scholar] [CrossRef] [Green Version]
- Dzamko, N.L. LRRK2 and the Immune System. Adv. Neurobiol. 2017, 14, 123–143. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.Y.; Fernandez-Hernandez, H.; Hu, J.; Schaffner, A.; Pankratz, N.; Hsu, N.-Y.; Chuang, L.-S.; Carmi, S.; Villaverde, N.; Li, X.; et al. Functional Variants in the LRRK2 Gene Confer Shared Effects on Risk for Crohn’s Disease and Parkinson’s Disease. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobbestael, E.; Civiero, L.; Wit, T.D.; Taymans, J.-M.; Greggio, E.; Baekelandt, V. Pharmacological LRRK2 Kinase Inhibition Induces LRRK2 Protein Destabilization and Proteasomal Degradation. Sci. Rep. 2016, 6, 33897. [Google Scholar] [CrossRef] [Green Version]
- Berger, A.; Lorain, S.; Joséphine, C.; Desrosiers, M.; Peccate, C.; Voit, T.; Garcia, L.; Sahel, J.-A.; Bemelmans, A.-P. Repair of Rhodopsin MRNA by Spliceosome-Mediated RNA Trans-Splicing: A New Approach for Autosomal Dominant Retinitis Pigmentosa. Mol. Ther. 2015, 23, 918–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aurnhammer, C.; Haase, M.; Muether, N.; Hausl, M.; Rauschhuber, C.; Huber, I.; Nitschko, H.; Busch, U.; Sing, A.; Ehrhardt, A.; et al. Universal Real-Time PCR for the Detection and Quantification of Adeno-Associated Virus Serotype 2-Derived Inverted Terminal Repeat Sequences. Hum. Gene Ther. Methods 2012, 23, 18–28. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cresto, N.; Gardier, C.; Gaillard, M.-C.; Gubinelli, F.; Roost, P.; Molina, D.; Josephine, C.; Dufour, N.; Auregan, G.; Guillermier, M.; et al. The C-Terminal Domain of LRRK2 with the G2019S Substitution Increases Mutant A53T α-Synuclein Toxicity in Dopaminergic Neurons In Vivo. Int. J. Mol. Sci. 2021, 22, 6760. https://doi.org/10.3390/ijms22136760
Cresto N, Gardier C, Gaillard M-C, Gubinelli F, Roost P, Molina D, Josephine C, Dufour N, Auregan G, Guillermier M, et al. The C-Terminal Domain of LRRK2 with the G2019S Substitution Increases Mutant A53T α-Synuclein Toxicity in Dopaminergic Neurons In Vivo. International Journal of Molecular Sciences. 2021; 22(13):6760. https://doi.org/10.3390/ijms22136760
Chicago/Turabian StyleCresto, Noémie, Camille Gardier, Marie-Claude Gaillard, Francesco Gubinelli, Pauline Roost, Daniela Molina, Charlène Josephine, Noëlle Dufour, Gwenaëlle Auregan, Martine Guillermier, and et al. 2021. "The C-Terminal Domain of LRRK2 with the G2019S Substitution Increases Mutant A53T α-Synuclein Toxicity in Dopaminergic Neurons In Vivo" International Journal of Molecular Sciences 22, no. 13: 6760. https://doi.org/10.3390/ijms22136760
APA StyleCresto, N., Gardier, C., Gaillard, M. -C., Gubinelli, F., Roost, P., Molina, D., Josephine, C., Dufour, N., Auregan, G., Guillermier, M., Bernier, S., Jan, C., Gipchtein, P., Hantraye, P., Chartier-Harlin, M. -C., Bonvento, G., Van Camp, N., Taymans, J. -M., Cambon, K., ... Brouillet, E. (2021). The C-Terminal Domain of LRRK2 with the G2019S Substitution Increases Mutant A53T α-Synuclein Toxicity in Dopaminergic Neurons In Vivo. International Journal of Molecular Sciences, 22(13), 6760. https://doi.org/10.3390/ijms22136760