
Product Distributions of Cytochrome P450 OleTJE with Phenyl-Substituted Fatty Acids: A Computational Study



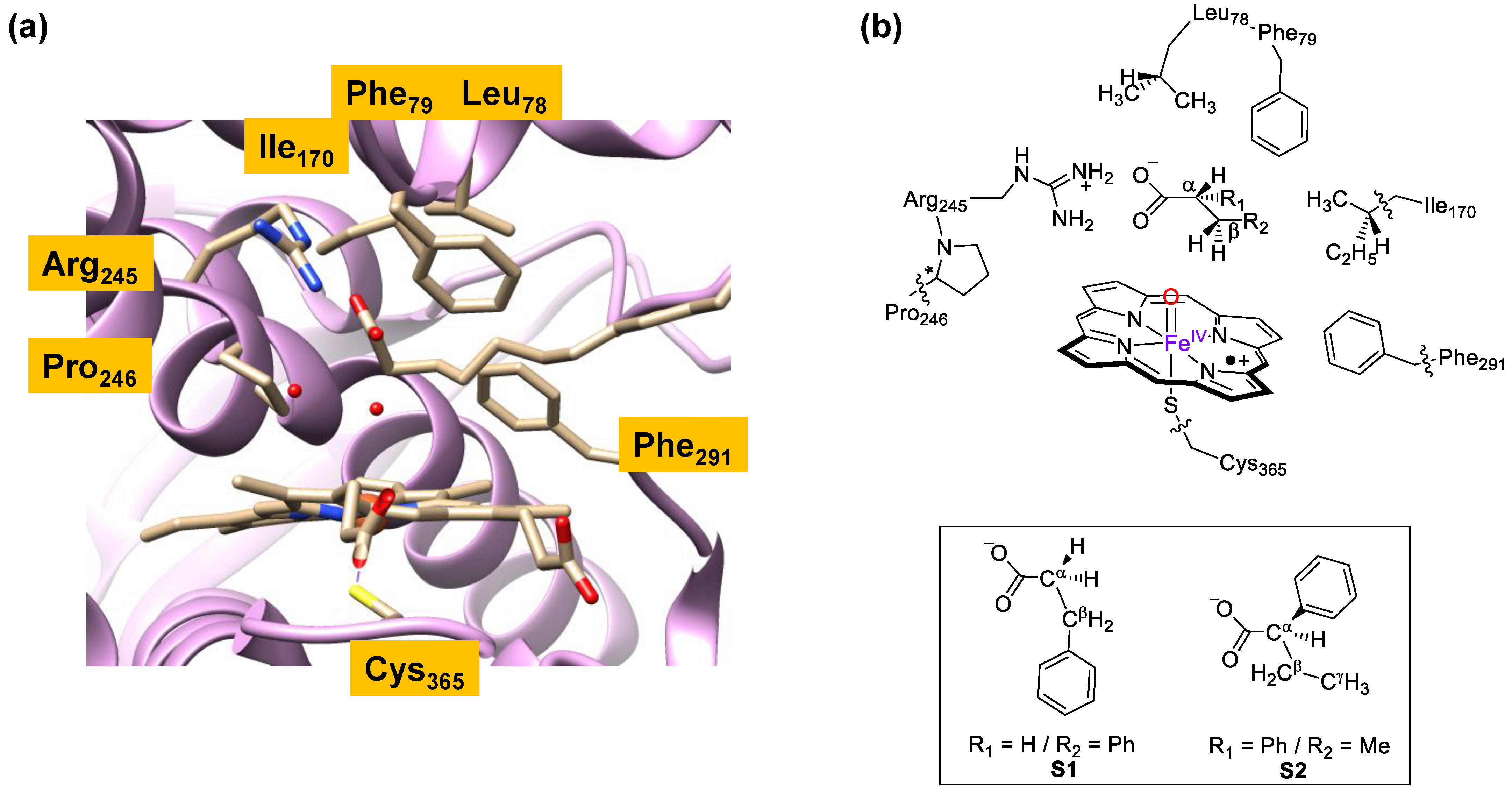{kind=link}
{kind=link}
{kind=link}
{kind=link}
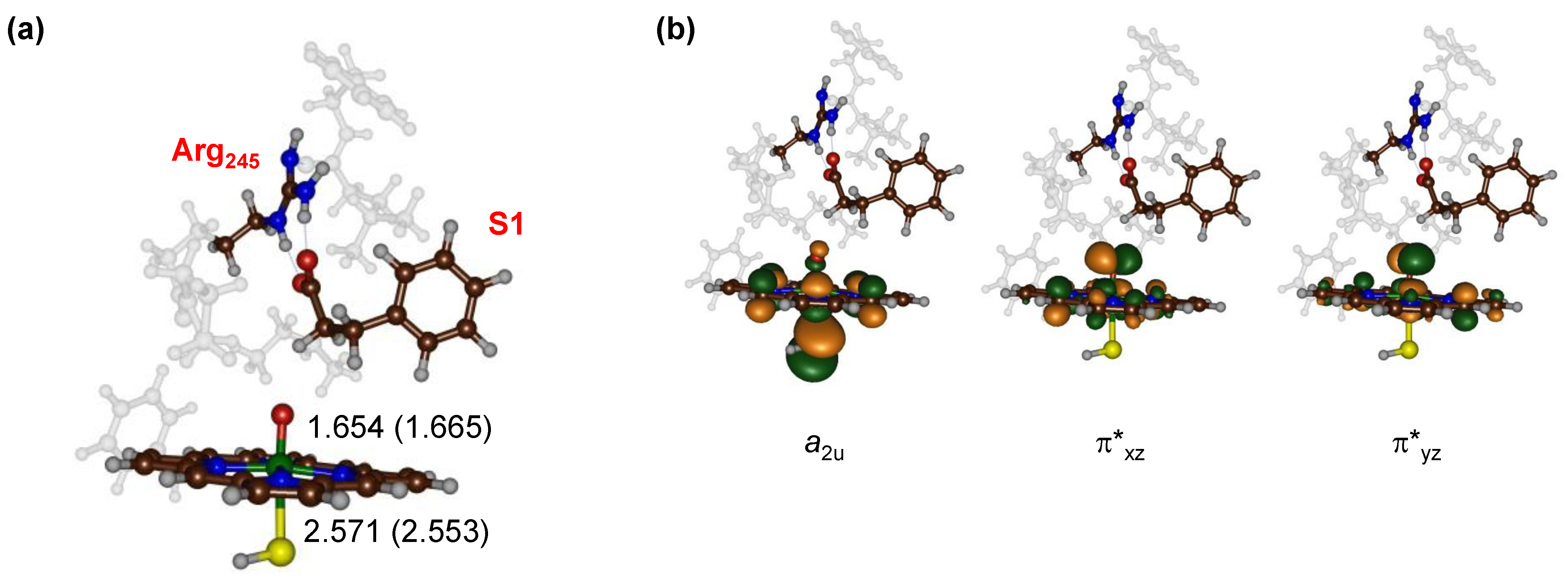
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
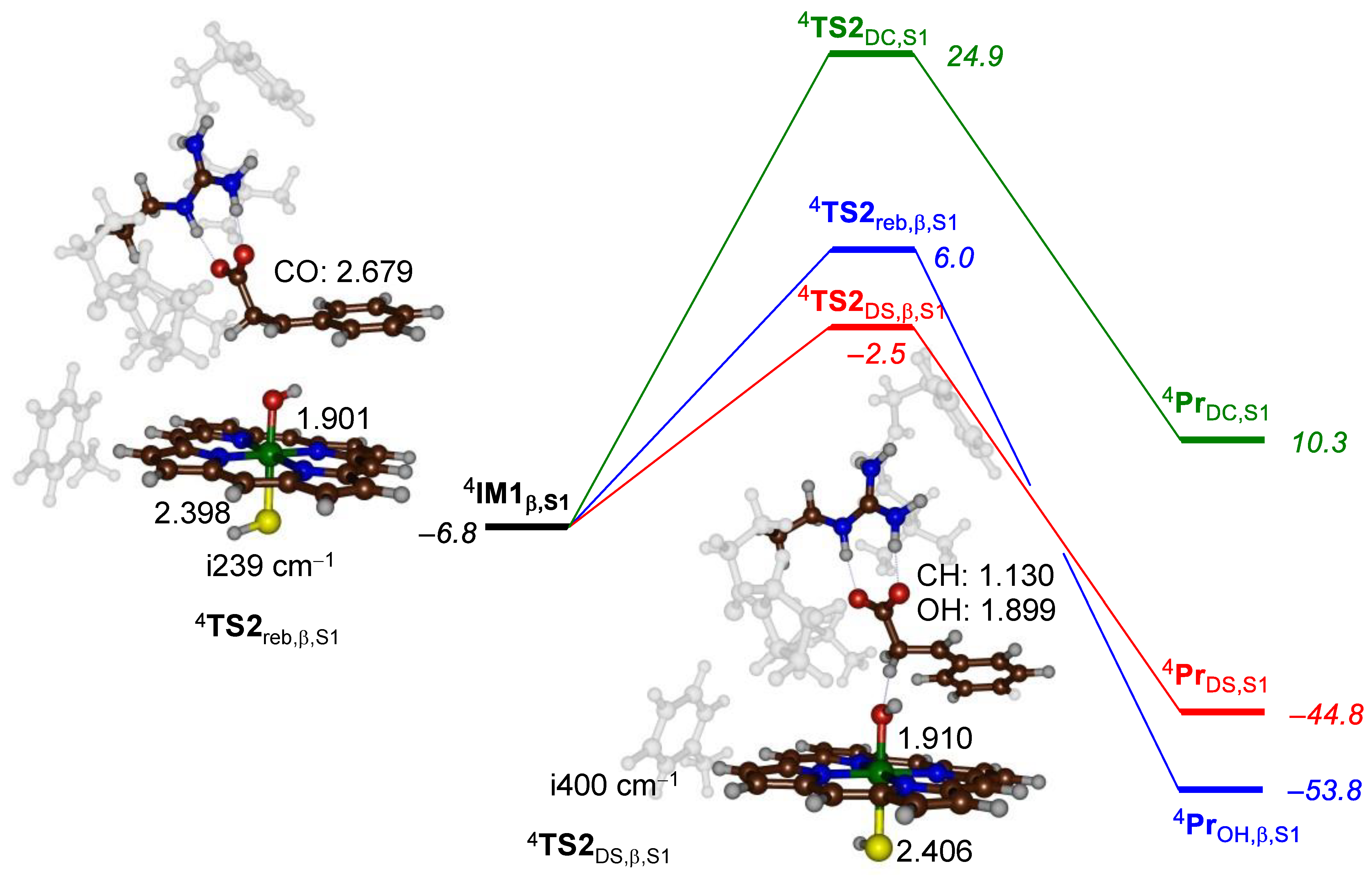
2.1. 3-Phenylpropionate Activation by CpdI of P450 OleTJE
2.2. 2-Phenylbutyrate Activation by CpdI of P450 OleTJE
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Beeson, W.T.; Vu, V.V.; Span, E.A.; Phillips, C.M.; Marletta, M.A. Cellulose degradation by polysaccharide monooxygenases. Ann. Rev. Biochem. 2015, 84, 923–946. [Google Scholar] [CrossRef]
- Kang, A.; Lee, T.S. Converting sugars to biofuels: Ethanol and beyond. Bioengineering 2015, 2, 184–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walton, P.H.; Davies, G.J. On the catalytic mechanisms of lytic polysaccharide monooxygenases. Curr. Opin. Chem. Biol. 2016, 31, 195–207. [Google Scholar] [CrossRef]
- Lup, A.N.K.; Abnisa, F.; Daud, W.M.A.W.; Aroua, M.K. A review on reaction mechanisms of metal-catalyzed deoxygenation process in bio-oil model compounds. Appl. Catal. A 2017, 541, 87–106. [Google Scholar] [CrossRef]
- Coines, J.; Raich, L.; Rovira, C. Modeling catalytic reaction mechanisms in glycoside hydrolases. Curr. Opin. Chem. Biol. 2019, 53, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, F.; Masgrau, L. Computational modeling of carbohydrate processing enzymes reactions. Curr. Opin. Chem. Biol. 2021, 61, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, A.G. Designer metalloenzymes for synthetic biology: Enzyme hybrids for catalysis. Curr. Opin. Chem. Biol. 2020, 58, 63–71. [Google Scholar] [CrossRef]
- Das, A.; Hessin, C.; Ren, Y.; Desage-El Murr, M. Biological concepts for catalysis and reactivity: Empowering bioinspiration. Chem. Soc. Rev. 2020, 49, 8840–8867. [Google Scholar] [CrossRef]
- Ghosh, A.C.; Duboc, C.; Gennari, M. Synergy between metals for small molecule activation: Enzymes and bioinspired complexes. Coord. Chem. Rev. 2021, 428, 213606. [Google Scholar] [CrossRef]
- Sono, M.; Roach, M.P.; Coulter, E.D.; Dawson, J.H. Heme-containing oxygenases. Chem. Rev. 1996, 96, 2841–2888. [Google Scholar] [CrossRef]
- Meunier, B.; de Visser, S.P.; Shaik, S. Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schlichting, I. Structure and chemistry of cytochrome P450. Chem. Rev. 2005, 105, 2253–2277. [Google Scholar] [CrossRef] [PubMed]
- Ortiz de Montellano, P.R. (Ed.) Cytochrome P450: Structure, Mechanism and Biochemistry, 3rd ed.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2005. [Google Scholar]
- Kadish, K.M.; Smith, K.M.; Guilard, R. (Eds.) Handbook of Porphyrin Science; World Scientific Publishing Co.: Hackensack, NJ, USA, 2010. [Google Scholar]
