
Coagulation Signaling through PAR1 as a Therapeutic Target in Pancreatic Ductal Adenocarcinoma

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Thrombosis in Cancer
3. Coagulation Activity as a Modifier of the PDAC Disease Progression
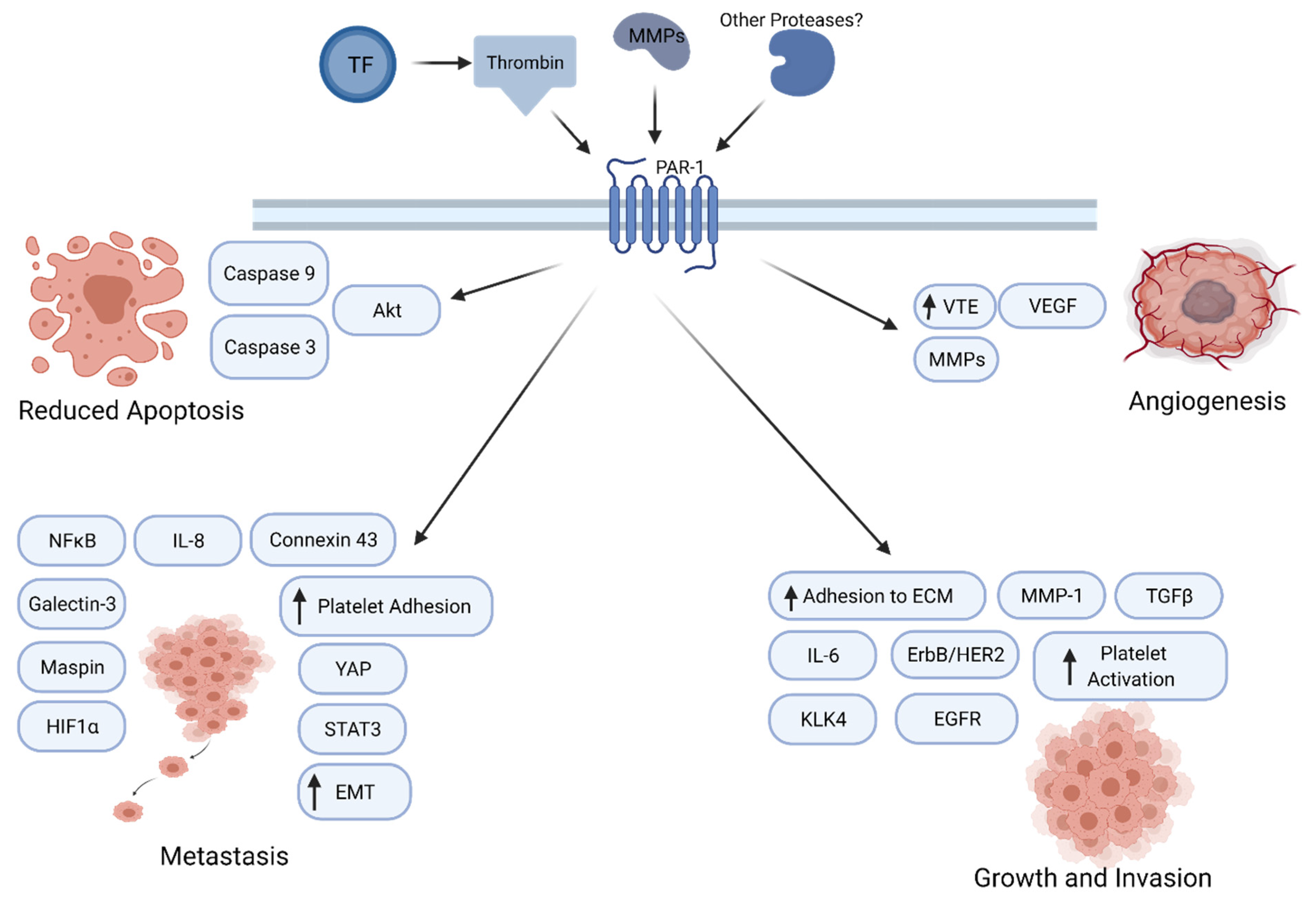
4. Protease-Activated Receptor-1 Signaling in Cancer Progression
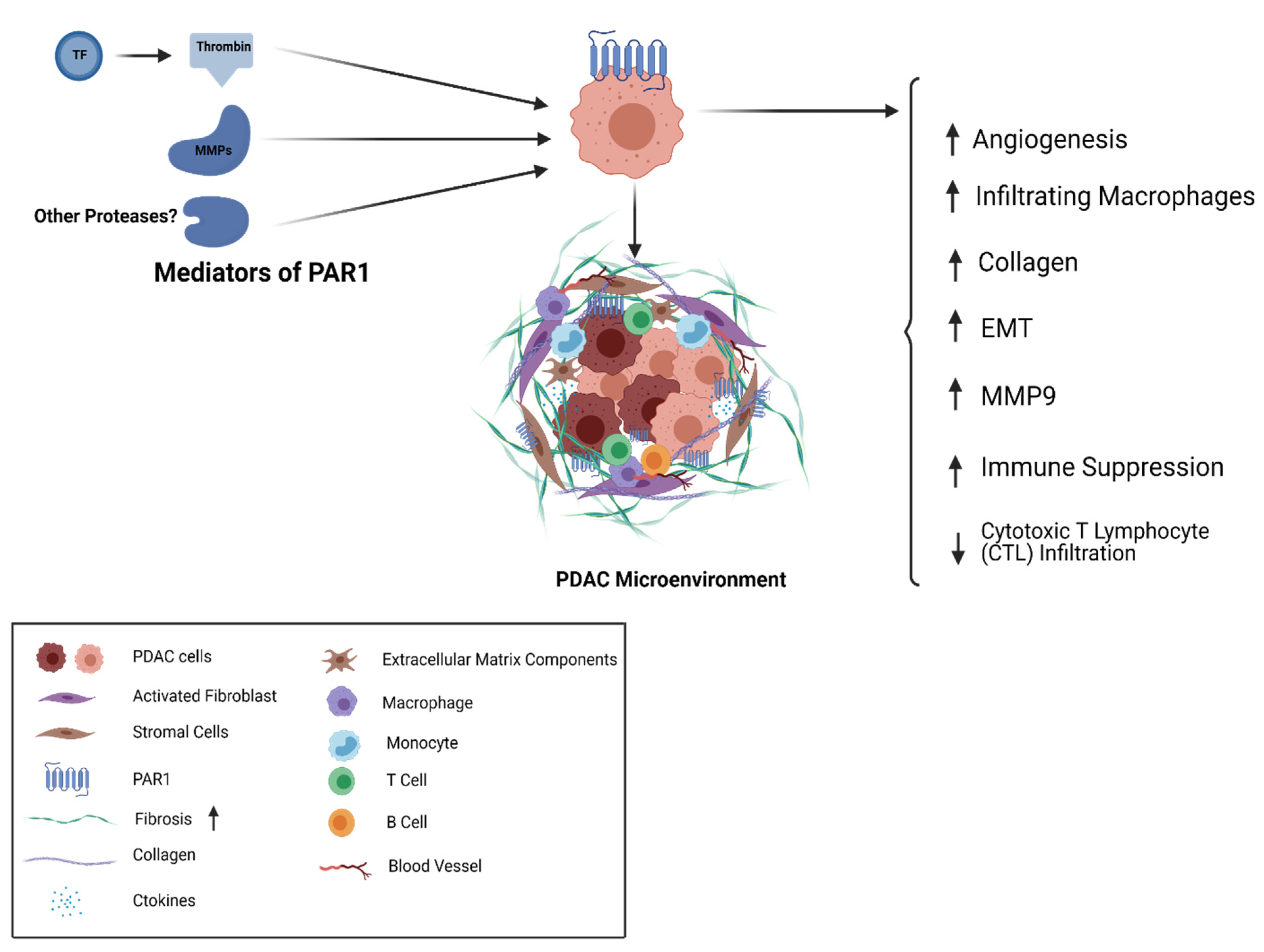
5. Thrombin/PAR1 Signaling in PDAC
6. Targeting PAR1 as a Therapeutic Approach in PDAC
7. Summary
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Pancreatic Cancer: Statistics | Cancer.Net. Available online: https://www.cancer.net/cancer-types/pancreatic-cancer/statistics (accessed on 30 December 2020).
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [Green Version]
- Common Cancer Sites–Cancer Stat Facts. Available online: https://seer.cancer.gov/statfacts/html/common.html (accessed on 29 December 2020).
- Pancreatic Cancer: Stages | Cancer.Net. Available online: https://www.cancer.net/cancer-types/pancreatic-cancer/stages (accessed on 30 December 2020).
- Miller, B. Racial/Ethnic Patterns of Cancer in the United States, 1988–1992; US Department of Health and Human Services, National Institutes of Health: Bethesda, MD, USA, 1998.
- Wong, M.C.S.; Jiang, J.Y.; Liang, M.; Fang, Y.; Yeung, M.S.; Sung, J.J.Y. Global Temporal Patterns of Pancreatic Cancer and Association with Socioeconomic Development. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wahi, M.M.; Shah, N.; Schrock, C.E.; Rosemurgy, A.S.; Goldin, S.B. Reproductive Factors and Risk of Pancreatic Cancer in Women: A Review of the Literature. Ann. Epidemiol. 2009, 19, 103–111. [Google Scholar] [CrossRef]
- Wolpin, B.M.; Kraft, P.; Gross, M.; Helzlsouer, K.; Bueno-De-Mesquita, H.B.; Steplowski, E.; Stolzenberg-Solomon, R.Z.; Arslan, A.A.; Jacobs, E.J.; Lacroix, A.; et al. Pancreatic Cancer Risk and ABO Blood Group Alleles: Results from the Pancreatic Cancer Cohort Consortium. Cancer Res. 2010, 70, 1015–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brentnall, T.A.; Kamineni, A.; Potter, J.D. Family History of Diabetes and Pancreatic Cancer as Risk Factors for Pancreatic Cancer: The PACIFIC Study. Cancer Epidemiol. Prev. Biomark. 2013, 22. [Google Scholar] [CrossRef]
- Demir, I.E.; Friess, H.; Ceyhan, G.O. Neural Plasticity in Pancreatitis and Pancreatic Cancer. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, C.; Huang, H.; Jiang, Q.; Zhao, D.; Tian, Y.; Ma, J.; Yuan, W.; Sun, Y.; Che, X.; et al. Effects of Alcohol Drinking and Smoking on Pancreatic Ductal Adenocarcinoma Mortality: A Retrospective Cohort Study Consisting of 1783 Patients. Sci. Rep. 2017, 7, 9572. [Google Scholar] [CrossRef] [Green Version]
- Esposito, I.; Konukiewitz, B.; Schlitter, A.M.; Klöppel, G. Pathology of Pancreatic Ductal Adenocarcinoma: Facts, Challenges and Future Developments. World J. Gastroenterol. 2014, 20, 13833–13841. [Google Scholar] [CrossRef] [PubMed]
- Matthaei, H.; Schulick, R.D.; Hruban, R.H.; Maitra, A. Cystic Precursors to Invasive Pancreatic Cancer. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, S.; Van Buren, G.I.I.; Fisher, E.W. Pancreatic Cancer: Advances in Treatment. World J. Gastroenterol. 2014, 20, 9354–9360. [Google Scholar]
- Brune, K.; Abe, T.; Canto, M.; O’Malley, L.; Klein, A.P.; Maitra, A.; Adsay, N.V.; Fishman, E.K.; Cameron, J.L.; Yeo, C.J.; et al. Multifocal Neoplastic Precursor Lesions Associated with Lobular Atrophy of the Pancreas in Patients Having a Strong Family History of Pancreatic Cancer. Am. J. Surg. Pathol. 2006, 30, 1067–1076. [Google Scholar]
