
Hops/Tmub1 Heterozygous Mouse Shows Haploinsufficiency Effect in Influencing p53-Mediated Apoptosis

 , ,
, ,  ,
,  and
and 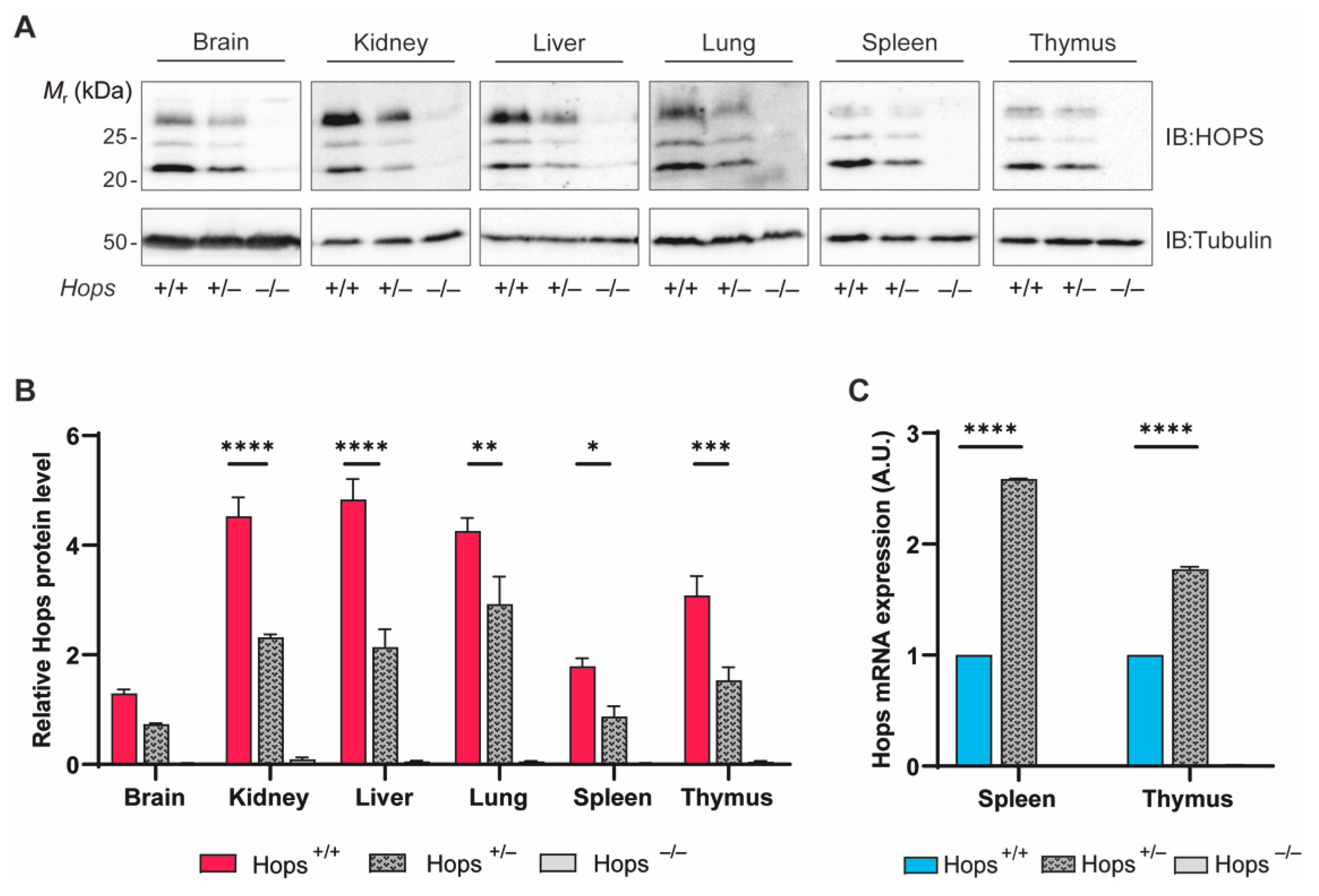{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. HOPS Expression and Characterization in Heterozygous Mice
2.2. Hops Heterozygous Mice Were Defective in DNA-Damage-Induced Repair
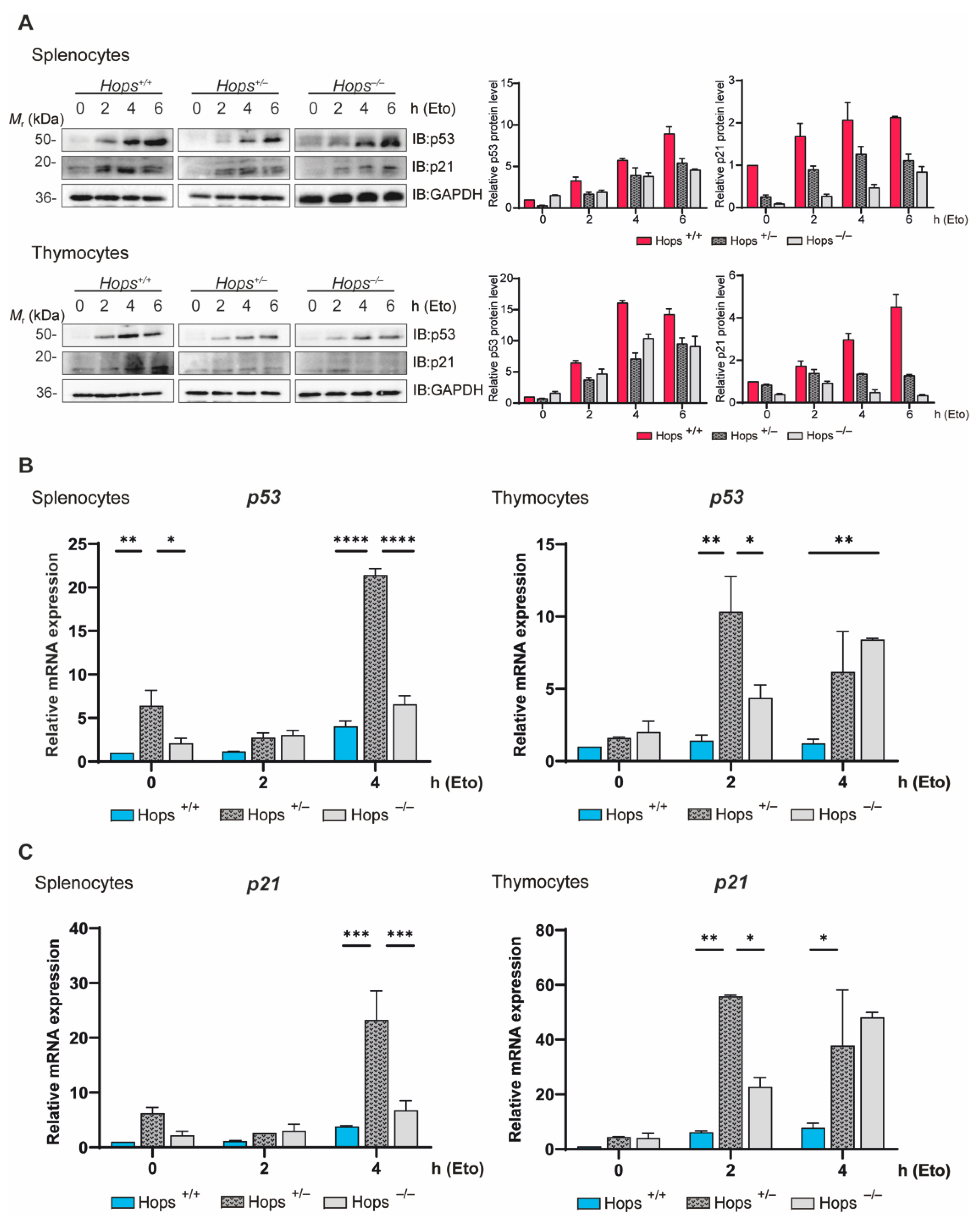
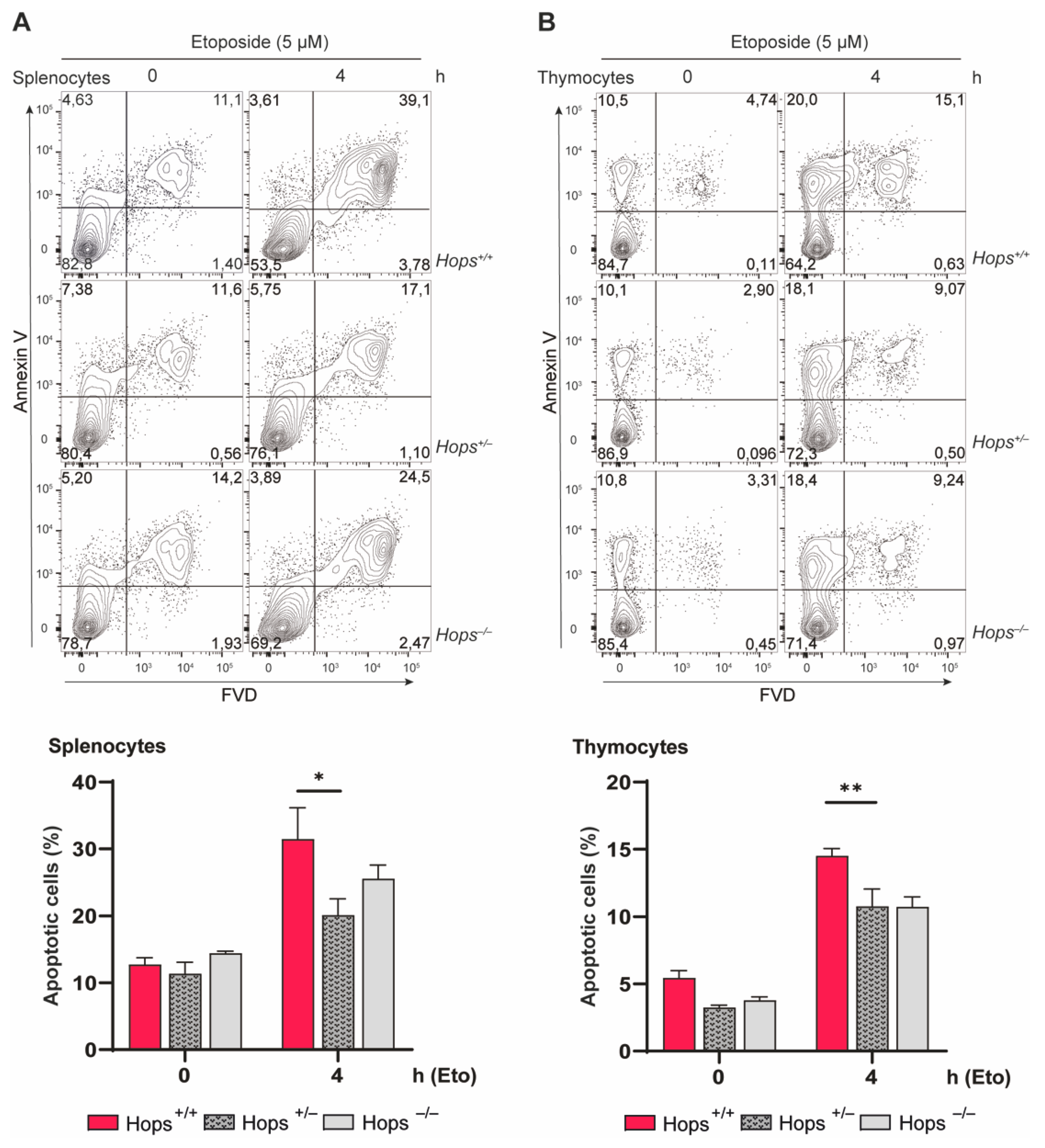
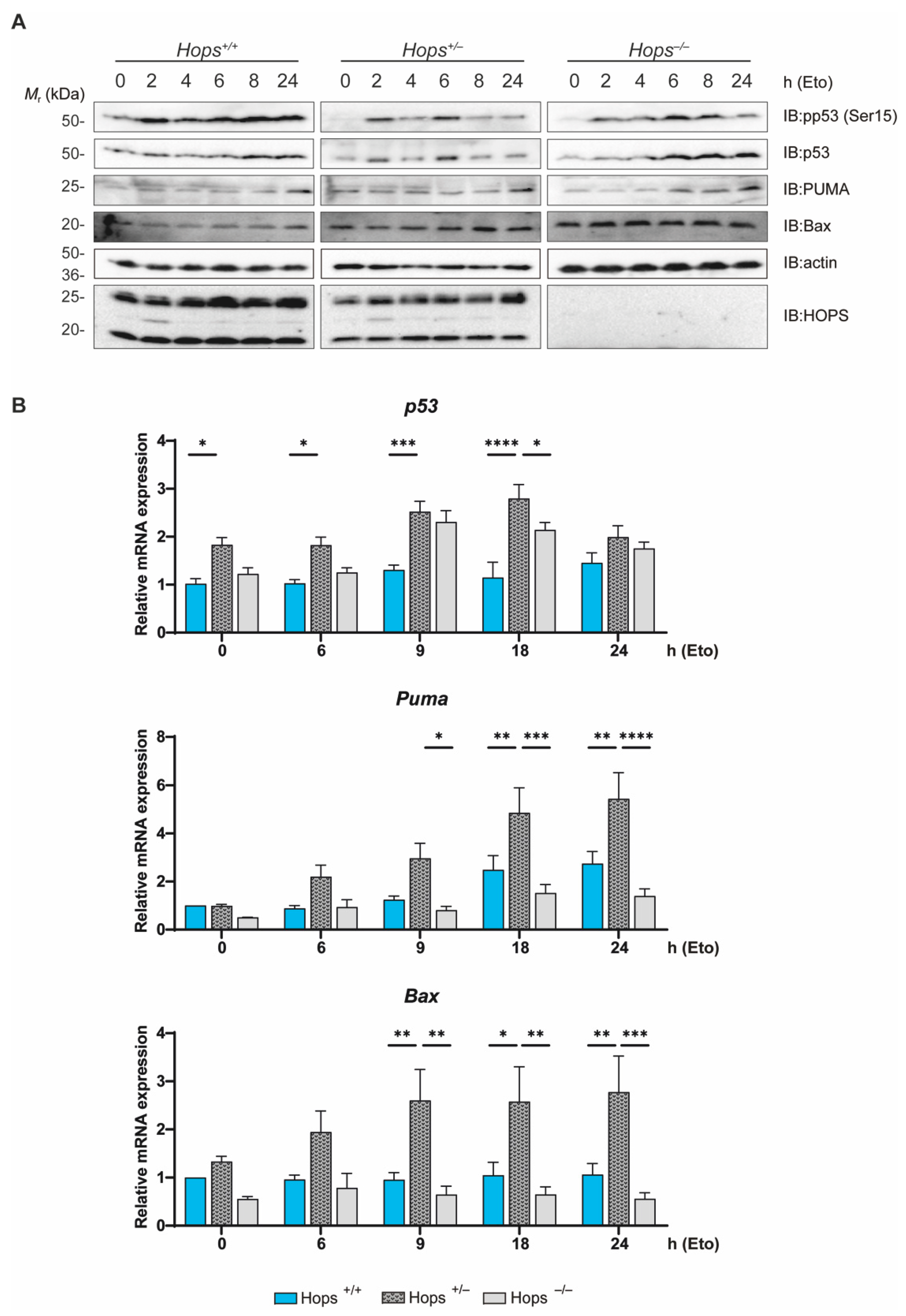
2.3. Hops Heterozygous Mice Showed Defective Apoptosis Induction
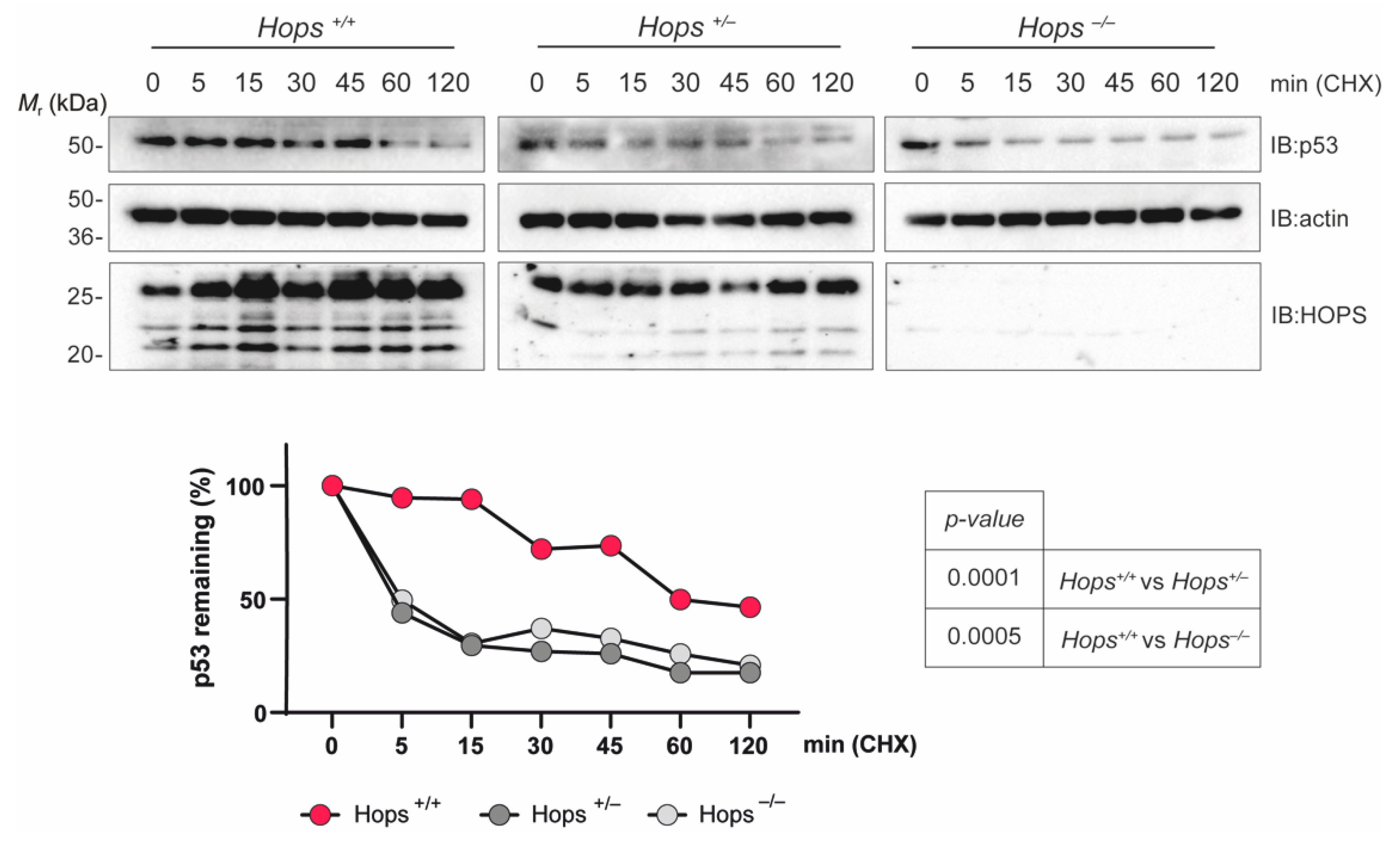
2.4. The p53 Protein Showed a Shortened Half-Life in Hops Heterozygous MEFs
2.5. Hops Heterozygous MEFs Exhibited Altered p53 Activation Following DNA Damage
3. Discussion
4. Materials and Methods
4.1. Mice, Models, and Treatments
4.2. Establishment of Immortalized MEFs
4.3. RNA Extraction and Real-Time PCR
4.4. Western Blot Analysis and Antibodies
4.5. Flow Cytometry Analysis
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Castelli, M.; Piobbico, D.; Bartoli, D.; Pieroni, S.; Brunacci, C.; Bellet, M.M.; Chiacchiaretta, M.; Della Fazia, M.A.; Servillo, G. Different Functions of HOPS Isoforms in the Cell: HOPS Shuttling Isoform Is Determined by RIP Cleavage System. Cell Cycle 2013, 13, 293–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Della