- Grogan, G. Cytochromes P450: Exploiting diversity and enabling application as biocatalysts. Curr. Opin. Chem. Biol. 2011, 15, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Fasan, R. Tuning P450 enzymes as oxidation catalysts. ACS Catal. 2012, 2, 647–666. [Google Scholar] [CrossRef]
- Poulos, T.L. Heme enzyme structure and function. Chem. Rev. 2014, 114, 3919–3962. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Groves, J.T. Oxygen activation and radical transformations in heme proteins and metalloporphyrins. Chem. Rev. 2018, 118, 2491–2553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schofield, C.J.; Zhang, Z. Structural and mechanistic studies on 2-oxoglutarate-dependent oxygenases and related enzymes. Curr. Opin. Struc. Biol. 1999, 9, 722–731. [Google Scholar] [CrossRef]
- Solomon, E.I.; Brunold, T.C.; Davis, M.I.; Kemsley, J.N.; Lee, S.K.; Lehnert, N.; Neese, F.; Skulan, A.J.; Yang, Y.S.; Zhou, J. Geometric and electronic structure/function correlations in non-heme iron enzymes. Chem. Rev. 2000, 100, 235–349. [Google Scholar] [CrossRef] [PubMed]
- Abu-Omar, M.M.; Loaiza, A.; Hontzeas, N. Reaction mechanisms of mononuclear non-heme iron oxygenases. Chem. Rev. 2005, 105, 2227–2252. [Google Scholar] [CrossRef]
- Krebs, C.; Galonić Fujimori, D.; Walsh, C.T.; Bollinger Jr, J.M. Non-heme Fe(IV)–oxo intermediates. Acc. Chem. Res. 2007, 40, 484–492. [Google Scholar] [CrossRef] [Green Version]
- Kovaleva, E.G.; Lipscomb, J.D. Versatility of biological non-heme Fe(II) centers in oxygen activation reactions. Nat. Chem. Biol. 2008, 4, 186–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Visser, S.P.; Kumar, D. (Eds.) Iron-Containing Enzymes: Versatile Catalysts of Hydroxylation Reactions in Nature; Royal Society of Chemistry Publishing: Cambridge, UK, 2011. [Google Scholar]
- White, M.D.; Flashman, E. Catalytic strategies of the non-heme iron dependent oxygenases and their roles in plant biology. Curr. Opin. Chem. Biol. 2016, 31, 126–135. [Google Scholar] [CrossRef] [Green Version]
- De Visser, S.P. Mechanistic insight on the activity and substrate selectivity of nonheme iron dioxygenases. Chem. Rec. 2018, 18, 1501–1516. [Google Scholar] [CrossRef] [Green Version]
- Shoji, O.; Fujishiro, T.; Nakajima, H.; Kim, M.; Nagano, S.; Shiro, Y.; Watanabe, Y. Hydrogen peroxide dependent monooxygenations by tricking the substrate recognition of cytochrome P450BSβ. Angew. Chem. Int. Ed. 2007, 46, 3656–3659. [Google Scholar] [CrossRef]
- Rude, M.A.; Baron, T.S.; Brubaker, S.; Alibhai, M.; Del Cardayre, S.B.; Schirmer, A. Terminal olefin (1-alkene) biosynthesis by a novel P450 fatty acid decarboxylase from Jeotgalicoccus Species. Appl. Environ. Microbiol. 2011, 77, 1718–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Wang, C.; Yan, J.; Zhang, W.; Guan, W.; Lu, X.; Li, S. Hydrogen peroxide-independent production of α-alkenes by OleTJE P450 fatty acid decarboxylase. Biotechnol. Biofuels 2014, 7, 28–40. [Google Scholar] [CrossRef] [Green Version]
- Shoji, O.; Watanabe, Y. Peroxygenase reactions catalyzed by cytochromes P450. J. Biol. Inorg. Chem. 2014, 19, 529–539. [Google Scholar] [CrossRef]
