
Satellitome Analysis of the Pacific Oyster Crassostrea gigas Reveals New Pattern of Satellite DNA Organization, Highly Scattered across the Genome


 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results
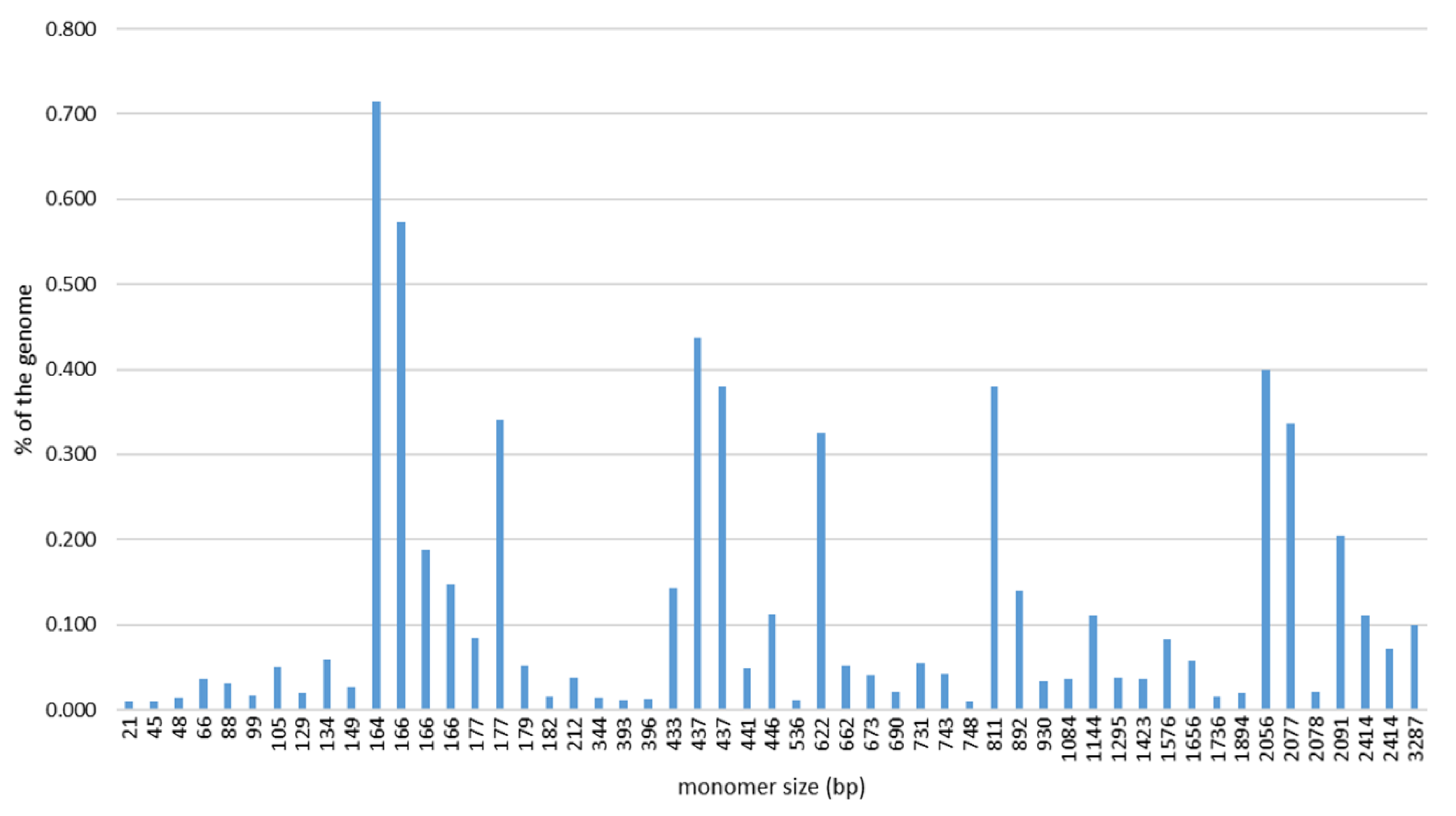
2.1. SatDNA Content of the C. gigas Genome
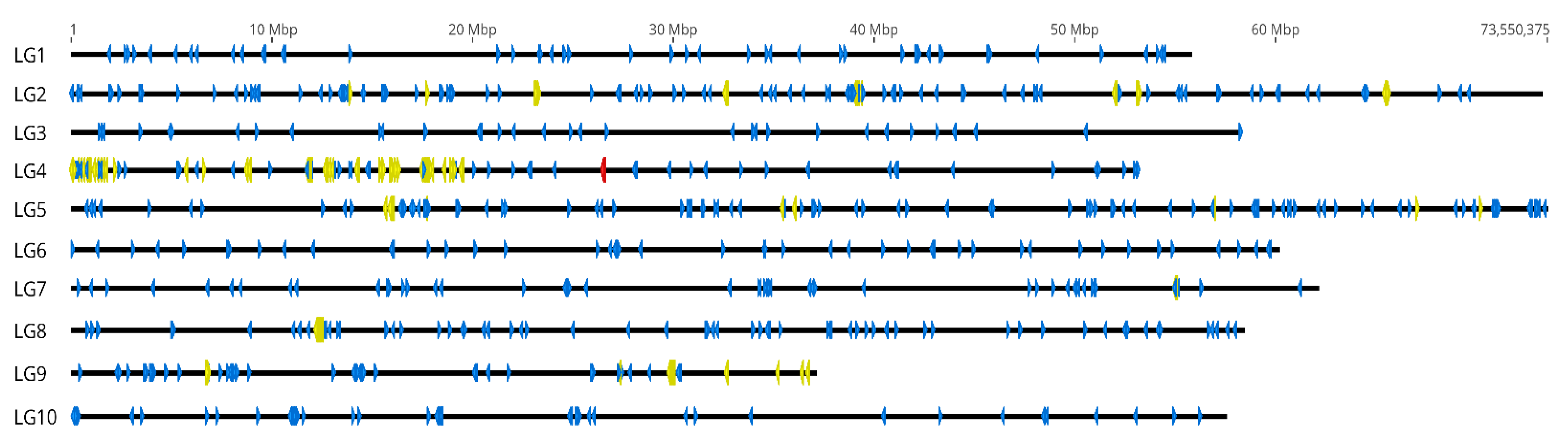
2.2. In Silico Chromosomal Localization of C. gigas satDNAs
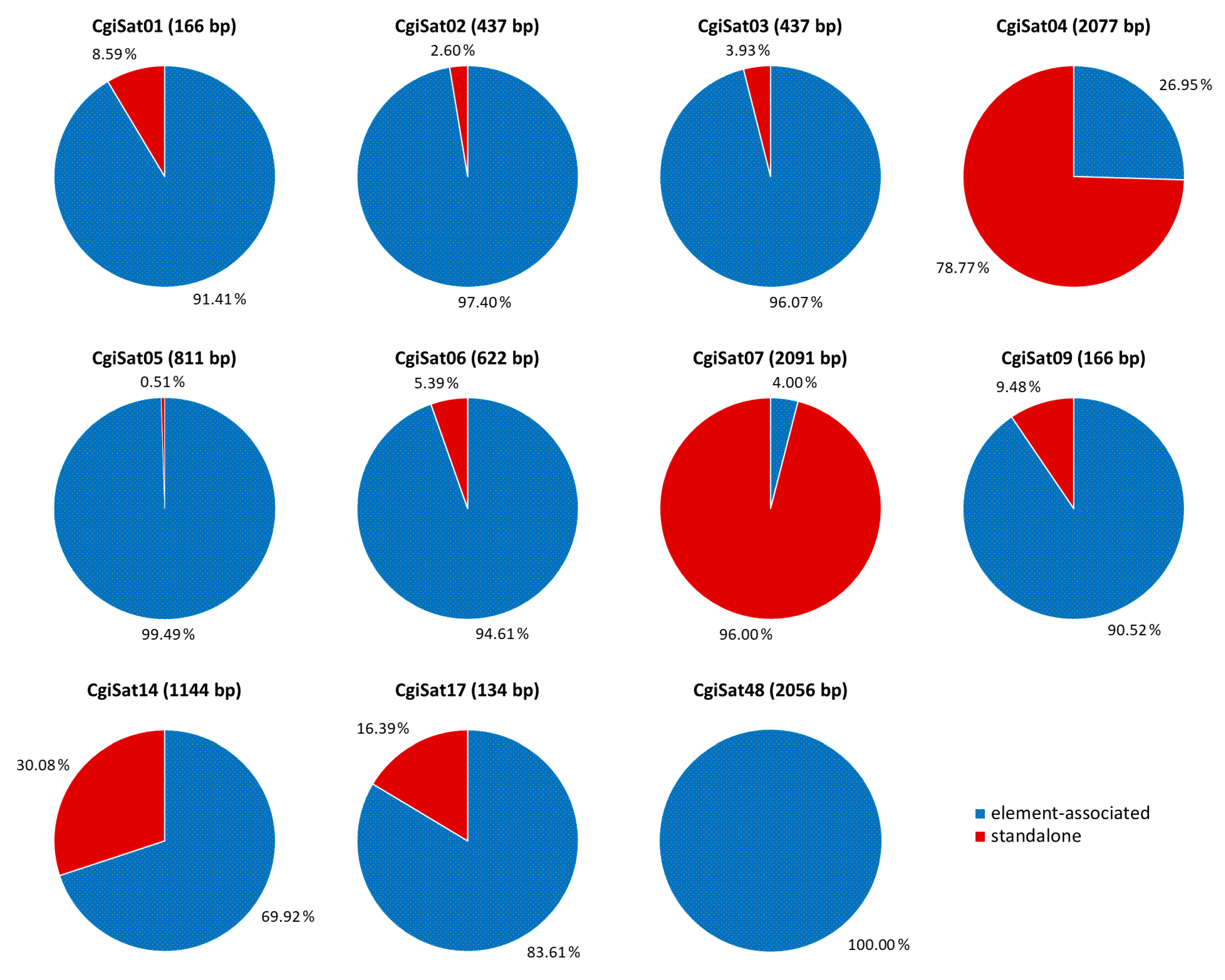
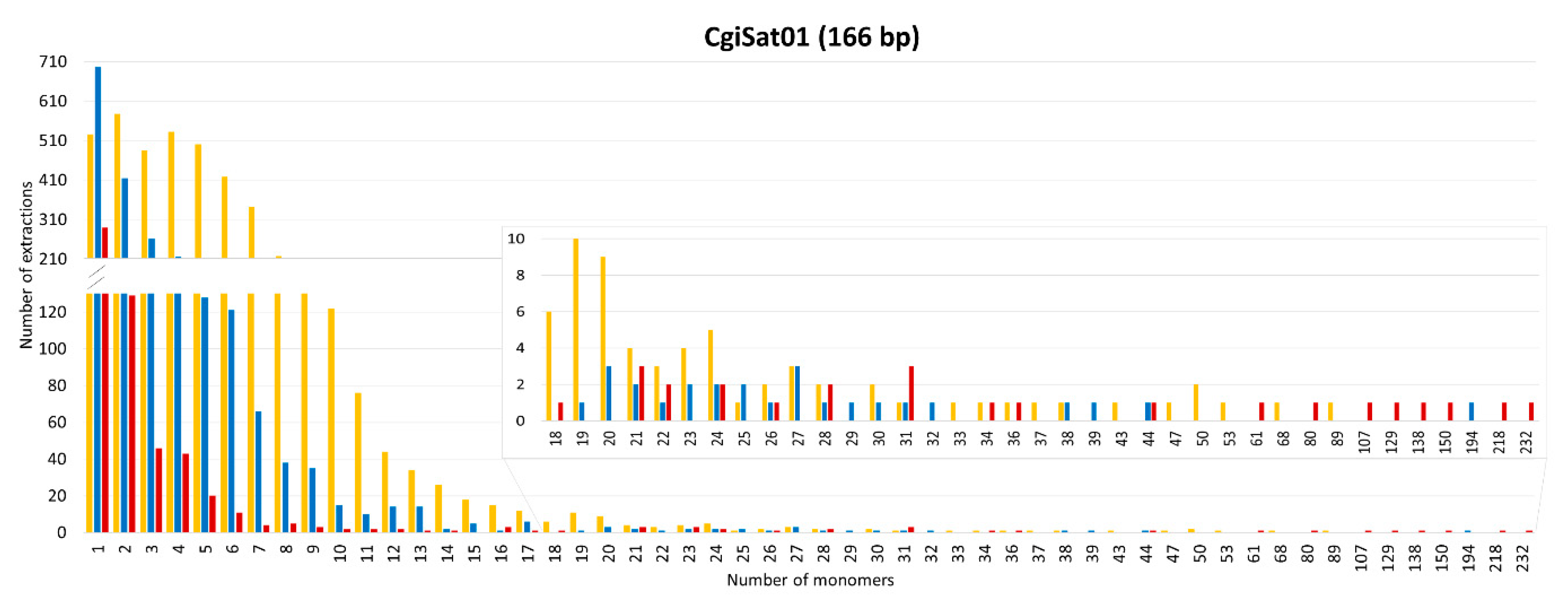
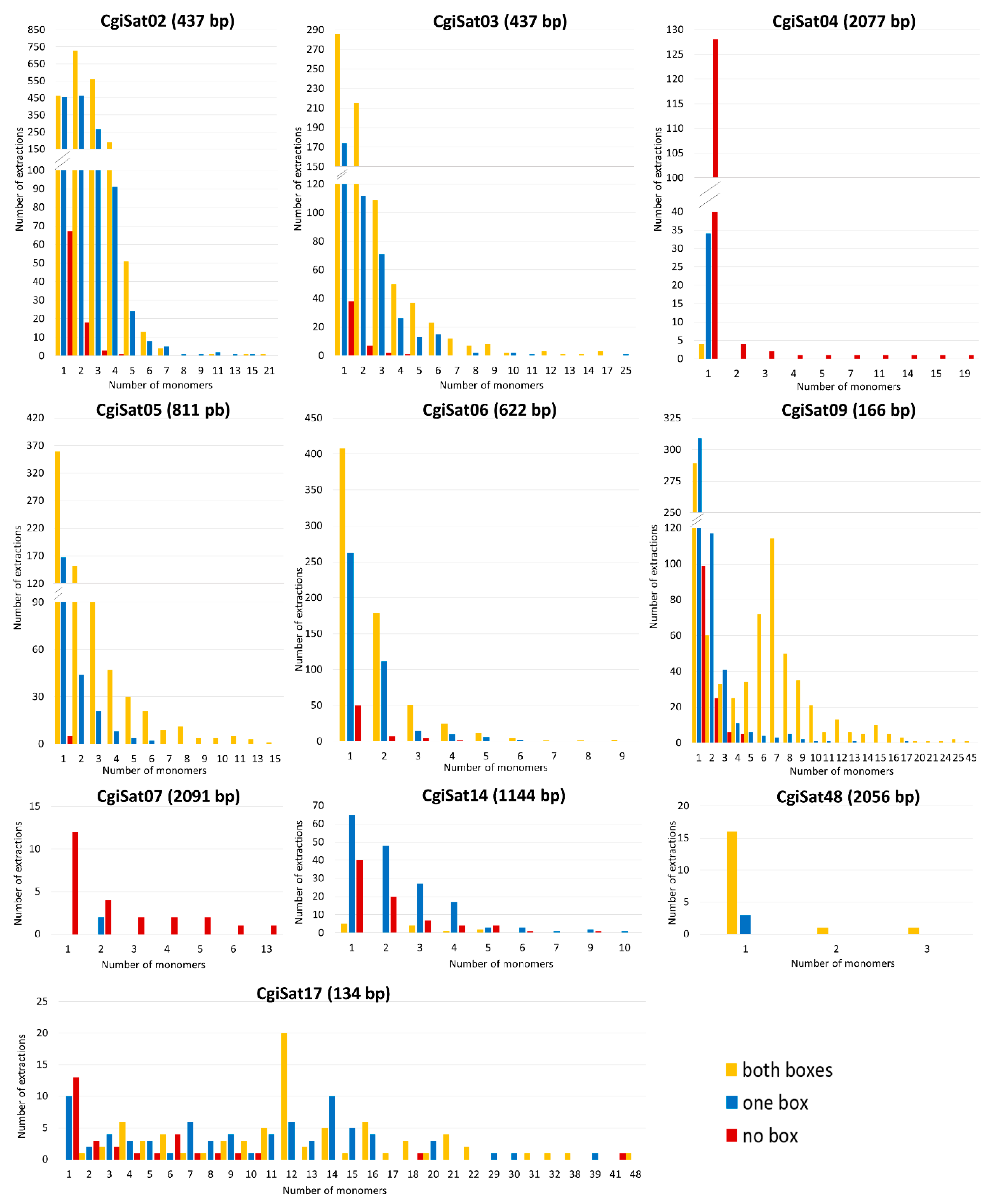
2.3. Deciphering the Dominant Organizational Forms of C. gigas Tandem Repeats
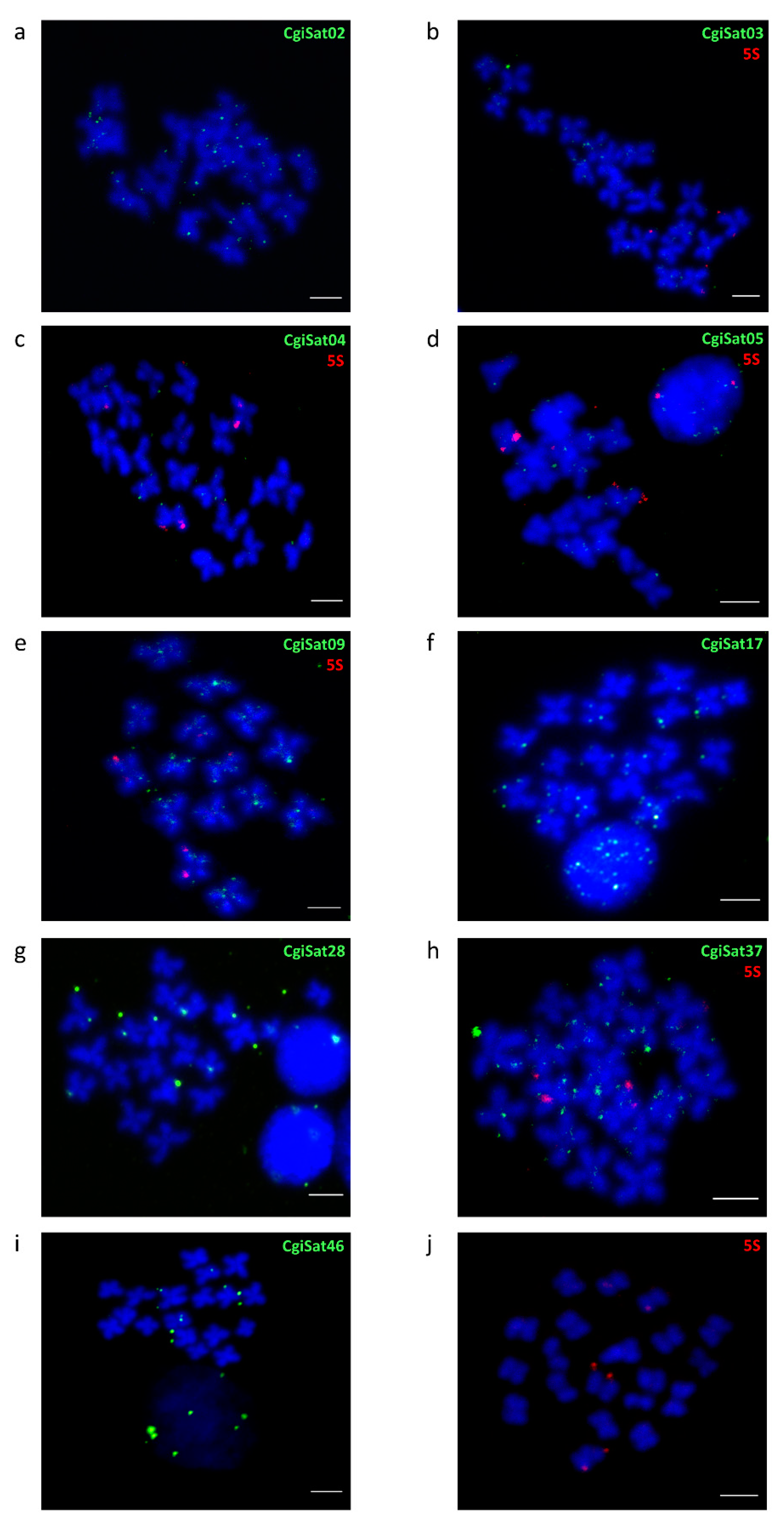
2.4. Fluorescence In Situ Hybridization
3. Discussion
4. Materials and Methods
4.1. Sequencing and Read Clustering
4.2. Satellite DNA Analysis
4.3. Analysis of the Flanking Regions of the satDNA Arrays
4.4. Mitotic Chromosomes Preparations
4.5. Probe Labelling
4.6. Fluorescence In Situ Hybridization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Charlesworth, B.; Sniegowski, P.; Stephan, W. The Evolutionary Dynamics of Repetitive DNA in Eukaryotes. Nature 1994, 371, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.; Heslop-Harrison, J.S. Genomes, Genes and Junk: The Large-Scale Organization of Plant Chromosomes. Trends Plant Sci. 1998, 3, 195–199. [Google Scholar] [CrossRef]
- Plohl, M.; Luchetti, A.; Meštrović, N.; Mantovani, B. Satellite DNAs between Selfishness and Functionality: Structure, Genomics and Evolution of Tandem Repeats in Centromeric (Hetero) Chromatin. Gene 2008, 409, 72–82. [Google Scholar] [CrossRef]
- Garrido-Ramos, M.A. Satellite DNA: An Evolving Topic. Genes 2017, 8, 230. [Google Scholar] [CrossRef] [PubMed]
- Hartley, G.; O’Neill, R.J. Centromere Repeats: Hidden Gems of the Genome. Genes 2019, 10, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tørresen, O.K.; Star, B.; Mier, P.; Andrade-Navarro, M.A.; Bateman, A.; Jarnot, P.; Gruca, A.; Grynberg, M.; Kajava, A.V.; Promponas, V.J.; et al. Tandem Repeats Lead to Sequence Assembly Errors and Impose Multi-Level Challenges for Genome and Protein Databases. Nucleic Acids Res. 2019, 47, 10994–11006. [Google Scholar] [CrossRef] [PubMed]
- Šatović, E.; Tunjić Cvitanić, M.; Plohl, M. Tools and Databases for Solving Problems in Detection and Identification of Repetitive DNA Sequences. Period. Biol. 2020, 121–122, 7–14. [Google Scholar] [CrossRef]
- Kim, Y.B.; Oh, J.H.; Mciver, L.J.; Rashkovetsky, E.; Michalak, K.; Garner, H.R.; Kang, L.; Nevo, E.; Korol, A.B.; Michalak, P. Divergence of Drosophila melanogaster Repeatomes in Response to a Sharp Microclimate Contrast in Evolution. Proc. Natl. Acad. Sci. USA 2014, 111, 10630–10635. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Ruano, F.J.; López-León, M.D.; Cabrero, J.; Camacho, J.P.M. High-Throughput Analysis of the Satellitome Illuminates Satellite DNA Evolution. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palacios-Gimenez, O.M.; Koelman, J.; Flores, M.P.; Bradford, T.M.; Jones, K.K.; Cooper, S.J.B.; Kawakami, T.; Suh, A. Comparative Analysis of Morabine Grasshopper Genomes Reveals Highly Abundant Transposable Elements and Rapidly Proliferating Satellite DNA Repeats. BMC Biol. 2020, 18. [Google Scholar] [CrossRef]
- Utsunomia, R.; de Silva, D.M.Z.; Ruiz-Ruano, F.J.; Goes, C.A.G.; Melo, S.; Ramos, L.P.; Oliveira, C.; Porto-Foresti, F.; Foresti, F.; Hashimoto, D.T. Satellitome Landscape Analysis of Megaleporinus macrocephalus (Teleostei, Anostomidae) Reveals Intense Accumulation of Satellite Sequences on the Heteromorphic Sex Chromosome. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sader, M.; Vaio, M.; Cauz-Santos, L.A.; Dornelas, M.C.; Vieira, M.L.C.; Melo, N.; Pedrosa-Harand, A. Large vs small genomes in Passiflora: The influence of the mobilome and the satellitome. Planta 2021, 253, 1–18. [Google Scholar] [CrossRef]