Fazia, M.A.; Castelli, M.; Bartoli, D.; Pieroni, S.; Pettirossi, V.; Piobbico, D.; Viola-Magni, M.; Servillo, G. HOPS: A Novel CAMP-Dependent Shutting Protein Involved in Protein Synthesis Regulation. J. Cell Sci. 2005, 118, 3185–3194. [Google Scholar] [CrossRef] [Green Version]
- Castelli, M.; Pieroni, S.; Brunacci, C.; Piobbico, D.; Bartoli, D.; Bellet, M.M.; Colombo, E.; Pelicci, P.G.; Della Fazia, M.A.; Servillo, G. Hepatocyte Odd Protein Shuttling (HOPS) Is a Bridging Protein in the Nucleophosmin-P19Arf Network. Oncogene 2013, 32, 3350–3358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pieroni, S.; Della Fazia, M.A.; Castelli, M.; Piobbico, D.; Bartoli, D.; Brunacci, C.; Bellet, M.M.; Viola-Magni, M.; Servillo, G. HOPS Is an Essential Constituent of Centrosome Assembly. Cell Cycle 2008, 7, 1462–1466. [Google Scholar] [CrossRef] [PubMed]
- Della Fazia, M.A.; Servillo, G.; Sassone-Corsi, P. Cyclic AMP Signalling and Cellular Proliferation: Regulation of CREB and CREM. FEBS Lett. 1997, 410, 22–24. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Liu, H.; Wang, X.; Chen, P.; Chen, H. IL-6 Induction of Hepatocyte Proliferation through the Tmub1-Regulated Gene Pathway. Int. J. Mol. Med. 2012, 29, 1106–1112. [Google Scholar] [CrossRef]
- Liu, M.; Yuan, T.; Liu, H.; Chen, P. CCAAT/Enhancer-Binding Protein β Regulates Interleukin-6-Induced Transmembrane and Ubiquitin-like Domain Containing 1 Gene Expression in Hepatocytes. Mol. Med. Rep. 2014, 10, 2177–2183. [Google Scholar] [CrossRef]