- Fahrmann, J.F.; Bantis, E.L.; Capello, M.; Scelo, G.; Dennison, J.B.; Patel, N.; Murage, E.; Vykoukal, J.; Kundnani, D.L.; Foretova, L.; et al. A Plasma-Derived Protein-Metabolite Multiplexed Panel for Early-Stage Pancreatic Cancer. J. Natl. Cancer Inst. 2019, 111, 372–379. [Google Scholar] [CrossRef]
- Ansari, D.; Gustafsson, A.; Andersson, R. Update on the Management of Pancreatic Cancer: Surgery is Not Enough. World J. Gastroenterol. 2015, 21, 3157–3165. [Google Scholar] [CrossRef] [PubMed]
- Fuerst, M.L. Treating Pancreatic Cancer. Oncol. Times 2003, 25, 70. [Google Scholar] [CrossRef]
- Müller, P.C.; Frey, M.C.; Ruzza, C.M.; Nickel, F.; Jost, C.; Gwerder, C.; Hackert, T.; Z’Graggen, K.; Kessler, U. Neoadjuvant Chemotherapy in Pancreatic Cancer: An Appraisal of the Current High-Level Evidence. Pharmacology 2020, 2020, 1–11. [Google Scholar] [CrossRef]
- Franke, A.J.; Rosati, L.M.; Pawlik, T.M.; Kumar, R.; Herman, J.M. The Role of Radiation Therapy in Pancreatic Ductal Adenocarcinoma in the Neoadjuvant and Adjuvant Settings. Semin. Oncol. 2015, 42, 144–162. [Google Scholar] [CrossRef]
- Metharom, P.; Falasca, M.; Berndt, M.C. The History of Armand Trousseau and Cancer-Associated Thrombosis. Cancers 2019, 11, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piazza, G. Venous Thromboembolism and Cancer. Circulation 2013, 128, 2614–2618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorensen, H.T.; Mellemkjaer, L.; Olsen, J.H.; Baron, J.A. Prognosis of Cancers Associated with Venous Thromboembolism. N. Engl. J. Med. 2000, 343, 1846–1850. [Google Scholar] [CrossRef] [PubMed]
- Vitale, F.V.; Longo-Sorbello, G.S.; Rotondo, S.; Ferrau, F. Understanding and Treating Solid Tumor–related Disseminated Intravascular Coagulation in the “Era” of Targeted Cancer Therapies. SAGE Open Med. 2017, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campello, E.; Ilich, A.; Simioni, P.; Key, N.S. The Relationship between Pancreatic Cancer and Hypercoagulability: A Comprehensive Review on Epidemiological and Biological Issues. Br. J. Cancer 2019, 121, 359–371. [Google Scholar] [CrossRef]
- Zalatnai, A.; Perjési, E.; Galambos, E. Much More than Trousseau Syndrome. The Broad Spectrum of the Pancreatic Paraneoplastic Syndromes. Pathol. Oncol. Res. 2017, 24, 1–10. [Google Scholar] [CrossRef]
- Khorana, A.A.; Fine, R.L. Pancreatic Cancer and Thromboembolic Disease. Lancet Oncol. 2004, 5, 655–663. [Google Scholar] [CrossRef]
- Khorana, A.A.; Ahrendt, S.A.; Ryan, C.K.; Francis, C.W.; Hruban, R.H.; Hu, Y.C.; Hostetter, G.; Harvey, J.; Taubman, M.B. Tissue Factor Expression, Angiogenesis, and Thrombosis in Pancreatic Cancer. Clin. Cancer Res. 2007, 13, 2870–2875. [Google Scholar] [CrossRef] [Green Version]
- Geddings, J.E.; Mackman, N. Tumor-derived Tissue Factor–positive Microparticles and Venous Thrombosis in Cancer Patients. Blood 2013, 122, 1873–1880. [Google Scholar] [CrossRef] [PubMed]
- Echrish, H.; Xiao, Y.; Madden, L.; Allgar, V.; Cooke, J.; Wedgwood, K.; Dasgupta, D.; Greenman, J.; Maraveyas, A. Effect of Resection of Localized Pancreaticobiliary Adenocarcinoma on Angiogenic Markers and Tissue Factor Related Pro-thrombotic and Pro-angiogenic Activity. Thromb. Res. 2014, 134, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Andrén-Sandberg, Å.; Lecander, I.; Martinsson, G.; Åstedt, B. Peaks in Plasma Plasminogen Activator Inhibitor-1 Concentration May Explain Thrombotic Events in Cases of Pancreatic Carcinoma. Cancer 1992, 69, 2884–2887. [Google Scholar] [CrossRef]
- Hisada, Y.; Mackman, N. Cancer-associated Pathways and Biomarkers of Venous Thrombosis. Blood 2017, 130, 1499–1506. [Google Scholar] [CrossRef]