- Macas, J.; Novak, P.; Pellicer, J.; Cizkova, J.; Koblizkova, A.; Neumann, P.; Fukova, I.; Dolezel, J.; Kelly, L.J.; Leitch, I.J. In Depth Characterization of Repetitive DNA in 23 Plant Genomes Reveals Sources of Genome Size Variation in the Legume Tribe Fabeae. PLoS ONE 2015, 10, e0143424. [Google Scholar] [CrossRef] [PubMed]
- Klemme, S.; Banaei-Moghaddam, A.M.; Macas, J.; Wicker, T.; Novák, P.; Houben, A. High-Copy Sequences Reveal Distinct Evolution of the Rye B Chromosome. New Phytol. 2013, 199, 550–558. [Google Scholar] [CrossRef]
- Palacios-Gimenez, O.M.; Dias, G.B.; De Lima, L.G.; Kuhn, G.C.E.S.; Ramos, É.; Martins, C.; Cabral-De-Mello, D.C. High-Throughput Analysis of the Satellitome Revealed Enormous Diversity of Satellite DNAs in the Neo-Y Chromosome of the Cricket Eneoptera surinamensis. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belyayev, A.; Josefiová, J.; Jandová, M.; Kalendar, R.; Krak, K.; Mandák, B. Natural History of a Satellite DNA Family: From the Ancestral Genome Component to Species-Specific Sequences, Concerted and Non-Concerted Evolution. Int. J. Mol. Sci. 2019, 20, 1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, G.B.; Heringer, P.; Svartman, M.; Kuhn, G.C.S.S. Helitrons Shaping the Genomic Architecture of Drosophila: Enrichment of DINE-TR1 in α- and β-Heterochromatin, Satellite DNA Emergence, and PiRNA Expression. Chromosome Res. 2015, 23, 597–613. [Google Scholar] [CrossRef]
- Brajković, J.; Feliciello, I.; Bruvo-Mađarić, B.; Ugarković, D.W. Satellite DNA-like Elements Associated with Genes within Euchromatin of the Beetle Tribolium castaneum. G3 Genes Genomes Genet. 2012, 2, 931–941. [Google Scholar] [CrossRef] [Green Version]
- Šatović, E.; Vojvoda Zeljko, T.; Luchetti, A.; Mantovani, B.; Plohl, M. Adjacent Sequences Disclose Potential for Intra-Genomic Dispersal of Satellite DNA Repeats and Suggest a Complex Network with Transposable Elements. BMC Genom. 2016, 17, 997. [Google Scholar] [CrossRef] [Green Version]
- Feliciello, I.; Pezer, Ž.; Kordiš, D.; Mađarić, B.B.; Ugarković, Đ. Evolutionary History of Alpha Satellite DNA Repeats Dispersed within Human Genome Euchromatin. Genome Biol. Evol. 2020, 14561197. [Google Scholar] [CrossRef]
- Vondrak, T.; Ávila Robledillo, L.; Novák, P.; Koblížková, A.; Neumann, P.; Macas, J. Characterization of repeat arrays in ultra-long nanopore reads reveals frequent origin of satellite DNA from retrotransposon-derived tandem repeats. Plant J. 2020, 101, 484–500. [Google Scholar] [CrossRef] [Green Version]
- Gaffney, P.M.; Pierce, J.C.; Mackinley, A.G.; Titchen, D.A.; Glenn, W.K. Pearl, a Novel Family of Putative Transposable Elements in Bivalve Mollusks. J. Mol. Evol. 2003, 56, 308–316. [Google Scholar] [CrossRef]
- Dias, G.B.; Svartman, M.; Delprat, A.; Ruiz, A.; Kuhn, G.C.S.S. Tetris Is a Foldback Transposon That Provided the Building Blocks for an Emerging Satellite DNA of Drosophila virilis. Genome Biol. Evol. 2014, 6, 1302–1313. [Google Scholar] [CrossRef] [Green Version]
- Luchetti, A. TerMITEs: Miniature Inverted-Repeat Transposable Elements (MITEs) in the Termite Genome (Blattodea: Termitoidae). Mol. Genet. Genomics 2015, 290, 1499–1509. [Google Scholar] [CrossRef]
- Wijsman, J.W.M.; Fang, J.; Roncarati, A. Global Production of Marine Bivalves. Trends and Challenges. In Goods and Services of Marine Bivalves; Smaal, A.C., Ferreira, J.G., Grant, J., Petersen, J.K., Strand, Ø., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 7–26. [Google Scholar] [CrossRef] [Green Version]
