
Gemcitabine-Based Chemoradiotherapy Enhanced by a PARP Inhibitor in Pancreatic Cancer Cell Lines


Abstract
:1. Introduction
2. Results
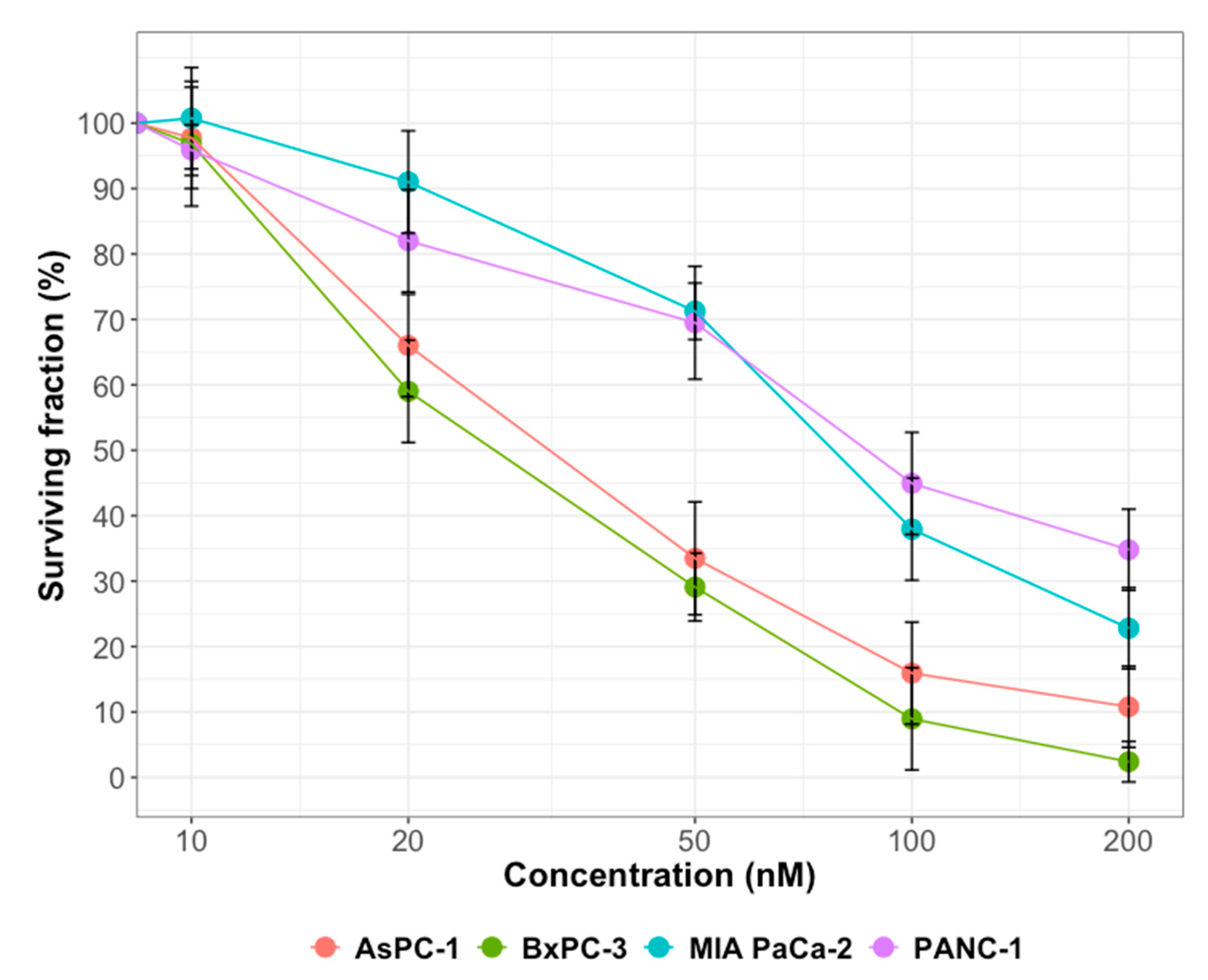
2.1. Cytotoxicity of Gemcitabine or Olaparib on PDAC Cell Lines
2.2. Effect of Olaparib on PARylation
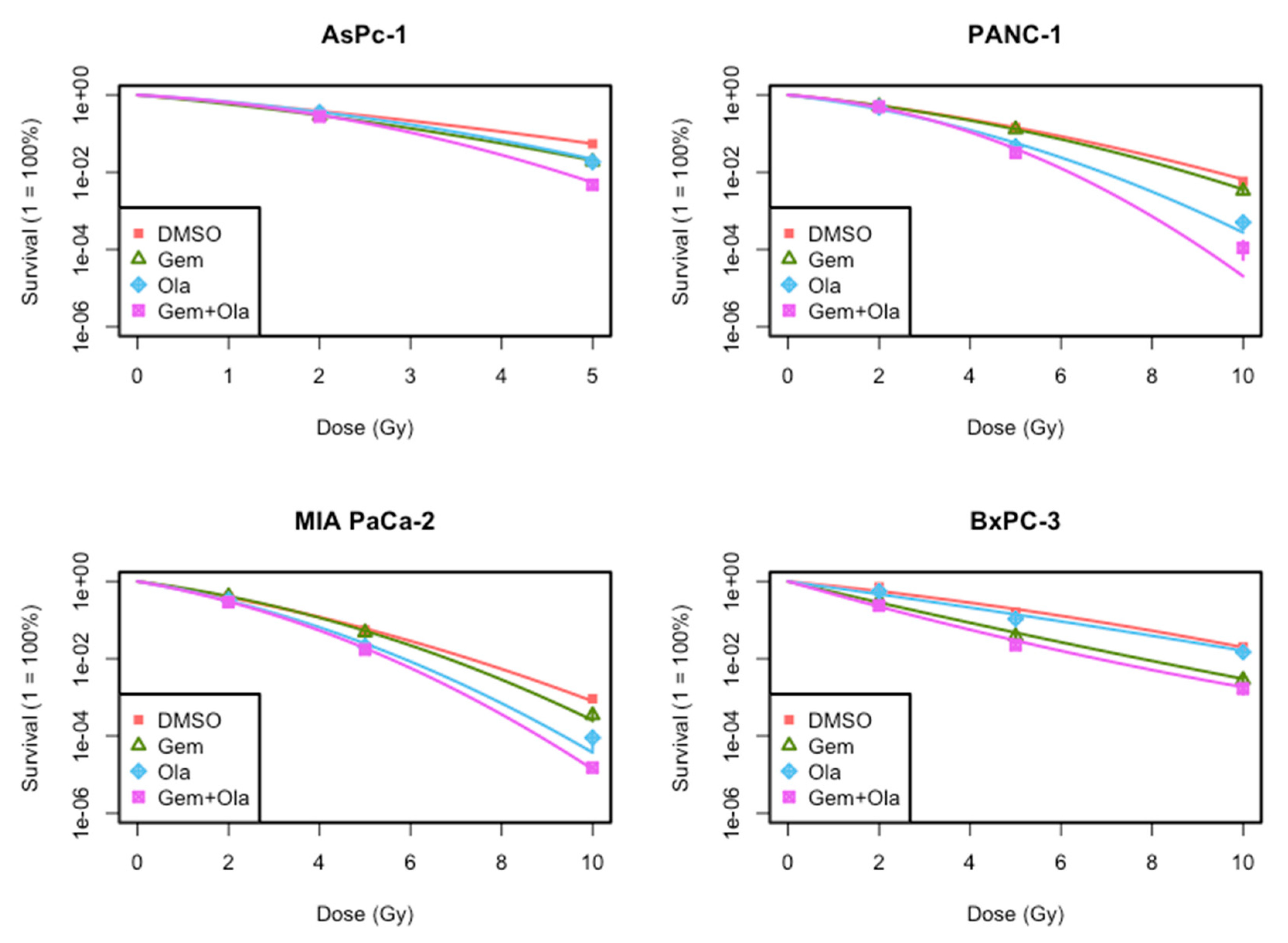
2.3. Radiosensitization of Gemcitabine and/or Olaparib on PDAC Cell Lines
2.4. Effect of the Treatments on Cell Cycle
2.5. Effect of the Treatments on Persistent γ-H2AX Foci
2.6. Effect of the Treatments on Cell Death Induction (Apoptosis and Autophagy)
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Drugs and Chemicals
4.3. Irradiation Exposure
4.4. Clonogenic Survival Assay
4.5. PARP Inhibitor Activity
4.6. Cell Cycle Distribution
4.7. Apoptotic and Necrotic Detection Assay
4.8. Autophagic Detection Assay
4.9. Determination of γ-H2AX Formation
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vincent, A.; Herman, J.; Schulick, R.; Hruban, R.H.; Goggins, M. Pancreatic cancer. Lancet 2011, 378, 607–620. [Google Scholar] [CrossRef]
- Zhu, C.-P.; Shi, J.; Chen, Y.-X.; Xie, W.-F.; Lin, Y. Gemcitabine in the chemoradiotherapy for locally advanced pancreatic cancer: A meta-analysis. Radiother. Oncol. 2011, 99, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.L.; Swisher, E.M.; Kaufmann, S.H. Poly (ADP-Ribose) Polymerase Inhibitors: Recent Advances and Future Development. J. Clin. Oncol. 2015, 33, 1397–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senra, J.M.; Telfer, B.A.; Cherry, K.E.; McCrudden, C.M.; Hirst, D.G.; O’Connor, M.J.; Wedge, S.R.; Stratford, I.J. Inhibition of PARP-1 by Olaparib (AZD2281) Increases the Radiosensitivity of a Lung Tumor Xenograft. Mol. Cancer Ther. 2011, 10, 1949–1958. [Google Scholar] [CrossRef] [Green Version]
- Vance, S.; Liu, E.; Zhao, L.; Parsels, J.D.; Parsels, L.A.; Brown, J.L.; Maybaum, J.; Lawrence, T.S.; Morgan, M.A. Selective radiosensitization of p53 mutant pancreatic cancer cells by combined inhibition of Chk1 and PARP1. Cell Cycle 2011, 10, 4321–4329. [Google Scholar] [CrossRef] [Green Version]