- Hisada, Y.; Garratt, K.B.; Maqsood, A.; Grover, S.P.; Kawano, T.; Cooley, B.C.; Erlich, J.; Moik, F.; Flick, M.J.; Pabinger, I.; et al. Plasminogen Activator Inhibitor 1 and Venous Thrombosis in Pancreatic Cancer. Blood Adv. 2021, 5, 487–495. [Google Scholar] [CrossRef]
- Elemary, M.; Moodley, O.; Pearson, D.; Goubran, H. Cancer-Associated Thrombosis (CAT). Precis. Anticoag. Med. 2020, 16, 127–145. [Google Scholar] [CrossRef]
- Su, Y.; Deng, X.; Ma, R.; Dong, Z.; Wang, F.; Shi, J. The Exposure of Phosphatidylserine Influences Procoagulant Activity in Retinal Vein Occlusion by Microparticles, Blood Cells, and Endothelium. Oxidative Med. Cell. Longev. 2018, 2018, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Beristain-Covarrubias, N.; Perez-Toledo, M.; Thomas, M.R.; Henderson, I.R.; Watson, S.P.; Cunningham, A.F. Understanding Infection-Induced Thrombosis: Lessons Learned from Animal Models. Front. Immunol. 2019, 10, 2569. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Thachil, J.; Asakura, H.; Levy, J.H.; Iba, T. Thrombomodulin in Disseminated Intravascular Coagulation and Other Critical Conditions–A Multi-faceted Anticoagulant Protein with Therapeutic Potential. Crit. Care 2019, 23, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alessi, M.-C.; Juhan-Vague, I. PAI-1 and the Metabolic Syndrome. Arter. Thromb. Vasc. Biol. 2006, 26, 2200–2207. [Google Scholar] [CrossRef]
- Huber, M.; Brehm, C.U.; Gress, T.M.; Buchholz, M.; Alhamwe, B.A.; Von Strandmann, E.P.; Slater, E.P.; Bartsch, J.W.; Bauer, C.; Lauth, M. The Immune Microenvironment in Pancreatic Cancer. Int. J. Mol. Sci. 2020, 21, 7307. [Google Scholar] [CrossRef]
- Felix, K.; Gaida, M.M. Neutrophil-Derived Proteases in the Microenvironment of Pancreatic Cancer -Active Players in Tumor Progression. Int. J. Biol. Sci. 2016, 12, 302–313. [Google Scholar] [CrossRef] [Green Version]
- Miller-Ocuin, J.L.; Liang, X.; Boone, B.A.; Doerfler, W.R.; Singhi, A.D.; Tang, D.; Kang, R.; Lotze, M.T.; Zeh, H.J. DNA Released from Neutrophil Extracellular Traps (NETs) Activates Pancreatic Stellate Cells and Enhances Pancreatic Tumor Growth. OncoImmunology 2019, 8, e1605822. [Google Scholar] [CrossRef]
- Snoderly, H.T.; Boone, B.A.; Bennewitz, M.F. Neutrophil Extracellular Traps in Breast Cancer and Beyond: Current Perspectives on NET Stimuli, Thrombosis and Metastasis, and Clinical Utility for Diagnosis and Treatment. Breast Cancer Res. 2019, 21, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Martinod, K.; Wagner, D.D. Thrombosis: Tangled up in NETs. Blood 2014, 123, 2768–2776. [Google Scholar] [CrossRef]
- Degen, J.L.; Palumbo, J.S. Hemostatic Factors, Innate Immunity and Malignancy. Thromb. Res. 2012, 129, S1–S5. [Google Scholar] [CrossRef]
- Rickles, F.R.; Edwards, R.L. Activation of blood coagulation in cancer: Trousseau’s syndrome revisited. Blood 1983, 62, 14–31. [Google Scholar] [CrossRef] [Green Version]
- Mueller, B.M.; Reisfeld, R.A.; Edgington, T.S.; Ruf, W. Expression of Tissue Factor by Melanoma Cells Promotes Efficient Hematogenous Metastasis. Proc. Natl. Acad. Sci. USA 1992, 89, 11832–11836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.L.; May, L.; Lhotak, V.; Shahrzad, S.; Shirasawa, S.; Weitz, J.I.; Coomber, B.L.; Mackman, N.; Rak, J.W. Oncogenic Events Regulate Tissue Factor Expression in Colorectal Cancer Cells: Implications for Tumor Progression and Angiogenesis. Blood 2005, 105, 1734–1741. [Google Scholar] [CrossRef]