- Dennig, A.; Kuhn, M.; Tassoti, S.; Thiessenhusen, A.; Gilch, S.; Bülter, T.; Haas, T.; Hall, M.; Faber, K. Oxidative decarboxylation of short-chain fatty acids to 1-alkenes. Angew. Chem. Int. Ed. 2015, 54, 8819–8822. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.L.; Hsieh, C.H.; Makris, T.M. Decarboxylation of fatty acids to terminal alkenes by cytochrome P450 Compound I. J. Am. Chem. Soc. 2015, 137, 4940–4943. [Google Scholar] [CrossRef]
- Grant, J.L.; Mitchell, M.E.; Makris, T.M. Catalytic strategy for carbon–carbon bond scission by the cytochrome P450 OleT. Proc. Natl. Acad. Sci. USA 2016, 113, 10049–10054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Lan, D.; Durrani, R.; Hollmann, F. Peroxygenases en route to becoming dream catalysts. What are the opportunities and challenges? Curr. Opin. Chem. Biol. 2017, 37, 1–9. [Google Scholar] [CrossRef]
- Munro, A.W.; McLean, K.J.; Grant, J.L.; Makris, T.M. Structure and function of the cytochrome P450 peroxygenase enzymes. Biochem. Soc. Trans. 2018, 46, 183–196. [Google Scholar] [CrossRef] [Green Version]
- Pickl, M.; Kurakin, S.; Cantú Reinhard, F.G.; Schmid, P.; Pöcheim, A.; Winkler, C.K.; Kroutil, W.; de Visser, S.P.; Faber, K. Mechanistic studies of fatty acid activation by CYP152 peroxygenases reveal unexpected desaturase activity. ACS Catal. 2019, 9, 565–577. [Google Scholar] [CrossRef] [Green Version]
- Bauer, D.; Zachos, I.; Sieber, V. Production of propene from n-butanol: A three-step cascade utilizing the cytochrome P450 fatty acid decarboxylase OleTJE. ChemBioChem 2020, 21, 3273–3281. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, A.S.; Ali, H.S.; Faponle, A.S.; de Visser, S.P. How external perturbations affect the chemoselectivity of substrate activation by cytochrome P450 OleTJE. Phys. Chem. Chem. Phys. 2020, 22, 27178–27190. [Google Scholar] [CrossRef]
- Derat, E.; Shaik, S. The Poulos-Kraut mechanism of Compound I formation in horseradish peroxidase: A QM/MM study. J. Phys. Chem. B 2006, 110, 10526–10533. [Google Scholar] [CrossRef]
- Cho, K.-B.; Derat, E.; Shaik, S. Compound I of nitric oxide synthase: The active site protonation state. J. Am. Chem. Soc. 2007, 129, 3182–3188. [Google Scholar] [CrossRef] [PubMed]
- de Visser, S.P.; Tan, L.S. Is the bound substrate in nitric oxide synthase protonated or neutral and what is the active oxidant that performs substrate hydroxylation? J. Am. Chem. Soc. 2008, 130, 12961–12974. [Google Scholar] [CrossRef]
- Vidossich, P.; Fiorin, G.; Alfonso-Prieto, M.; Derat, E.; Shaik, S.; Rovira, C. On the role of water in peroxidase catalysis: A theoretical investigation of HRP Compound I formation. J. Phys. Chem. B 2010, 114, 5161–5169. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-X.; Postils, V.; Sun, W.; Faponle, A.S.; Solà, M.; Wang, Y.; Nam, W.; de Visser, S.P. Reactivity patterns of (protonated) Compound II and Compound I of Cytochrome P450: Which is the better oxidant? Chem. Eur. J. 2017, 23, 6406–6418. [Google Scholar] [CrossRef]