- Fu, H.; Dong, R.; Zhang, Y.; Xu, J.; Liu, M.; Chen, P. Tmub1 Negatively Regulates Liver Regeneration via Inhibiting STAT3 Phosphorylation. Cell. Signal. 2019, 55, 65–72. [Google Scholar] [CrossRef]
- Chen, Y.; Fu, H.; Zhang, Y.; Chen, P. Transmembrane and Ubiquitin-Like Domain Containing 1 Protein (TMUB1) Negatively Regulates Hepatocellular Carcinoma Proliferation via Regulating Signal Transducer and Activator of Transcription 1 (STAT1). Med. Sci. Monit. 2019, 25, 9471–9482. [Google Scholar] [CrossRef]
- Fu, H.; Zhang, Y.; Chen, J.; Zhou, B.; Chen, G.; Chen, P. Tmub1 Suppresses Hepatocellular Carcinoma by Promoting the Ubiquitination of ΔNp63 Isoforms. Mol. Ther. Oncolytics 2020, 18, 126–136. [Google Scholar] [CrossRef]
- Castelli, M.; Piobbico, D.; Chiacchiaretta, M.; Brunacci, C.; Pieroni, S.; Bartoli, D.; Gargaro, M.; Fallarino, F.; Puccetti, P.; Soddu, S.; et al. HOPS/TMUB1 Retains P53 in the Cytoplasm and Sustains P53-dependent Mitochondrial Apoptosis. EMBO Rep. 2019, 21, e48073. [Google Scholar] [CrossRef]
- Bellet, M.M.; Pieroni, S.; Castelli, M.; Piobbico, D.; Fallarino, F.; Romani, L.; Della-Fazia, M.A.; Servillo, G. HOPS/Tmub1 Involvement in the NF-KB-Mediated Inflammatory Response through the Modulation of TRAF6. Cell Death Dis. 2020, 11, 865. [Google Scholar] [CrossRef]