- Nitori, N.; Ino, Y.; Nakanishi, Y.; Yamada, T.; Honda, K.; Yanagihara, K.; Kosuge, T.; Kanai, Y.; Kitajima, M.; Hirohashi, S. Prognostic Significance of Tissue Factor in Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2005, 11, 2531–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.W.; Komar, C.A.; Bengsch, F.; Graham, K.; Beatty, G.L. Genetically Engineered Mouse Models of Pancreatic Cancer: The KPC Model (LSL-Kras G12D/+; LSL-Trp53 R172H/+; Pdx-1-Cre), Its Variants, and Their Application in Immuno-oncology Drug Discovery. Curr. Protoc. Pharmacol. 2016, 73, 14.39.1–14.39.20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, Y.W.V.D.; Hengel, L.G.V.D.; Myers, H.R.; Ayachi, O.; Jordanova, E.; Ruf, W.; Spek, C.A.; Reitsma, P.H.; Bogdanov, V.Y.; Versteeg, H.H. Alternatively Spliced Tissue Factor Induces Angiogenesis through Integrin Ligation. Proc. Natl. Acad. Sci. USA 2009, 106, 19497–19502. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, R.; Ozhegov, E.; Berg, Y.W.V.D.; Aronow, B.J.; Franco, R.S.; Palascak, M.B.; Fallon, J.T.; Ruf, W.; Versteeg, H.; Bogdanov, V. Splice Variants of Tissue Factor Promote Monocyte-endothelial Interactions by Triggering the Expression of Cell Adhesion Molecules via Integrin-mediated Signaling. J. Thromb. Haemost. 2011, 9, 2087–2096. [Google Scholar] [CrossRef]
- Unruh, D.; Turner, K.; Srinivasan, R.; Kocatürk, B.; Qi, X.; Chu, Z.; Aronow, B.J.; Plas, D.R.; Gallo, C.A.; Kalthoff, H.; et al. Alternatively Spliced Tissue Factor Contributes to Tumor Spread and Activation of Coagulation in Pancreatic Ductal Adenocarcinoma. Int. J. Cancer 2013, 134, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Unruh, D.; Ünlü, B.; Lewis, C.S.; Qi, X.; Chu, Z.; Sturm, R.; Keil, R.; Ahmad, S.A.; Sovershaev, T.; Adam, M.; et al. Antibody-based Targeting of Alternatively Spliced Tissue Factor: A New Approach to Impede the Primary Growth and Spread of Pancreatic Ductal Adenocarcinoma. Oncotarget 2016, 7, 25264–25275. [Google Scholar] [CrossRef] [Green Version]
- Riewald, M.; Ruf, W. Mechanistic Coupling of Protease Signaling and Initiation of Coagulation by Tissue Factor. Proc. Natl. Acad. Sci. USA 2001, 98, 7742–7747. [Google Scholar] [CrossRef] [Green Version]
- Henrikson, K.P.; Salazar, S.L.; Ii, J.W.F.; Pentecost, B.T. Role of Thrombin Receptor in Breast Cancer Invasiveness. Br. J. Cancer 1999, 79, 401–406. [Google Scholar] [CrossRef] [Green Version]
- Adams, G.N.; Sharma, B.K.; Rosenfeldt, L.; Frederick, M.; Flick, M.J.; Witte, D.P.; Mosnier, L.O.; Harmel-Laws, E.; Steinbrecher, K.A.; Palumbo, J.S. Protease-activated Receptor-1 Impedes Prostate and Intestinal Tumor Progression in Mice. J. Thromb. Haemost. 2018, 16, 2258–2269. [Google Scholar] [CrossRef] [Green Version]
- Versteeg, H.H.; Schaffner, F.; Kerver, M.; Ellies, L.G.; Andrade-Gordon, P.; Mueller, B.M.; Ruf, W. Protease-Activated Receptor (PAR) 2, but not PAR1, Signaling Promotes the Development of Mammary Adenocarcinoma in Polyoma Middle T Mice. Cancer Res. 2008, 68, 7219–7227. [Google Scholar] [CrossRef] [Green Version]
- Martin, C.B.; Mahon, G.M.; Klinger, M.B.; Kay, R.J.; Symons, M.; Der, C.J.; Whitehead, I.P. The Thrombin Receptor, PAR-1, Causes Transformation by Activation of Rho-mediated Signaling Pathways. Oncogene 2001, 20, 1953–1963. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.-J.; Katz, V.; Salah, Z.; Maoz, M.; Cohen, I.; Uziely, B.; Turm, H.; Grisaru-Granovsky, S.; Suzuki, H.; Bar-Shavit, R. Mammary Gland Tissue Targeted Overexpression of Human Protease-Activated Receptor 1 Reveals a Novel Link to β-Catenin Stabilization. Cancer Res. 2006, 66, 5224–5233. [Google Scholar] [CrossRef] [Green Version]
- Even-Ram, S.; Uziely, B.; Cohen, P.; Grisaru-Granovsky, S.; Maoz, M.; Ginzburg, Y.; Reich, R.; Vlodavsky, I.; Bar-Shavit, R. Thrombin Receptor Overexpression in Malignant and Physiological Invasion Processes. Nat. Med. 1998, 4, 909–914. [Google Scholar] [CrossRef]