- Karnak, D.; Engelke, C.G.; Parsels, L.A.; Kausar, T.; Wei, D.; Robertson, J.R.; Marsh, K.B.; Davis, M.A.; Zhao, L.; Maybaum, J.; et al. Combined Inhibition of Wee1 and PARP1/2 for Radiosensitization in Pancreatic Cancer. Clin. Cancer Res. 2014, 20, 5085–5096. [Google Scholar] [CrossRef] [Green Version]
- Pauwels, B.; Korst, A.E.C.; Pattyn, G.G.O.; Lambrechts, H.A.J.; Van Bockstaele, D.R.; Vermeulen, K.; Lenjou, M.; de Pooter, C.M.J.; Vermorken, J.B.; Lardon, F. Cell cycle effect of gemcitabine and its role in the radiosensitizing mechanism in vitro. Int. J. Radiat. Oncol. Biol. Phys. 2003, 57, 1075–1083. [Google Scholar] [CrossRef]
- Porcelli, L.; Quatrale, A.E.; Mantuano, P.; Leo, M.G.; Silvestris, N.; Rolland, J.F.; Carioggia, E.; Lioce, M.; Paradiso, A.; Azzariti, A. Optimize radiochemotherapy in pancreatic cancer: PARP inhibitors a new therapeutic opportunity. Mol. Oncol. 2013, 7, 308–322. [Google Scholar] [CrossRef]
- Hirai, T.; Shirai, H.; Fujimori, H.; Okayasu, R.; Sasai, K.; Masutani, M. Radiosensitization effect of poly(ADP-ribose) polymerase inhibition in cells exposed to low and high liner energy transfer radiation. Cancer Sci. 2012, 103, 1045–1050. [Google Scholar] [CrossRef]
- Pauwels, B.; Korst, A.E.; Lardon, F.; Vermorken, J.B. Combined modality therapy of gemcitabine and radiation. Oncologist 2005, 10, 34–51. [Google Scholar] [CrossRef]
- Morgan, M.A.; Parsels, L.A.; Zhao, L.; Parsels, J.D.; Davis, M.A.; Hassan, M.C.; Arumugarajah, S.; Hylander-Gans, L.; Morosini, D.; Simeone, D.M.; et al. Mechanism of Radiosensitization by the Chk1/2 Inhibitor AZD7762 Involves Abrogation of the G2 Checkpoint and Inhibition of Homologous Recombinational DNA Repair. Cancer Res. 2010, 70, 4972–4981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Im, M.M.; Flanagan, S.A.; Ackroyd, J.J.; Shewach, D.S. Drug Metabolism and Homologous Recombination Repair in Radiosensitization with Gemcitabine. Radiat. Res. 2015, 183, 114–123. [Google Scholar] [CrossRef] [Green Version]