- Levine, A.J.; Momand, J.; Finlay, C.A. The P53 Tumour Suppressor Gene. Nature 1991, 351, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Brady, C.A.; Attardi, L.D. P53 at a Glance. J. Cell Sci. 2010, 123, 2527–2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of P53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oren, M. Decision Making by P53: Life, Death and Cancer. Cell Death Differ. 2003, 10, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.L.; Gu, W. New Insights into P53 Activation. Cell Res. 2010, 20, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D. P53—Master and Commander. N. Engl. J. Med. 2007, 357, 2539–2541. [Google Scholar] [CrossRef]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. P53 Mutations in Human Cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meek, D.W. Tumour Suppression by P53: A Role for the DNA Damage Response? Nat. Rev. Cancer 2009, 9, 714–723. [Google Scholar] [CrossRef]
- Delia, D.; Mizutani, S.; Panigone, S.; Tagliabue, E.; Fontanella, E.; Asada, M.; Yamada, T.; Taya, Y.; Prudente, S.; Saviozzi, S.; et al. ATM Protein and P53-Serine 15 Phosphorylation in Ataxia-Telangiectasia (AT) Patients and at Heterozygotes. Br. J. Cancer 2000, 82, 1938–1945. [Google Scholar] [CrossRef]
- Lakin, N.D.; Jackson, S.P. Regulation of P53 in Response to DNA Damage. Oncogene 1999, 18, 7644–7655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Sheikh, M.S.; Huang, Y. Decision Making by P53: Life versus Death. Mol. Cell. Pharmacol. 2010, 2, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, T.; Krajewski, S.; Krajewska, M.; Wang, H.G.; Lin, H.K.; Liebermann, D.A.; Hoffman, B.; Reed, J.C. Tumor Suppressor P53 Is a Regulator of Bcl-2 and Bax Gene Expression in vitro and in vivo. Oncogene 1994, 9, 1799–1805. [Google Scholar] [PubMed]
- Zhan, Q.; Bieszczad, C.K.; Bae, I.; Fornace, A.J.; Craig, R.W. Induction of BCL2 Family Member MCL1 as an Early Response to DNA Damage. Oncogene 1997, 14, 1031–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional Control of Human P53-Regulated Genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How Does P53 Induce Apoptosis and How Does This Relate to P53-Mediated Tumour Suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Norbury, C.J.; Hickson, I.D. Cellular Responses to DNA Damage. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 367–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, C.A.; Fridman, J.S.; Yang, M.; Baranov, E.; Hoffman, R.M.; Lowe, S.W. Dissecting P53 Tumor Suppressor Functions in Vivo. Cancer Cell 2002, 1, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Godefroy, N.; Lemaire, C.; Renaud, F.; Rincheval, V.; Perez, S.; Parvu-Ferecatu, I.; Mignotte, B.; Vayssière, J.L. P53 Can Promote Mitochondria- and Caspase-Independent Apoptosis. Cell Death Differ. 2004, 11, 785–787. [Google Scholar] [CrossRef]