- Vaughn, C.C.; Hoellein, T.J. Bivalve Impacts in Freshwater and Marine Ecosystems. Annu. Rev. Ecol. Evol. Syst. 2018, 49, 183–208. [Google Scholar] [CrossRef] [Green Version]
- Suárez-Ulloa, V.; Fernández-Tajes, J.; Manfrin, C.; Gerdol, M.; Venier, P.; Eirín-López, J.M. Bivalve Omics: State of the Art and Potential Applications for the Biomonitoring of Harmful Marine Compounds. Mar. Drugs 2013, 11, 4370–4389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes-dos-Santos, A.; Lopes-Lima, M.; Castro, L.F.C.; Froufe, E. Molluscan Genomics: The Road so Far and the Way Forward. Hydrobiologia 2020, 847, 1705–1726. [Google Scholar] [CrossRef]
- Fernández Robledo, J.A.; Yadavalli, R.; Allam, B.; Gerdol, M.; Greco, S.; Stevick, R.J.; Zhang, Y.; Heil, C.A.; Tracy, A.N.; Bishop-bailey, D.; et al. From the Raw Bar to the Bench: Bivalves as Models for Human Health. Dev. Comp. Immunol. 2020, 92, 260–282. [Google Scholar] [CrossRef] [PubMed]
- Murgarella, M.; Puiu, D.; Novoa, B.; Figueras, A.; Posada, D.; Canchaya, C. A First Insight into the Genome of the Filter- Feeder Mussel Mytilus galloprovincialis. PLoS ONE 2016, 11, e0151561. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Fang, X.; Guo, X.; Li, L.; Luo, R.; Xu, F.; Yang, P.; Zhang, L.; Wang, X.; Qi, H.; et al. The Oyster Genome Reveals Stress Adaptation and Complexity of Shell Formation. Nature 2012, 490, 49–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, T.; Kawashima, T.; Koyanagi, R.; Gyoja, F.; Tanaka, M.; Ikuta, T.; Shoguchi, E.; Fujiwara, M.; Shinzato, C.; Hisata, K.; et al. Draft Genome of the Pearl Oyster Pinctada fucata: A Platform for Understanding Bivalve Biology. DNA Res. 2012, 19, 117–130. [Google Scholar] [CrossRef] [Green Version]
- Mun, S.; Kim, Y.J.; Markkandan, K.; Shin, W.; Oh, S.; Woo, J.; Yoo, J.; An, H.; Han, K. The Whole-Genome and Transcriptome of the Manila Clam (Ruditapes Philippinarum). Genome Biol. Evol. 2017, 9, 1487–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šatović, E.; Vojvoda Zeljko, T.; Plohl, M. Characteristics and Evolution of Satellite DNA Sequences in Bivalve Mollusks. Eur. Zool. J. 2018, 85, 95–104. [Google Scholar] [CrossRef] [Green Version]
- Tunjić Cvitanić, M.; Vojvoda Zeljko, T.; Pasantes, J.J.; García-Souto, D.; Gržan, T.; Despot-Slade, E.; Plohl, M.; Šatović, E. Sequence Composition Underlying Centromeric and Heterochromatic Genome Compartments of the Pacific Oyster Crassostrea gigas. Genes 2020, 11, 695. [Google Scholar] [CrossRef]
- Clabby, C.; Goswami, U.; Flavin, F.; Wilkins, N.P.; Houghton, J.A.; Powell, R. Cloning, Characterization and Chromosomal Location of a Satellite DNA from the Pacific Oyster, Crassostrea gigas. Gene 1996, 168, 205–209. [Google Scholar] [CrossRef]
- Vojvoda Zeljko, T.; Pavlek, M.; Meštrović, N.; Plohl, M. Satellite DNA—like Repeats Are Dispersed throughout the Genome of the Pacific Oyster Crassostrea gigas Carried by Helentron Non—Autonomous Mobile Elements. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Thomas, J.; Pritham, E.J. Helitrons, the Eukaryotic Rolling-Circle Transposable Elements. Microbiol. Spectr. 2015, 3, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Peñaloza, C.; Gutierrez, A.P.; Eory, L.; Wang, S.; Guo, X.; Archibald, A.L.; Bean, T.P.; Houston, R.D. A Chromosome-Level Genome Assembly for the Pacific Oyster (Crassostrea gigas). GigaScience 2021, 10, 1–9. [Google Scholar] [CrossRef]
- López-Flores, I.; de la Herrán, R.; Garrido-Ramos, M.A.; Boudry, P.; Ruiz-Rejón, C.; Ruiz-Rejón, M. The Molecular Phylogeny of Oysters Based on a Satellite DNA Related to Transposons. Gene 2004, 339, 181–188. [Google Scholar] [CrossRef] [Green Version]