- Faponle, A.S.; Seebeck, F.P.; de Visser, S.P. Sulfoxide synthase versus cysteine dioxygenase reactivity in a nonheme iron enzyme. J. Am. Chem. Soc. 2017, 139, 9259–9270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmins, A.; Saint-André, M.; de Visser, S.P. Understanding how prolyl-4-hydroxylase structure steers a ferryl oxidant toward scission of a strong C–H bond. J. Am. Chem. Soc. 2017, 139, 9855–9866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.-T.; Ali, H.S.; de Visser, S.P. Electrostatic perturbations from the protein affect C–H bond strengths of the substrate and enable negative catalysis in the TmpA biosynthesis enzyme. Chem. Eur. J. 2021, 27, 8851–8864. [Google Scholar] [CrossRef] [PubMed]
- Belcher, J.; McLean, K.J.; Matthews, S.; Woodward, L.S.; Fisher, K.; Rigby, S.E.J.; Nelson, D.R.; Potts, D.; Baynham, M.T.; Parker, D.A.; et al. Structure and biochemical properties of the alkene producing cytochrome P450 OleTJE (CYP152L1) from the Jeotgalicoccus sp. 8456 bacterium. J. Biol. Chem. 2014, 289, 6535–6550. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein databank. Nucl. Acids Res. 2000, 28, 235–243. [Google Scholar] [CrossRef] [Green Version]
- Quesne, M.G.; Borowski, T.; de Visser, S.P. Quantum mechanics/molecular mechanics modelling of enzymatic processes: Caveats and breakthroughs. Chem. Eur. J. 2016, 22, 2562–2581. [Google Scholar] [CrossRef]
- Mubarak, M.Q.E.; Gérard, E.F.; Blanford, C.F.; Hay, S.; de Visser, S.P. How do vanadium chloroperoxidases generate hypochlorite from hydrogen peroxide and chloride? A computational study. ACS Catal. 2020, 10, 14067–14079. [Google Scholar] [CrossRef]
- Ji, L.; Faponle, A.S.; Quesne, M.G.; Sainna, M.A.; Zhang, J.; Franke, A.; Kumar, D.; van Eldik, R.; Liu, W.; de Visser, S.P. Drug metabolism by cytochrome P450 enzymes: What distinguishes the pathways leading to substrate hydroxylation over desaturation? Chem. Eur. J. 2015, 21, 9083–9092. [Google Scholar] [CrossRef]
- Faponle, A.S.; Quesne, M.G.; de Visser, S.P. Origin of the regioselective fatty acid hydroxylation versus decarboxylation by a cytochrome P450 peroxygenase: What drives the reaction to biofuel production? Chem. Eur. J. 2016, 22, 5478–5483. [Google Scholar] [CrossRef]
- Cantú Reinhard, F.G.; Lin, Y.-T.; Stańczak, A.; de Visser, S.P. Bioengineering of cytochrome P450 OleTJE: How does substrate positioning affect the product distributions? Molecules 2020, 25, 2675–2697. [Google Scholar] [CrossRef]
- Green, M.T. Evidence for sulfur-based radicals in thiolate compound I intermediates. J. Am. Chem. Soc. 1999, 121, 7939–7940. [Google Scholar] [CrossRef]
- Ogliaro, F.; Cohen, S.; de Visser, S.P.; Shaik, S. Medium polarization and hydrogen bonding effects on Compound I of cytochrome P450: What kind of a radical is it really? J. Am. Chem. Soc. 2000, 122, 12892–12893. [Google Scholar] [CrossRef]