- Carr, A.M. Piecing Together the P53 Puzzle. Science 2000, 287, 1765–1766. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lu, X. Live or Let Die: The Cell’s Response to P53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the P53 Network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Osborne, B.A. Apoptosis and the Maintenance of Homoeostasis in the Immune System. Curr. Opin. Immunol. 1996, 8, 245–254. [Google Scholar] [CrossRef]
- El-Deiry, W.S.; Harper, J.W.; Connor, P.M.; Velculescu, V.E.; Canman, C.E.; Jackman, J.; Pietenpol, J.A.; Burrell, M.; Hill, D.E.; Wang, Y.; et al. WAF1/CIP1 Is Induced in P53-Mediated G1 Arrest and Apoptosis. Cancer Res. 1994, 54, 1169–1174. [Google Scholar] [PubMed]
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a Potential Mediator of P53 Tumor Suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Bunz, F.; Dutriaux, A.; Lengauer, C.; Waldman, T.; Zhou, S.; Brown, J.P.; Sedivy, J.M.; Kinzler, K.W.; Vogelstein, B. Requirement for P53 and P21 to Sustain G2 Arrest after DNA Damage. Science 1998, 282, 1497–1501. [Google Scholar] [CrossRef]
- Clarke, A.R.; Purdie, C.A.; Harrison, D.J.; Morris, R.G.; Bird, C.C.; Hooper, M.L.; Wyllie, A.H. Thymocyte Apoptosis Induced by P53-Dependent and Independent Pathways. Nature 1993, 362, 849–852. [Google Scholar] [CrossRef]