- Jurka, J.; Kapitonov, V.V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase Update, a Database of Eukaryotic Repetitive Elements. Cytogenet. Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef]
- Yang, H.P.; Barbash, D.A. Abundant and Species-Specific DINE-1 Transposable Elements in 12 Drosophila Genomes. Genome Biol. 2008, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Šatović, E.; Plohl, M. Tandem Repeat-Containing MITE Elements in the Clam Donax trunculus. Genome Biol. Evol. 2013, 5, 2549–2559. [Google Scholar] [CrossRef] [Green Version]
- Xiong, W.; Dooner, H.K.; Du, C. Rolling-Circle Amplification of Centromeric Helitrons in Plant Genomes. Plant J. 2016, 88, 1038–1045. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, Z.; Guo, X. A Centromeric Satellite Sequence in the Pacific Oyster (Crassostrea gigas Thunberg) Identified by Fluorescence In Situ Hybridization. Mar. Biotechnol. 2001, 3, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Cross, I.; Díaz, E.; Sánchez, I.; Rebordinos, L. Molecular and Cytogenetic Characterization of Crassostrea angulata Chromosomes. Aquaculture 2005, 247, 135–144. [Google Scholar] [CrossRef]
- Louzada, S.; Lopes, M.; Ferreira, D.; Adega, F.; Escudeiro, A.; Gama-carvalho, M.; Chaves, R. Decoding the role satellite DNA in genome Architecture and Plasticity—An Evolutionary and Clinical Affair. Genes 2020, 11, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouilly, K.; Chaves, R.; Leitao, A.; Benabdelmouna, A.; Guedes-Pinto, H. Chromosomal Organization of Simple Sequence Repeats in Chromosome Patterns. J. Genet. 2008, 87, 119–125. [Google Scholar] [CrossRef]
- Van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The Third Revolution in Sequencing Technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Sedlazeck, F.J.; Lee, H.; Darby, C.A.; Schatz, M.C. Piercing the Dark Matter: Bioinformatics of Long-Range Sequencing and Mapping. Nat. Rev. Genet. 2018, 19, 329–346. [Google Scholar] [CrossRef]
- Wang, X.; Xu, W.; Wei, L.; Zhu, C.; He, C.; Song, H.; Cai, Z.; Yu, W.; Jiang, Q.; Li, L.; et al. Nanopore Sequencing and De Novo Assembly of a Black-Shelled Pacific Oyster (Crassostrea gigas) Genome. Front. Genet. 2019, 10, 1211. [Google Scholar] [CrossRef] [Green Version]
- Pita, S.; Panzera, F.; Mora, P.; Vela, J.; Cuadrado, Á.; Sánchez, A.; Palomeque, T.; Lorite, P. Comparative Repeatome Analysis on Triatoma infestans Andean and Non-Andean Lineages, Main Vector of Chagas Disease. PLoS ONE 2017, 12, e0181635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boštjančić, L.L.; Bonassin, L.; Anušić, L.; Lovrenčić, L.; Besendorfer, V.; Maguire, I.; Grandjean, F.; Austin, C.M.; Greve, C.; Hamadou, A.B.; et al. The Pontastacus leptodactylus (Astacidae) Repeatome Provides Insight Into Genome Evolution and Reveals Remarkable Diversity of Satellite DNA. Front. Genet. 2021, 11, 611745. [Google Scholar] [CrossRef] [PubMed]
- Heslop-Harrison, J.S.P.; Schwarzacher, T. Nucleosomes and Centromeric DNA Packaging. Proc. Natl. Acad. Sci. USA 2013, 110, 19974–19975. [Google Scholar] [CrossRef] [Green Version]
- Meštrović, N.; Mravinac, B.; Pavlek, M.; Vojvoda-Zeljko, T.; Šatović, E.; Plohl, M. Structural and Functional Liaisons between Transposable Elements and Satellite DNAs. Chromosome Res. 2015, 23, 583–596. [Google Scholar] [CrossRef]
- Alkan, C.; Ventura, M.; Archidiacono, N.; Rocchi, M.; Sahinalp, S.C.; Eichler, E.E. Organization and Evolution of Primate Centromeric DNA from Whole-Genome Shotgun Sequence Data. PLoS Comput. Biol. 2007, 3, 1807–1818. [Google Scholar] [CrossRef]