- Ewald, B.; Sampath, D.; Plunkett, W. H2AX phosphorylation marks gemcitabine-induced stalled replication forks and their collapse upon S-phase checkpoint abrogation. Mol. Cancer Ther. 2007, 6, 1239–1248. [Google Scholar] [CrossRef] [Green Version]
- Bryant, H.E.; Petermann, E.; Schultz, N.; Jemth, A.-S.; Loseva, O.; Issaeva, N.; Johansson, F.; Fernandez, S.; McGlynn, P.; Helleday, T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009, 28, 2601–2615. [Google Scholar] [CrossRef] [Green Version]
- Parsels, L.A.; Engelke, C.G.; Parsels, J.; Flanagan, S.A.; Zhang, Q.; Tanska, D.; Wahl, D.R.; Canman, C.E.; Lawrence, T.S.; Morgan, M.A. Combinatorial Efficacy of Olaparib with Radiation and ATR Inhibitor Requires PARP1 Protein in Homologous Recombination–Proficient Pancreatic Cancer. Mol. Cancer Ther. 2021, 20, 263–273. [Google Scholar] [CrossRef]
- Chen, S.; Wang, G.; Niu, X.; Zhao, J.; Tan, W.; Wang, H.; Zhao, L.; Ge, Y. Combination of AZD2281 (Olaparib) and GX15-070 (Obatoclax) results in synergistic antitumor activities in preclinical models of pancreatic cancer. Cancer Lett. 2014, 348, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Pandita, A.; Kumar, B.; Manvati, S.; Vaishnavi, S.; Singh, S.K.; Bamezai, R.N.K. Synergistic Combination of Gemcitabine and Dietary Molecule Induces Apoptosis in Pancreatic Cancer Cells and Down Regulates PKM2 Expression. PLoS ONE 2014, 9, e107154. [Google Scholar] [CrossRef] [PubMed]
- Fiorini, C.; Cordani, M.; Padroni, C.; Blandino, G.; Di Agostino, S.; Donadelli, M. Mutant p53 stimulates chemoresistance of pancreatic adenocarcinoma cells to gemcitabine. Biochim. Biophys. Acta BBA Mol. Cell Res. 2015, 1853, 89–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papademetrio, D.L.; Cavaliere, V.; Simunovich, T.; Costantino, S.; Campos, M.D.; Lombardo, T.; Kaiser, C.M.F.; Álvarez, É. Interplay between autophagy and apoptosis in pancreatic tumors in response to gemcitabine. Target. Oncol. 2014, 9, 123–134. [Google Scholar] [CrossRef]