- Lowe, S.W.; Schmitt, E.M.; Smith, S.W.; Osborne, B.A.; Jacks, T. P53 Is Required for Radiation-Induced Apoptosis in Mouse Thymocytes. Nature 1993, 362, 847–849. [Google Scholar] [CrossRef]
- Toledo, F.; Wahl, G.M. Regulating the P53 Pathway: In Vitro Hypotheses, in Vivo Veritas. Nat. Rev. Cancer 2006, 6, 909–923. [Google Scholar] [CrossRef]
- Vousden, K.H.; Lane, D.P. P53 in Health and Disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Loughery, J.; Cox, M.; Smith, L.M.; Meek, D.W. Critical Role for P53-Serine 15 Phosphorylation in Stimulating Transactivation at P53-Responsive Promoters. Nucleic Acids Res. 2014, 42, 7666–7680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Traganos, F.; Darzynkiewicz, Z. Phosphorylation of P53 on Ser15 during Cell Cycle Caused by Topo I and Topo II Inhibitors in Relation to ATM and Chk2 Activation. Cell Cycle 2008, 7, 3048–3055. [Google Scholar] [CrossRef] [Green Version]
- Saito, S.; Yamaguchi, H.; Higashimoto, Y.; Chao, C.; Xu, Y.; Fornace, A.J.; Appella, E.; Anderson, C.W. Phosphorylation Site Interdependence of Human P53 Post-Translational Modifications in Response to Stress. J. Biol. Chem. 2003, 278, 37536–37544. [Google Scholar] [CrossRef] [Green Version]