- Scalvenzi, T.; Pollet, N. Insights on Genome Size Evolution from a Miniature Inverted Repeat Transposon Driving a Satellite DNA. Mol. Phylogenet. Evol. 2014, 81, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Paço, A.; Freitas, R.; Vieira-Da-Silva, A. Conversion of DNA Sequences: From a Transposable Element to a Tandem Repeat or to a Gene. Genes 2019, 10, 1014. [Google Scholar] [CrossRef] [Green Version]
- Hikosaka, A.; Kawahara, A. Lineage-Specific Tandem Repeats Riding on a Transposable Element of MITE in Xenopus Evolution: A New Mechanism for Creating Simple Sequence Repeats. J. Mol. Evol. 2004, 59, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Izsvák, Z.; Ivics, Z.; Shimoda, N.; Mohn, D.; Okamoto, H.; Hackett, P.B. Short Inverted-Repeat Transposable Elements in Teleost Fish and Implications for a Mechanism of Their Amplification. J. Mol. Evol. 1999, 48, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.J.; Nagel, A.; Bachmann, J.; Bachmann, L. Evolutionary Dynamics of the SGM Transposon Family in the Drosophila obscura Species Group. Mol. Biol. Evol. 2000, 17, 1597–1609. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, G.C.S.; Heslop-Harrison, J.S. Characterization and Genomic Organization of PERI, a Repetitive DNA in the Drosophila buzzatii Cluster Related to DINE-1 Transposable Elements and Highly Abundant in the Sex Chromosomes. Cytogenet. Genome Res. 2011, 132, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Vadnagara, K.; Pritham, E.J. DINE-1, the Highest Copy Number Repeats in Drosophila Melanogaster Are Non-Autonomous Endonuclease-Encoding Rolling-Circle Transposable Elements (Helentrons). Mob. DNA 2014, 5, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novák, P.; Robledillo, L.Á.; Koblížková, A.; Vrbová, I.; Neumann, P.; Macas, J. TAREAN: A Computational Tool for Identification and Characterization of Satellite DNA from Unassembled Short Reads. Nucleic Acids Res. 2017, 45, e111. [Google Scholar] [CrossRef] [PubMed]
- Novák, P.; Neumann, P.; Pech, J.; Steinhaisl, J.; Macas, J. RepeatExplorer: A Galaxy-Based Web Server for Genome-Wide Characterization of Eukaryotic Repetitive Elements from Next-Generation Sequence Reads. Bioinformatics 2013, 29, 792–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Expósito, M.J.; Pasantes, J.J.; Méndez, J. NOR Activity in Larval and Juvenile Mussels (Mytilus galloprovincialis Lmk.). J. Exp. Mar. Bio. Ecol. 1994, 175, 155–165. [Google Scholar] [CrossRef]
- Pérez-García, C.; Morán, P.; Pasantes, J.J. Cytogenetic Characterization of the Invasive Mussel Species Xenostrobus securis Lmk. (Bivalvia: Mytilidae). Genome 2011, 54, 771–778. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| satDNA | Monomer Length (bp) | % of the Genome (Average) | % of the Satellitome | % AT | Repbase |
|---|---|---|---|---|---|
| CgiSat01a | 164 | 0.72 | 11.29 | 59.8 | DNA/Helitron |
| CgiSat01b | 166 | 0.57 | 9.04 | 57.2 | DNA/Helitron |
| CgiSat02 | 437 | 0.44 | 6.91 | 65.7 | DNA/Helitron |
| CgiSat03 | 437 | 0.38 | 6.00 | 67.5 | DNA/Helitron |
| CgiSat04 | 2077 | 0.34 | 5.32 | 67.3 | DNA/Helitron |
| CgiSat05 | 811 | 0.38 | 6.00 | 67.6 | DNA/Helitron |
| CgiSat06 | 622 | 0.33 | 5.13 | 67.4 | DNA/Helitron |
| CgiSat07 | 2091 | 0.21 | 3.24 | 67.0 | DNA/Helitron |
| CgiSat08 | 166 | 0.19 | 2.96 | 67.5 | DNA/Helitron |
| CgiSat09 | 166 | 0.15 | 2.33 | 63.3 | DNA/Helitron |
| CgiSat10 | 433 | 0.14 | 2.26 | 63.0 | DNA/Helitron |
| CgiSat11 | 892 | 0.14 | 2.21 | 64.6 | LTR/BEL |
| CgiSat12 | 446 | 0.11 | 1.78 | 64.8 | - |
| CgiSat13 | 2414 | 0.11 | 1.74 | 67.4 | DNA/Kolobok |
| CgiSat14 | 1144 | 0.11 | 1.74 | 68.9 | DNA/Helitron |
| CgiSat15 | 177 | 0.08 | 1.32 | 68.9 | - |
| CgiSat16 | 2414 | 0.07 | 1.12 | 66.8 | DNA/Kolobok |