- Won, J.H.; Zhang, Y.; Ji, B.; Logsdon, C.D.; Yule, D.I. Phenotypic Changes in Mouse Pancreatic Stellate Cell Ca2+ Signaling Events Following Activation in Culture and in a Disease Model of Pancreatitis. Mol. Biol. Cell 2011, 22, 421–436. [Google Scholar] [CrossRef]
- Kim, S.-J.; Shin, J.-Y.; Lee, K.-D.; Bae, Y.-K.; Choi, I.-J.; Park, S.H.; Chun, K.-H. Galectin-3 Facilitates Cell Motility in Gastric Cancer by Up-Regulating Protease-Activated Receptor-1(PAR-1) and Matrix Metalloproteinase-1(MMP-1). PLoS ONE 2011, 6, e25103. [Google Scholar] [CrossRef]
- Wang, W.; Mize, G.J.; Zhang, X.; Takayama, T.K. Kallikrein-related Peptidase-4 initiates Tumor–Stroma Interactions in Prostate Cancer through Protease-activated Receptor-1. Int. J. Cancer 2010, 126, 599–610. [Google Scholar] [CrossRef]
- Coughlin, S.R. Thrombin Signalling and Protease-activated Receptors. Nat. Cell Biol. 2000, 407, 258–264. [Google Scholar] [CrossRef]
- Nierodzik, M.L.; Karpatkin, S. Thrombin Induces Tumor Growth, Metastasis, and Angiogenesis: Evidence for a Thrombin-regulated Dormant Tumor Phenotype. Cancer Cell 2006, 10, 355–362. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, V.B.; Pai, K.S.; Lau, A.; Cunningham, D.D. Creatine Kinase, an ATP-generating Enzyme, is Required for Thrombin Receptor Signaling to the Cytoskeleton. Proc. Natl. Acad. Sci. USA 2000, 97, 12062–12067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Even-Ram, S.C.; Maoz, M.; Pokroy, E.; Reich, R.; Katz, B.-Z.; Gutwein, P.; Altevogt, P.; Bar-Shavit, R. Tumor Cell Invasion Is Promoted by Activation of Protease Activated Receptor-1 in Cooperation with the αvβ5 Integrin. J. Biol. Chem. 2001, 276, 10952–10962. [Google Scholar] [CrossRef] [Green Version]
- Amador, M.A.T.; Cavalcante, G.C.; Santos, N.P.C.; Gusmão, L.; Guerreiro, J.F.; Ribeiro-Dos-Santo, Â.; Santos, S. Distribution of Allelic and Genotypic Frequencies of IL1A, IL4, NFKB1 and PAR1 Variants in Native American, African, European and Brazilian Populations. BMC Res. Notes 2016, 9, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, S.; Nakajima, K.; Tohyama, Y.; Kohsaka, S. Characteristic response of astrocytes to plasminogen/plasmin to upregulate transforming growth factor beta 3 (TGFβ3) production/secretion through proteinase-activated receptor-1 (PAR-1) and the downstream phosphatidylinositol 3-kinase (PI3K)-Akt/PKB signaling cascade. Brain Res. 2009, 1305, 1–13. [Google Scholar] [CrossRef]
- Melnikova, V.O.; Balasubramanian, K.; Villares, G.J.; Dobroff, A.S.; Zigler, M.; Wang, H.; Petersson, F.; Price, J.E.; Schroit, A.; Prieto, V.G.; et al. Crosstalk between Protease-activated Receptor 1 and Platelet-activating Factor Receptor Regulates Melanoma Cell Adhesion Molecule (MCAM/MUC18) Expression and Melanoma Metastasis. J. Biol. Chem. 2009, 284, 28845–28855. [Google Scholar] [CrossRef] [Green Version]
- Villares, G.J.; Zigler, M.; Wang, H.; Melnikova, V.O.; Wu, H.; Friedman, R.; Leslie, M.C.; Vivas-Mejia, P.E.; Lopez-Berestein, G.; Sood, A.K.; et al. Targeting Melanoma Growth and Metastasis with Systemic Delivery of Liposome-Incorporated Protease-Activated Receptor-1 Small Interfering RNA. Cancer Res. 2008, 68, 9078–9086. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Yu, J.; Song, S.; Yue, X.; Li, Q. Protease-activated Receptor-1 (PAR-1): A Promising Molecular Target for Cancer. Oncotarget 2017, 8, 107334–107345. [Google Scholar] [CrossRef] [Green Version]
- Gratio, V.; Beaufort, N.; Seiz, L.; Maier, J.; Virca, G.D.; Debela, M.; Grebenchtchikov, N.; Magdolen, V.; Darmoul, D. Kallikrein-Related Peptidase 4: A New Activator of the Aberrantly Expressed Protease-Activated Receptor 1 in Colon Cancer Cells. Am. J. Pathol. 2010, 176, 1452–1461. [Google Scholar] [CrossRef] [Green Version]