- Ogliaro, F.; de Visser, S.P.; Cohen, S.; Kaneti, J.; Shaik, S. The experimentally elusive oxidant of cytochrome P450: A theoretical “trapping” defining more closely the “real” species. ChemBioChem 2001, 2, 848–851. [Google Scholar] [CrossRef]
- de Visser, S.P.; Shaik, S.; Sharma, P.K.; Kumar, D.; Thiel, W. Active species of horseradish peroxidase (HRP) and cytochrome P450: Two electronic chameleons. J. Am. Chem. Soc. 2003, 125, 15779–15788. [Google Scholar] [CrossRef] [PubMed]
- Hersleth, H.-P.; Ryde, U.; Rydberg, P.; Görbitz, C.H.; Andersson, K.K. Structures of the high-valent metal-ion haem–oxygen intermediates in peroxidases, oxygenases and catalases. J. Inorg. Biochem. 2006, 100, 460–476. [Google Scholar] [CrossRef] [PubMed]
- Groenhof, A.R.; Ehlers, A.W.; Lammertsma, K. Proton assisted oxygen-oxygen bond splitting in cytochrome P450. J. Am. Chem. Soc. 2007, 129, 6204–6209. [Google Scholar] [CrossRef]
- Bathelt, C.M.; Zurek, J.; Mulholland, A.J.; Harvey, J.N. Electronic structure of compound I in human isoforms of cytochrome P450 from QM/MM modeling. J. Am. Chem. Soc. 2005, 127, 12900–12908. [Google Scholar] [CrossRef]
- Shaik, S.; Kumar, D.; de Visser, S.P.; Altun, A.; Thiel, W. Theoretical perspective on the structure and mechanism of cytochrome P450 enzymes. Chem. Rev. 2005, 105, 2279–2328. [Google Scholar] [CrossRef]
- Rydberg, P.; Sigfridsson, E.; Ryde, U. On the role of the axial ligand in heme proteins: A theoretical study. J. Biol. Inorg. Chem. 2004, 9, 203–223. [Google Scholar] [CrossRef]
- Harvey, J.N.; Bathelt, C.M.; Mulholland, A.J. QM/MM modeling of compound I active species in cytochrome P450, cytochrome c peroxidase, and ascorbate peroxidase. J. Comput. Chem. 2006, 27, 1352–1362. [Google Scholar] [CrossRef]
- Li, D.; Wang, Y.; Han, K.; Zhan, C.-G. Fundamental reaction pathways for cytochrome P450-catalyzed 5′-hydroxylation and N-demethylation of nicotine. J. Phys. Chem. B 2010, 114, 9023–9030. [Google Scholar] [CrossRef] [Green Version]
- Hirao, H.; Chuanprasit, P.; Cheong, Y.Y.; Wang, X. How is a metabolic intermediate formed in the mechanism-based inactivation of cytochrome P450 by using 1,1-dimethylhydrazine: Hydrogen abstraction or nitrogen oxidation? Chem. Eur. J. 2013, 19, 7361–7369. [Google Scholar] [CrossRef]
- Hirao, H.; Cheong, Z.C.; Wang, X. Pivotal role of water in terminating enzymatic function: A density functional theory study of the mechanism-based inactivation of cytochromes P450. J. Phys. Chem. B 2012, 116, 7787–7794. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.; Li, H. Hydrogen abstraction of camphor catalyzed by cytochrome P450cam: A QM/MM study. J. Phys. Chem. B 2016, 120, 12312–12320. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shi, J.; Liu, Y. Oxidative rearrangement mechanism of pentalenolactone F catalyzed by cytochrome P450 CYP161C2 (PntM). Inorg. Chem. 2018, 57, 8933–8941. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Y. Mechanical insights into the enzymatic cleavage of double C–C bond in poly(cis-1,4-isoprene) by the latex clearing protein. Inorg. Chem. 2020, 59, 9627–9637. [Google Scholar] [CrossRef]