| CgiSat17 | 134 | 0.06 | 0.92 | 76.1 | DNA/Helitron |
| CgiSat18 | 731 | 0.05 | 0.86 | 63.9 | Interspersed Repeat |
| CgiSat19 | 212 | 0.04 | 0.60 | 72.6 | - |
| CgiSat20 | 662 | 0.05 | 0.81 | 65.4 | - |
| CgiSat21 | 441 | 0.05 | 0.77 | 69.2 | DNA/Helitron |
| CgiSat22 | 743 | 0.04 | 0.66 | 53.3 | LTR/Gypsy |
| CgiSat23 | 1295 | 0.04 | 0.60 | 64.9 | DNA/MuDR |
| CgiSat24 | 66 | 0.04 | 0.58 | 57.6 | - |
| CgiSat25 | 88 | 0.03 | 0.49 | 61.4 | NonLTR/R1 |
| CgiSat26 | 1894 | 0.02 | 0.30 | 60.4 | LTR/Gypsy |
| CgiSat27 | 99 | 0.02 | 0.26 | 60.6 | - |
| CgiSat28 | 182 | 0.02 | 0.25 | 44.0 | DNA/MuDR |
| CgiSat29 | 344 | 0.01 | 0.23 | 52.9 | DNA/DNA4-44 |
| CgiSat30 | 48 | 0.01 | 0.22 | 68.7 | - |
| CgiSat31 | 393 | 0.01 | 0.18 | 52.2 | IntegratedVirus/DNAV |
| CgiSat32 | 536 | 0.01 | 0.17 | 66.6 | DNA/Mariner |
| CgiSat33 | 105 | 0.05 | 0.80 | 46.7 | - |
| CgiSat34 | 1423 | 0.04 | 0.57 | 67.3 | DNA/DNA3-8 |
| CgiSat35 | 690 | 0.02 | 0.32 | 68.8 | DNA/IS3EU |
| CgiSat36 | 45 | 0.01 | 0.16 | 60.0 | - |
| CgiSat37 | 177 | 0.34 | 5.37 | 56.5 | DNA/Helitron |
| CgiSat38 | 3287 | 0.10 | 1.58 | 68.0 | DNA/DNA2-7 |
| CgiSat39 | 179 | 0.05 | 0.82 | 52.0 | - |
| CgiSat40 | 673 | 0.04 | 0.65 | 62.9 | DNA/Crypton |
| CgiSat41 | 1736 | 0.02 | 0.24 | 66.5 | DNA/Ginger1 |
| CgiSat42 | 930 | 0.03 | 0.52 | 66.9 | DNA/DNA4-31 |
| CgiSat43 | 21 | 0.01 | 0.16 | 66.7 | - |
| CgiSat44 | 748 | 0.01 | 0.16 | 68.6 | LTR/DIRS |
| CgiSat45 | 1576 | 0.08 | 1.31 | 64.1 | DNA/DNA4-2 |
| CgiSat46 | 149 | 0.03 | 0.41 | 54.4 | - |
| CgiSat47 | 129 | 0.02 | 0.30 | 69.8 | DNA/IS3EU |
| CgiSat48 | 2056 | 0.40 | 6.32 | 64.8 | DNA/Helitron |
| CgiSat49 | 1084 | 0.04 | 0.57 | 69.4 | DNA/DNA2-12 |
| CgiSat50 | 2078 | 0.02 | 0.33 | 64.1 | - |
| CgiSat51 | 396 | 0.01 | 0.19 | 63.1 | LTR/Gypsy |
| CgiSat52 | 1656 | 0.06 | 0.92 | 61.5 | DNA/Polinton |
| Assembly Accession: GCA_902806645.1 | Assembly Accession: GCA_011032805.1 | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| satDNA | LINKAGE GROUP | No. of Chromosomes Occupied | No. of Monomers on Chromosomes | No. of Monomers on Unplaced Scaffolds | Average No. of Monomers Per Chromosome | satDNA | CHROMOSOME | No. of Chromosomes Occupied | No. of Monomers on Chromosomes | Average No. of Monomers Per Chromosome | ||||||||||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |||||||||
| CgiSat01 | + | + | + | + | + | + | + | + | + | + | 10 | 28,120 | 3946 | 2812 | CgiSat01 | + | + | + | + | + | + | + | + | + | + | 10 | 28,486 | 2849 |
| CgiSat02 | + | + | + | + | + | + | + | + | + | + | 10 | 6499 | 1249 | 650 | CgiSat02 | + | + | + | + | + | + | + | + | + | + | 10 | 5563 | 556 |
| CgiSat03 | + | + | + | + | + | + | + | + | + | + | 10 | 2502 | 411 | 250 | CgiSat03 | + | + | + | + | + | + | + | + | + | + | 10 | 2116 | 212 |
| CgiSat04 | + | + | + | + | + | + | + | + | + | + | 10 | 223 | 32 | 22 | CgiSat04 | + | + | + | + | + | + | + | + | + | + | 10 | 198 | 20 |
| CgiSat05 | + | + | + | + | + | + | + | + | + | + | 10 | 1811 | 312 | 181 | CgiSat05 | + | + | + | + | + | + | + | + | + | + | 10 | 1553 | 155 |
| CgiSat06 | + | + | + | + | + | + | + | + | + | + | 10 | 1628 | 202 | 163 | CgiSat06 | + | + | + | + | + | + | + | + | + | + | 10 | 1574 | 157 |
| CgiSat07 | + | + | + | − | + | + | + | + | + | + | 9 | 55 | 11 | 6 | CgiSat07 | + | + | + | + | + | + | + | + | + | + | 10 | 48 | 5 |
| CgiSat08 | + | + | + | + | + | + | + | + | + | + | 10 | 5171 | 1030 | 517 | CgiSat08 | + | + | + | + | + | + | + | + | + | + | 10 | 4949 | 495 |
| CgiSat09 | + | + | + | + | + | + | + | + | + | + | 10 | 4411 | 406 | 441 | CgiSat09 | + | + | + | + | + | + | + | + | + | + | 10 | 4609 | 461 |