- Robinson, R.L.; Sharma, A.; Bai, S.; Heneidi, S.; Lee, T.J.; Kodeboyina, S.K.; Patel, N.; Sharma, S. Comparative STAT3-Regulated Gene Expression Profile in Renal Cell Carcinoma Subtypes. Front. Oncol. 2019, 9, 10–12. [Google Scholar] [CrossRef] [Green Version]
- Smoktunowicz, N.; Platé, M.; Stern, A.O.; D’Antongiovanni, V.; Robinson, E.; Chudasama, V.; Caddick, S.; Scotton, C.J.; Jarai, G.; Chambers, R.C. TGFβ Upregulates PAR-1 Expression and Signalling Responses in A549 Lung Adenocarcinoma Cells. Oncotarget 2016, 7, 65471–65484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellone, M.D.; Laukkanen, M.O.; Teramoto, H.; Bellelli, R.; Alì, G.; Fontanini, G.; Santoro, M.; Gutkind, J.S. Cross Talk between the Bombesin Neuropeptide Receptor and Sonic Hedgehog Pathways in Small Cell Lung Carcinoma. Oncogene 2015, 34, 1679–1687. [Google Scholar] [CrossRef] [Green Version]
- Mahadevan, D.; Von Hoff, D.D. Tumor-stroma Interactions in Pancreatic Ductal Adenocarcinoma. Mol. Cancer Ther. 2007, 6, 1186–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Queiroz, K.C.S.; Shi, K.; Duitman, J.; Aberson, H.L.; Wilmink, J.W.; Van Noesel, C.J.M.; Richel, D.J.; Spek, C.A. Protease-activated Receptor-1 Drives Pancreatic Cancer Progression and Chemoresistance. Int. J. Cancer 2014, 135, 2294–2304. [Google Scholar] [CrossRef] [PubMed]
- Tekin, C.; Shi, K.; Daalhuisen, J.B.; Brink, M.S.T.; Bijlsma, M.F.; Spek, C.A. PAR1 Signaling on Tumor Cells Limits Tumor Growth by Maintaining a Mesenchymal Phenotype in Pancreatic Cancer. Oncotarget 2018, 9, 32010–32023. [Google Scholar] [CrossRef] [Green Version]
- Griffin, C.T.; Srinivasan, Y.; Zheng, Y.-W.; Huang, W.; Coughlin, S.R. A Role for Thrombin Receptor Signaling in Endothelial Cells During Embryonic Development. Science 2001, 293, 1666–1670. [Google Scholar] [CrossRef]
- Yin, Y.; Salah, Z.; Maoz, M.; Ram, S.C.E.; Ochayon, S.; Neufeld, G.; Katzav, S.; Bar-Shavit, R. Oncogenic Transformation Induces Tumor Angiogenesis: A Role for PAR1 Activation. FASEB J. 2002, 17, 163–174. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Angiogenesis in Cancer and Other Diseases. Nat. Cell Biol. 2000, 407, 249–257. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [Green Version]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-Associated Stromal Fibroblasts Promote Pancreatic Tumor Progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Mo, C.; Wang, Y.; Wei, D.; Xiao, H. Anti-tumour Strategies Aiming to Target Tumour-associated Macrophages. Immunology 2013, 138, 93–104. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Liu, Q.; Liao, Q. Tumor-Associated Macrophages in Pancreatic Ductal Adenocarcinoma: Origin, Polarization, Function, and Reprogramming. Front Cell Dev Biol. 2021, 8, 607209. [Google Scholar] [CrossRef]
- Weizman, N.; Krelin, Y.; Shabtayorbach, A.; Amit, M.; Binenbaum, Y.; Wong, R.J.; Gil, Z. Macrophages Mediate Gemcitabine Resistance of Pancreatic Adenocarcinoma by Upregulating Cytidine Deaminase. Oncogene 2013, 33, 3812–3819. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.Y.; Xu, J.Y.; Shi, X.Y.; Huang, W.; Ruan, T.Y.; Xie, P.; Ding, J.L. M2-polarized Tumor-associated Macrophages Promoted Epithelial–mesenchymal Transition in Pancreatic Cancer Cells, Partially through TLR4/IL-10 Signaling Pathway. Lab. Investig. 2013, 93, 844–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting Tumor-infiltrating Macrophages Decreases Tumor-initiating Cells, Relieves Immunosuppression, and Improves Chemotherapeutic Responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef] [Green Version]
- Tekin, C.; Aberson, H.L.; Waasdorp, C.; Hooijer, G.K.J.; De Boer, O.J.; Dijk, F.; Bijlsma, M.F.; Spek, C.A. Macrophage-secreted MMP9 Induces Mesenchymal Transition in Pancreatic Cancer Cells via PAR1 Activation. Cell. Oncol. 2020, 43, 1161–1174. [Google Scholar] [CrossRef] [PubMed]