- Phung, Q.M.; Pierloot, K. Low-lying electromeric states in chloro-ligated iron(IV)-oxo porphyrin as a model for compound I, studied with second-order perturbation theory based on density matrix renormalization group. J. Chem. Theory Comput. 2019, 15, 3033–3043. [Google Scholar] [CrossRef] [PubMed]
- Altarsha, M.; Benighaus, T.; Kumar, D.; Thiel, W. How is the reactivity of cytochrome P450cam affected by Thr252X mutation? A QM/MM study for X = serine, valine, alanine, glycine. J. Am. Chem. Soc. 2009, 131, 4755–4763. [Google Scholar] [CrossRef]
- Li, D.; Wang, Y.; Han, K. Recent density functional theory model calculations of drug metabolism by cytochrome P450. Coord. Chem. Rev. 2012, 256, 1137–1150. [Google Scholar] [CrossRef]
- Sainna, M.A.; Kumar, S.; Kumar, D.; Fornarini, S.; Crestoni, M.E.; de Visser, S.P. A comprehensive test set of epoxidation rate constants by iron(IV)-oxo porphyrin complexes. Chem. Sci. 2015, 6, 1516–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kepp, K.P. Heme isomers substantially affect heme’s electronic structure and function. Phys. Chem. Chem. Phys. 2017, 19, 22355–22362. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.; de Visser, S.P.; Shaik, S. Multistate reactivity in styrene epoxidation by Compound I of cytochrome P450: Mechanisms of products and side products formation. Chem. Eur. J. 2005, 11, 2825–2835. [Google Scholar] [CrossRef]
- Kumar, D.; Karamzadeh, B.; Sastry, G.N.; de Visser, S.P. What factors influence the rate constant of substrate epoxidation by Compound I of cytochrome P450 and analogous iron(IV)-oxo oxidants. J. Am. Chem. Soc. 2010, 132, 7656–7667. [Google Scholar] [CrossRef]
- Quesne, M.G.; Senthilnathan, D.; Singh, D.; Kumar, D.; Maldivi, P.; Sorokin, A.B.; de Visser, S.P. Origin of the enhanced reactivity of μ-nitrido-bridged diiron(IV)-oxo porphyrinoid complexes over cytochrome P450 Compound I. ACS Catal. 2016, 6, 2230–2243. [Google Scholar] [CrossRef] [Green Version]
- Colomban, C.; Tobing, A.H.; Mukherjee, G.; Sastri, C.V.; Sorokin, A.B.; de Visser, S.P. Mechanism of oxidative activation of fluorinated aromatic compounds by N-bridged diiron-phthalocyanine. What determines the reactivity? Chem. Eur. J. 2019, 25, 14320–14331. [Google Scholar] [CrossRef]
- Shaik, S.; Cohen, S.; de Visser, S.P.; Sharma, P.K.; Kumar, D.; Kozuch, S.; Ogliaro, F.; Danovich, D. The “rebound controversy”: An overview and theoretical modeling of the rebound step in C–H hydroxylation by cytochrome P450. Eur. J. Inorg. Chem. 2004, 207–226. [Google Scholar] [CrossRef]
- Ali, H.S.; Henchman, R.H.; de Visser, S.P. Lignin biodegradation by a cytochrome P450 enzyme: A computational study into syringol activation by GcoA. Chem. Eur. J. 2020, 26, 13093–13102. [Google Scholar] [CrossRef]