| CgiSat10 | + | + | + | + | + | + | + | + | + | + | 10 | 1082 | 328 | 108 | CgiSat10 | + | + | + | + | + | + | + | + | + | + | 10 | 1463 | 146 |
| CgiSat11 | − | + | − | − | + | − | + | − | − | − | 3 | 83 | 50 | 28 | CgiSat11 | + | − | + | + | + | − | + | − | − | − | 5 | 150 | 30 |
| CgiSat12 | − | + | + | − | + | + | − | + | + | − | 6 | 244 | 21 | 41 | CgiSat12 | + | + | + | + | + | − | + | + | − | + | 8 | 672 | 84 |
| CgiSat13 | − | + | + | + | + | + | + | + | + | + | 9 | 39 | 2 | 4 | CgiSat13 | + | + | − | + | + | − | + | + | + | − | 7 | 39 | 6 |
| CgiSat14 | + | + | + | + | + | + | + | + | + | + | 10 | 441 | 119 | 44 | CgiSat14 | + | + | + | + | + | + | + | + | + | + | 10 | 257 | 26 |
| CgiSat15 | + | + | + | + | + | + | + | + | + | + | 10 | 1768 | 163 | 177 | CgiSat15 | + | + | + | + | + | + | + | + | + | + | 10 | 1897 | 190 |
| CgiSat16 | − | + | + | + | + | + | + | + | + | + | 9 | 66 | 1 | 7 | CgiSat16 | + | + | + | + | − | + | − | − | + | − | 6 | 34 | 6 |
| CgiSat17 | + | + | + | + | + | + | + | + | + | + | 10 | 1619 | 309 | 162 | CgiSat17 | + | + | + | + | + | + | + | + | + | + | 10 | 1153 | 115 |
| CgiSat18 | + | + | + | + | + | + | + | + | + | + | 10 | 363 | 25 | 36 | CgiSat18 | + | + | + | + | + | + | + | + | + | + | 10 | 372 | 37 |
| CgiSat19 | + | + | + | + | + | + | + | + | + | + | 10 | 947 | 352 | 95 | CgiSat19 | + | + | + | + | + | + | + | + | + | + | 10 | 1304 | 130 |
| CgiSat20 | − | + | + | − | + | + | + | + | + | − | 7 | 148 | 54 | 21 | CgiSat20 | + | − | − | − | + | + | + | + | − | − | 5 | 245 | 49 |
| CgiSat21 | + | + | + | + | + | + | + | + | + | + | 10 | 542 | 109 | 54 | CgiSat21 | + | + | + | + | + | + | + | + | + | + | 10 | 505 | 51 |
| CgiSat22 | − | − | − | + | − | − | − | − | − | − | 1 | 45 | 0 | 45 | CgiSat22 | − | − | − | − | − | − | − | + | − | − | 1 | 31 | 31 |
| CgiSat23 | + | + | + | + | + | + | + | + | + | − | 9 | 78 | 14 | 9 | CgiSat23 | + | + | + | + | + | + | + | + | + | + | 10 | 148 | 15 |
| CgiSat24 | + | + | + | + | + | + | + | + | + | + | 10 | 2740 | 128 | 274 | CgiSat24 | + | + | + | + | + | + | + | + | + | + | 10 | 2829 | 283 |
| CgiSat25 | + | + | + | + | + | + | + | + | + | + | 10 | 2468 | 67 | 247 | CgiSat25 | + | + | + | + | + | + | + | + | + | + | 10 | 2983 | 298 |
| CgiSat26 | − | − | − | − | − | − | − | + | − | − | 1 | 13 | 0 | 13 | CgiSat26 | − | − | − | + | − | − | − | − | − | − | 1 | 15 | 15 |
| CgiSat27 | − | + | + | + | + | − | + | + | + | − | 7 | 2030 | 57 | 290 | CgiSat27 | + | − | + | + | + | − | + | + | − | + | 7 | 1554 | 222 |
| CgiSat28 | + | + | + | + | + | + | + | + | + | + | 10 | 487 | 167 | 49 | CgiSat28 | + | + | + | + | + | + | + | + | + | + | 10 | 548 | 55 |
| CgiSat29 | + | + | + | + | + | + | + | + | + | + | 10 | 277 | 19 | 28 | CgiSat29 | + | + | + | + | + | + | + | + | + | + | 10 | 267 | 27 |
| CgiSat30 | − | − | − | + | − | + | − | − | − | − | 2 | 2 | 1 | 1 | CgiSat30 | − | − | − | − | + | − | − | + | − | − | 2 | 4 | 2 |
| CgiSat31 | + | − | − | − | − | − | − | − | − | − | 1 | 52 | 0 | 52 | CgiSat31 | − | − | − | − | − | − | + | − | − | − | 1 | 61 | 61 |
| CgiSat32 | − | + | + | − | + | + | − | − | − | − | 4 | 147 | 0 | 37 | CgiSat32 | + | − | − | − | − | − | + | − | − | − | 2 | 75 | 38 |
| CgiSat33 | − | + | + | + | + | + | + | + | + | + | 9 | 3191 | 254 | 355 | CgiSat33 | − | + | − | + | + | + | + | + | + | + | 8 | 2789 | 349 |
| CgiSat34 | − | + | − | − | − | − | − | + | + | + | 4 | 12 | 2 | 3 | CgiSat34 | + | + | − | + | + | + | + | + | + | + | 9 | 43 | 5 |
| CgiSat35 | − | + | − | − | + | + | − | − | − | + | 4 | 6 | 0 | 2 | CgiSat35 | − | − | + | − | − | − | + | − | − | − | 2 | 4 | 2 |
| CgiSat36 | + | − | − | − | − | − | − | − | − | − | 1 | 2 | 57 | 2 | CgiSat36 | − | − | − | − | + | + | + | − | − | + | 4 | 96 | 24 |