- Austin, K.M.; Covic, L.; Kuliopulos, A. Matrix Metalloproteases and PAR1 Activation. Blood 2013, 121, 431–439. [Google Scholar] [CrossRef] [Green Version]
- Kuwada, K.; Kagawa, S.; Yoshida, R.; Sakamoto, S.; Ito, A.; Watanabe, M.; Ieda, T.; Kuroda, S.; Kikuchi, S.; Tazawa, H.; et al. The Epithelial-to-mesenchymal Transition Induced by Tumor-associated Macrophages Confers Chemoresistance in Peritoneally Disseminated Pancreatic Cancer. J. Exp. Clin. Cancer Res. 2018, 37, 1–10. [Google Scholar] [CrossRef]
- Yang, Y.; Stang, A.; Schweickert, P.G.; Lanman, N.A.; Paul, E.N.; Monia, B.P.; Revenko, A.S.; Palumbo, J.S.; Mullins, E.S.; Elzey, B.D.; et al. Thrombin Signaling Promotes Pancreatic Adenocarcinoma through PAR-1–Dependent Immune Evasion. Cancer Res. 2019, 79, 3417–3430. [Google Scholar] [CrossRef]
- Schweickert, P.G.; Yang, Y.; White, E.E.; Cresswell, G.M.; Elzey, B.D.; Ratliff, T.L.; Arumugam, P.; Antoniak, S.; Mackman, N.; Flick, M.J.; et al. Thrombin-PAR1 Signaling in Pancreatic Cancer Promotes an Immunosuppressive Microenvironment. J. Thromb. Haemost. 2021, 19, 161–172. [Google Scholar] [CrossRef]
- Ezhao, P.; Emetcalf, M.; Bunnett, N.W. Biased Signaling of Protease-Activated Receptors. Front. Endocrinol. 2014, 5, 67. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi-Matsui, M.; Zheng, Y.-W.; Sulciner, D.J.; Weiss, E.J.; Ludeman, M.J.; Coughlin, S.R. PAR3 is a Cofactor for PAR4 Activation by Thrombin. Nat. Cell Biol. 2000, 404, 609–613. [Google Scholar] [CrossRef]
- Kabacaoglu, D.; Ciecielski, K.J.; Ruess, D.A.; Algül, H. Immune Checkpoint Inhibition for Pancreatic Ductal Adenocarcinoma: Current Limitations and Future Options. Front. Immunol. 2018, 9, 1878. [Google Scholar] [CrossRef]
- Nomi, T.; Sho, M.; Akahori, T.; Hamada, K.; Kubo, A.; Kanehiro, H.; Nakamura, S.; Enomoto, K.; Yagita, H.; Azuma, M.; et al. Clinical Significance and Therapeutic Potential of the Programmed Death-1 Ligand/Programmed Death-1 Pathway in Human Pancreatic Cancer. Clin. Cancer Res. 2007, 13, 2151–2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamath, S.D.; Kalyan, A.; Kircher, S.; Nimeiri, H.; Fought, A.J.; Benson, A.; Mulcahy, M. Ipilimumab and Gemcitabine for Advanced Pancreatic Cancer: A Phase Ib Study. Oncology 2019, 25, e808–e815. [Google Scholar] [CrossRef] [Green Version]
- Thind, K.; Padrnos, L.J.; Ramanathan, R.K.; Borad, M.J. Immunotherapy in Pancreatic Cancer Treatment: A New Frontier. Ther. Adv. Gastroenterol. 2017, 10, 168–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, A.M.; Santarsiero, L.M.; Lutz, E.R.; Armstrong, T.D.; Chen, Y.-C.; Huang, L.-Q.; Laheru, D.A.; Goggins, M.; Hruban, R.H.; Jaffee, E.M. Mesothelin-specific CD8+ T Cell Responses Provide Evidence of in vivo Cross-Priming by Antigen-Presenting Cells in Vaccinated Pancreatic Cancer Patients. J. Exp. Med. 2004, 200, 297–306. [Google Scholar] [CrossRef]
- Li, T.; Li, H.; Li, S.; Xu, S.; Zhang, W.; Gao, H.; Xu, H.; Wu, C.; Wang, W.; Yu, X.; et al. Research Progress and Design Optimization of CAR-T Therapy for Pancreatic Ductal Adenocarcinoma. Cancer Med. 2019, 8, 5223–5231. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.I.; Oliver, A.J.; Samiei, T.; Chan, J.D.; Kershaw, M.H.; Slaney, C.Y. Genetic Redirection of T Cells for the Treatment of Pancreatic Cancer. Front. Oncol. 2019, 9, 56. [Google Scholar] [CrossRef] [Green Version]
- Le, K.; Wang, J.; Zhang, T.; Guo, Y.; Chang, H.; Wang, S.; Zhu, B. Overexpression of Mesothelin in Pancreatic Ductal Adenocarcinoma (PDAC). Int. J. Med Sci. 2020, 17, 422–427. [Google Scholar] [CrossRef] [Green Version]