- de Visser, S.P.; Ogliaro, F.; Shaik, S. How does ethene inactivate cytochrome P450 en route to its epoxidation? A density functional study. Angew. Chem. Int. Ed. 2001, 40, 2871–2874. [Google Scholar] [CrossRef]
- Kumar, D.; de Visser, S.P.; Shaik, S. How does product isotope effect prove the operation of a two-state “rebound” mechanism in C–H hydroxylation by cytochrome P450? J. Am. Chem. Soc. 2003, 125, 13024–13025. [Google Scholar] [CrossRef]
- Kumar, D.; de Visser, S.P.; Sharma, P.K.; Cohen, S.; Shaik, S. Radical clock substrates, their C–H hydroxylation mechanism by cytochrome P450 and other reactivity patterns: What does theory reveal about the clocks’ behavior? J. Am. Chem. Soc. 2004, 126, 1907–1920. [Google Scholar] [CrossRef]
- Kumar, D.; de Visser, S.P.; Shaik, S. Oxygen economy of cytochrome P450: What is the origin of the mixed functionality as a dehydrogenase–oxidase enzyme compared with its normal function? J. Am. Chem. Soc. 2004, 126, 5072–5073. [Google Scholar] [CrossRef]
- Rétey, J. Enzymic reaction selectivity by negative catalysis or how do enzymes deal with highly reactive intermediates? Angew. Chem. Int. Ed. 1990, 29, 355–361. [Google Scholar] [CrossRef]
- Vögeli, B.; Erb, T.J. ‘Negative’ and ‘positive catalysis’: Complementary principles that shape the catalytic landscape of enzymes. Curr. Opin. Chem. Biol. 2018, 47, 94–100. [Google Scholar] [CrossRef] [PubMed]
- de Visser, S.P.; Lin, Y.-T.; Ali, H.S.; Bagh, U.K.; Mukherjee, G.; Sastri, C.V. Negative catalysis or non-Bell-Evans-Polanyi reactivity by metalloenzymes: Examples from mononuclear heme and non-heme iron oxygenases. Coord. Chem. Rev. 2021, 439, 213914. [Google Scholar] [CrossRef]
- Pangia, T.M.; Yadav, V.; Gérard, E.F.; Lin, Y.-T.; de Visser, S.P.; Jameson, G.N.L.; Goldberg, D.P. Mechanistic investigation of oxygen rebound in a mononuclear nonheme iron complex. Inorg. Chem. 2019, 58, 9557–9561. [Google Scholar] [CrossRef] [PubMed]
- Cantú Reinhard, F.G.; de Visser, S.P. Oxygen atom transfer using an iron(IV)-oxo embedded in a tetracyclic N-heterocyclic carbene system: How does the reactivity compare to Cytochrome P450 Compound I? Chem. Eur. J. 2017, 23, 2935–2944. [Google Scholar] [CrossRef] [Green Version]
- Cummins, D.C.; Alvarado, J.G.; Zaragoza, J.P.T.; Mubarak, M.Q.E.; Lin, Y.-T.; de Visser, S.P.; Goldberg, D.P. Hydroxyl transfer to carbon radicals by Mn(OH) versus Fe(OH) corrole complexes. Inorg. Chem. 2020, 59, 16053–16064. [Google Scholar] [CrossRef]
- Codola, Z.; Gamba, I.; Acuña-Pares, F.; Casadevall, C.; Clemancey, M.; Latour, J.-M.; Luis, J.M.; Lloret-Fillol, J.; Costas, M. Design of iron coordination complexes as highly active homogenous water oxidation catalysts by deuteration of oxidation-sensitive sites. J. Am. Chem. Soc. 2019, 141, 323–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Visser, S.P.; Kumar, D.; Cohen, S.; Shacham, R.; Shaik, S. A predictive pattern of computed barriers for C–H hydroxylation by Compound I of cytochrome P450. J. Am. Chem. Soc. 2004, 126, 8362–8363. [Google Scholar] [CrossRef]