| CgiSat37 | + | + | + | + | + | + | + | + | + | + | 10 | 6367 | 516 | 637 | CgiSat37 | + | + | + | + | + | + | + | + | + | + | 10 | 6242 | 624 |
| CgiSat38 | + | + | − | + | − | + | − | + | − | − | 5 | 6 | 0 | 1 | CgiSat38 | + | − | + | − | − | + | − | + | + | − | 5 | 10 | 2 |
| CgiSat39 | + | + | + | + | + | + | + | + | + | + | 10 | 1617 | 64 | 162 | CgiSat39 | + | + | + | + | + | + | + | + | + | + | 10 | 1235 | 124 |
| CgiSat40 | + | + | + | + | + | + | + | + | + | + | 10 | 402 | 31 | 40 | CgiSat40 | + | + | + | + | + | + | + | + | + | + | 10 | 402 | 40 |
| CgiSat41 | − | + | − | − | − | − | − | − | − | − | 1 | 11 | 0 | 11 | CgiSat41 | + | − | − | − | − | − | − | − | − | − | 1 | 7 | 7 |
| CgiSat42 | + | + | + | − | + | + | + | + | − | − | 7 | 124 | 31 | 18 | CgiSat42 | + | − | + | + | + | − | + | − | − | − | 5 | 104 | 21 |
| CgiSat43 | + | + | + | + | + | + | + | + | + | + | 10 | 3145 | 380 | 315 | CgiSat43 | + | + | + | + | + | + | + | + | + | + | 10 | 3416 | 342 |
| CgiSat44 | + | + | + | + | + | + | + | + | + | + | 10 | 54 | 2 | 5 | CgiSat44 | + | + | + | + | + | + | + | + | + | + | 10 | 80 | 8 |
| CgiSat45 | − | + | − | − | − | − | − | − | − | − | 1 | 1 | 11 | 1 | CgiSat45 | + | − | − | − | − | − | − | − | − | − | 1 | 4 | 4 |
| CgiSat46 | + | − | − | − | + | − | − | − | − | + | 3 | 25 | 0 | 8 | CgiSat46 | − | + | − | + | + | + | − | + | + | − | 6 | 359 | 60 |
| CgiSat47 | + | + | + | − | + | + | − | + | + | − | 7 | 251 | 79 | 36 | CgiSat47 | + | − | + | + | + | + | + | + | + | + | 9 | 340 | 38 |
| CgiSat48 | − | − | + | + | + | + | + | + | − | + | 7 | 20 | 4 | 3 | CgiSat48 | + | + | + | + | − | + | − | − | − | + | 6 | 10 | 2 |
| CgiSat49 | + | + | + | + | + | + | + | + | + | + | 10 | 77 | 4 | 8 | CgiSat49 | + | + | + | + | + | + | + | + | + | + | 10 | 79 | 8 |
| CgiSat50 | − | − | − | − | − | − | − | − | − | − | 0 | 0 | 0 | 0 | CgiSat50 | − | − | − | − | − | − | − | − | − | − | 0 | 0 | 0 |
| CgiSat51 | − | − | − | + | − | + | − | − | − | − | 2 | 122 | 0 | 61 | CgiSat51 | − | + | − | − | − | − | − | + | − | − | 2 | 87 | 44 |
| CgiSat52 | + | − | − | − | − | − | − | − | − | − | 1 | 17 | 0 | 17 | CgiSat52 | − | − | − | − | − | − | + | − | − | − | 1 | 6 | 6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tunjić-Cvitanić, M.; Pasantes, J.J.; García-Souto, D.; Cvitanić, T.; Plohl, M.; Šatović-Vukšić, E. Satellitome Analysis of the Pacific Oyster Crassostrea gigas Reveals New Pattern of Satellite DNA Organization, Highly Scattered across the Genome. Int. J. Mol. Sci. 2021, 22, 6798. https://doi.org/10.3390/ijms22136798
Tunjić-Cvitanić M, Pasantes JJ, García-Souto D, Cvitanić T, Plohl M, Šatović-Vukšić E. Satellitome Analysis of the Pacific Oyster Crassostrea gigas Reveals New Pattern of Satellite DNA Organization, Highly Scattered across the Genome. International Journal of Molecular Sciences. 2021; 22(13):6798. https://doi.org/10.3390/ijms22136798
Chicago/Turabian StyleTunjić-Cvitanić, Monika, Juan J. Pasantes, Daniel García-Souto, Tonči Cvitanić, Miroslav Plohl, and Eva Šatović-Vukšić. 2021. "Satellitome Analysis of the Pacific Oyster Crassostrea gigas Reveals New Pattern of Satellite DNA Organization, Highly Scattered across the Genome" International Journal of Molecular Sciences 22, no. 13: 6798. https://doi.org/10.3390/ijms22136798
APA StyleTunjić-Cvitanić, M., Pasantes, J. J., García-Souto, D., Cvitanić, T., Plohl, M., & Šatović-Vukšić, E. (2021). Satellitome Analysis of the Pacific Oyster Crassostrea gigas Reveals New Pattern of Satellite DNA Organization, Highly Scattered across the Genome. International Journal of Molecular Sciences, 22(13), 6798. https://doi.org/10.3390/ijms22136798