- Shaw, A.R.; Porter, C.E.; Yip, T.; Mah, W.-C.; McKenna, M.K.; Dysthe, M.; Jung, Y.; Parihar, R.; Brenner, M.K.; Suzuki, M. Oncolytic Adeno-immunotherapy Modulates the Immune System Enabling CAR T-cells to Cure Pancreatic Tumors. Commun. Biol. 2021, 4, 1–13. [Google Scholar] [CrossRef]
- Beatty, G.L.; Torigian, D.A.; Chiorean, E.G.; Saboury, B.; Brothers, A.; Alavi, A.; Troxel, A.B.; Sun, W.; Teitelbaum, U.R.; Vonderheide, R.H.; et al. A Phase I Study of an Agonist CD40 Monoclonal Antibody (CP-870,893) in Combination with Gemcitabine in Patients with Advanced Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2013, 19, 6286–6295. [Google Scholar] [CrossRef] [Green Version]
- Khoufache, K.; Berri, F.; Nacken, W.; Vogel, A.B.; Delenne, M.; Camerer, E.; Coughlin, S.R.; Carmeliet, P.; Lina, B.; Rimmelzwaan, G.F.; et al. PAR1 Contributes to Influenza A Virus Pathogenicity in Mice. J. Clin. Investig. 2012, 123, 206–214. [Google Scholar] [CrossRef]
- Saban, R.; D’Andrea, M.R.; Andrade-Gordon, P.; Derian, C.K.; Dozmorov, I.; Ihnat, M.A.; Hurst, R.E.; Davis, C.A.; Simpson, C.; Saban, M.R. Mandatory Role of Proteinase-activated Receptor 1 in Experimental Bladder Inflammation. BMC Physiol. 2007, 7, 4. [Google Scholar] [CrossRef] [Green Version]
- Baker, N.C.; Lipinski, M.J.; Lhermusier, T.; Waksman, R. Overview of the 2014 Food and Drug Administration Cardiovascular and Renal Drugs Advisory Committee Meeting About Vorapaxar. Circulation 2014, 130, 1287–1294. [Google Scholar] [CrossRef] [Green Version]
- Chanakira, A.; Westmark, P.R.; Ong, I.M.; Sheehan, J.P. Tissue factor-factor VIIa complex triggers protease activated receptor 2-dependent growth factor release and migration in ovarian cancer. Gynecol. Oncol. 2017, 145, 167–175. [Google Scholar] [CrossRef] [Green Version]
- Goto, S.; Ogawa, H.; Takeuchi, M.; Flather, M.D.; Bhatt, D.L. J-LANCELOT (Japanese-Lesson from Antagonizing the Cellular Effect of Thrombin) Investigators. Double-blind, Placebo-controlled Phase II Studies of the Protease-activated Receptor 1 Antagonist E5555 (Atopaxar) in Japanese Patients with Acute Coronary Syndrome or High-risk Coronary Artery Disease. Eur. Hear. J. 2010, 31, 2601–2613. [Google Scholar] [CrossRef] [Green Version]
- O’Donoghue, M.L.; Bhatt, D.L.; Wiviott, S.D.; Goodman, S.G.; Fitzgerald, D.J.; Angiolillo, D.J.; Goto, S.; Montalescot, G.; Zeymer, U.; Aylward, P.E.; et al. Safety and Tolerability of Atopaxar in the Treatment of Patients with Acute Coronary Syndromes. Circulation 2011, 123, 1843–1853. [Google Scholar] [CrossRef] [Green Version]
- Cros, J.; Raffenne, J.; Couvelard, A.; Poté, N. Tumor Heterogeneity in Pancreatic Adenocarcinoma. Pathobiology 2017, 85, 64–71. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kothari, A.; Flick, M.J. Coagulation Signaling through PAR1 as a Therapeutic Target in Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2021, 22, 5138. https://doi.org/10.3390/ijms22105138
Kothari A, Flick MJ. Coagulation Signaling through PAR1 as a Therapeutic Target in Pancreatic Ductal Adenocarcinoma. International Journal of Molecular Sciences. 2021; 22(10):5138. https://doi.org/10.3390/ijms22105138
Chicago/Turabian StyleKothari, Aditi, and Matthew J. Flick. 2021. "Coagulation Signaling through PAR1 as a Therapeutic Target in Pancreatic Ductal Adenocarcinoma" International Journal of Molecular Sciences 22, no. 10: 5138. https://doi.org/10.3390/ijms22105138
APA StyleKothari, A., & Flick, M. J. (2021). Coagulation Signaling through PAR1 as a Therapeutic Target in Pancreatic Ductal Adenocarcinoma. International Journal of Molecular Sciences, 22(10), 5138. https://doi.org/10.3390/ijms22105138