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Alotaibi, M.; Sharma, K.; Saleh, T.; Povirk, L.F.; Hendrickson, E.A.; Gewirtz, D.A. Radiosensitization by PARP Inhibition in DNA Repair Proficient and Deficient Tumor Cells: Proliferative Recovery in Senescent Cells. Radiat. Res. 2016, 185, 229–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Darzynkiewicz, Z. Cytometric Assessment of Histone H2AX Phosphorylation. DNA Repair Protoc. 2006, 314, 73–80. [Google Scholar] [CrossRef]
- Ben-Josef, E.; Schipper, M.; Francis, I.R.; Hadley, S.; Ten-Haken, R.; Lawrence, T.; Normolle, D.; Simeone, D.M.; Sonnenday, C.; Abrams, R.; et al. A Phase I/II Trial of Intensity Modulated Radiation (IMRT) Dose Escalation With Concurrent Fixed-dose Rate Gemcitabine (FDR-G) in Patients With Unresectable Pancreatic Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2012, 84, 1166–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | AsPC-1 | BxPC-3 | MIA PaCa-2 | PANC-1 |
|---|---|---|---|---|
| Olaparib | 1.05 ± 0.06 | 0.93 ± 0.05 | 1.02 ± 0.03 | 0.97 ± 0.02 |
| Gemcitabine | 0.98 ± 0.03 | 0.96 ± 0.03 | 1.01 ± 0.02 | 0.95 ± 0.02 |
| Cell Line | Treatment | α (Gy-1) | ß (Gy-2) | SER |
|---|---|---|---|---|
| MIA PaCa-2 | DMSO | 0.454 | 0.025 | 1.0 |
| Gemcitabine | 0.404 | 0.040 | 1.04 ± 0.01 | |
| Olaparib * | 0.515 | 0.048 | 1.24 ± 0.001 | |
| Olaparib + Gemcitabine *,• | 0.590 | 0.041 | 1.30 ± 0.02 | |
| Expected combined SER | (1.29 ± 0.02) | |||
| PANC-1 | DMSO | 0.273 | 0.024 | 1.0 |
| Gemcitabine | 0.250 | 0.032 | 1.04 ± 0.01 | |
| Olaparib * | 0.402 | 0.041 | 1.38 ± 0.01 | |
| Olaparib + Gemcitabine *,• | 0.291 | 0.077 | 1.48 ± 0.05 | |
| Expected combined SER | (1.45 ± 0.001) | |||
| AsPC-1 | DMSO | 0.438 | 0.033 | 1.0 |
| Gemcitabine | 0.520 | 0.053 | 1.22 ± 0.01 | |
| Olaparib * | 0.319 | 0.098 | 1.21 ± 0.06 | |
| Olaparib + Gemcitabine *,• | 0.367 | 0.138 | 1.45 ± 0.08 | |
| Expected combined SER | (1.47 ± 0.09) | |||
| BxPC-3 | DMSO | 0.267 | 0.013 | 1.0 |