- Shaik, S.; Kumar, D.; de Visser, S.P. A valence bond modeling of trends in hydrogen abstraction barriers and transition states of hydroxylation reactions catalyzed by cytochrome P450 enzymes. J. Am. Chem. Soc. 2008, 130, 10128–10140. [Google Scholar] [CrossRef] [PubMed]
- Latifi, R.; Bagherzadeh, M.; de Visser, S.P. Origin of the correlation of the rate constant of substrate hydroxylation by nonheme iron(IV)-oxo complexes with the bond-dissociation energy of the C–H bond of the substrate. Chem. Eur. J. 2009, 15, 6651–6662. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–272. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—consistent molecular orbital methods. XII. Further extensions of Gaussian—Type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Kumar, D.; Sastry, G.N.; de Visser, S.P. Effect of the axial ligand on substrate sulfoxidation mediated by iron(IV)-oxo porphyrin cation radical oxidants. Chem. Eur. J. 2011, 17, 6196–6205. [Google Scholar] [CrossRef]
- Yang, T.; Quesne, M.G.; Neu, H.M.; Cantú Reinhard, F.G.; Goldberg, D.P.; de Visser, S.P. Singlet versus triplet reactivity in an Mn(V)-Oxo species: Testing theoretical predictions against experimental evidence. J. Am. Chem. Soc. 2016, 138, 12375–12386. [Google Scholar] [CrossRef] [Green Version]
- Cantú Reinhard, F.G.; Faponle, A.S.; de Visser, S.P. Substrate sulfoxidation by an iron(IV)-oxo complex: Benchmarking computationally calculated barrier heights to experiment. J. Phys. Chem. A 2016, 120, 9805–9814. [Google Scholar] [CrossRef] [PubMed]
- Cantú Reinhard, F.G.; Sainna, M.A.; Upadhyay, P.; Balan, G.A.; Kumar, D.; Fornarini, S.; Crestoni, M.E.; de Visser, S.P. A systematic account on aromatic hydroxylation by a cytochrome P450 model Compound I: A low-pressure mass spectrometry and computational study. Chem. Eur. J. 2016, 22, 18608–18619. [Google Scholar] [CrossRef] [PubMed]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, Y.-T.; de Visser, S.P. Product Distributions of Cytochrome P450 OleTJE with Phenyl-Substituted Fatty Acids: A Computational Study. Int. J. Mol. Sci. 2021, 22, 7172. https://doi.org/10.3390/ijms22137172
Lin Y-T, de Visser SP. Product Distributions of Cytochrome P450 OleTJE with Phenyl-Substituted Fatty Acids: A Computational Study. International Journal of Molecular Sciences. 2021; 22(13):7172. https://doi.org/10.3390/ijms22137172
Chicago/Turabian StyleLin, Yen-Ting, and Sam P. de Visser. 2021. "Product Distributions of Cytochrome P450 OleTJE with Phenyl-Substituted Fatty Acids: A Computational Study" International Journal of Molecular Sciences 22, no. 13: 7172. https://doi.org/10.3390/ijms22137172
APA StyleLin, Y. -T., & de Visser, S. P. (2021). Product Distributions of Cytochrome P450 OleTJE with Phenyl-Substituted Fatty Acids: A Computational Study. International Journal of Molecular Sciences, 22(13), 7172. https://doi.org/10.3390/ijms22137172