| Gemcitabine | 0.640 | −0.006 | 1.75 ± 0.01 | |
| Olaparib * | 0.374 | 0.004 | 1.02 ± 0.02 | |
| Olaparib + Gemcitabine *,• | 0.779 | −0.015 | 2.07 ± 0.07 | |
| Expected combined SER | (1.79 ± 0.05) |
| Cell line | CTL | Gemcitabine |
|---|---|---|
| AsPC-1 | 22.29 ± 2.23 | 28.17 ± 0.80 |
| MIA PaCa-2 | 22.73 ± 1.29 | 32.50 ± 3.59 * |
| PANC-1 | 22.26 ± 1.69 | 25.03 ± 1.56 |
| BxPC-3 | 21.23 ± 2.23 | 26.83 ± 0.77 |
| Cell Line | Treatment | Necrotic Cells (%) at 10 Gy |
|---|---|---|
| MIA PaCa-2 | DMSO | 45.1 |
| Gemcitabine | 47.3 | |
| Olaparib | 55.7 | |
| Olaparib + Gemcitabine | 53.8 | |
| PANC-1 | DMSO | 17.1 |
| Gemcitabine | 25.3 | |
| Olaparib | 28.6 | |
| Olaparib + Gemcitabine | 36.8 | |
| AsPC-1 | DMSO | 29.3 |
| Gemcitabine | 39.1 | |
| Olaparib | 34.9 | |
| Olaparib + Gemcitabine | 47.6 | |
| BxPC-3 | DMSO | 29.2 |
| Gemcitabine | 27.6 | |
| Olaparib | 32.4 | |
| Olaparib + Gemcitabine | 35.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waissi, W.; Amé, J.-C.; Mura, C.; Noël, G.; Burckel, H. Gemcitabine-Based Chemoradiotherapy Enhanced by a PARP Inhibitor in Pancreatic Cancer Cell Lines. Int. J. Mol. Sci. 2021, 22, 6825. https://doi.org/10.3390/ijms22136825
Waissi W, Amé J-C, Mura C, Noël G, Burckel H. Gemcitabine-Based Chemoradiotherapy Enhanced by a PARP Inhibitor in Pancreatic Cancer Cell Lines. International Journal of Molecular Sciences. 2021; 22(13):6825. https://doi.org/10.3390/ijms22136825
Chicago/Turabian StyleWaissi, Waisse, Jean-Christophe Amé, Carole Mura, Georges Noël, and Hélène Burckel. 2021. "Gemcitabine-Based Chemoradiotherapy Enhanced by a PARP Inhibitor in Pancreatic Cancer Cell Lines" International Journal of Molecular Sciences 22, no. 13: 6825. https://doi.org/10.3390/ijms22136825
APA StyleWaissi, W., Amé, J. -C., Mura, C., Noël, G., & Burckel, H. (2021). Gemcitabine-Based Chemoradiotherapy Enhanced by a PARP Inhibitor in Pancreatic Cancer Cell Lines. International Journal of Molecular Sciences, 22(13), 6825. https://doi.org/10.3390/ijms22136825