- Dumaz, N.; Meek, D.W. Serine 15 Phosphorylation Stimulates P53 Transactivation but Does Not Directly Influence Interaction with HDM2. EMBO J. 1999, 18, 7002–7010. [Google Scholar] [CrossRef] [Green Version]
- Kannan, K.; Amariglio, N.; Rechavi, G.; Jakob-Hirsch, J.; Kela, I.; Kaminski, N.; Getz, G.; Domany, E.; Givol, D. DNA Microarrays Identification of Primary and Secondary Target Genes Regulated by P53. Oncogene 2001, 20, 2225–2234. [Google Scholar] [CrossRef] [Green Version]
- Kruse, J.P.; Gu, W. Modes of P53 Regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef] [Green Version]
- Espinosa, J.M. Mechanisms of Regulatory Diversity within the P53 Transcriptional Network. Oncogene 2008, 27, 4013–4023. [Google Scholar] [CrossRef] [Green Version]
- Laptenko, O.; Prives, C. Transcriptional Regulation by P53: One Protein, Many Possibilities. Cell Death Differ. 2006, 13, 951–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prives, C.; Hall, P.A. The P53 Pathway. J. Pathol. 1999, 187, 112–126. [Google Scholar] [CrossRef]
- Ko, L.J.; Prives, C. P53: Puzzle and Paradigm. Genes Dev. 1996, 10, 1054–1072. [Google Scholar] [CrossRef] [Green Version]
- Muller, P.A.J.; Vousden, K.H. P53 Mutations in Cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Della-Fazia, M.A.; Castelli, M.; Piobbico, D.; Pieroni, S.; Servillo, G. The Ins and Outs of HOPS/TMUB1 in Biology and Pathology. FEBS J. 2021, 288, 2773–2783. [Google Scholar] [CrossRef]
- Della-Fazia, M.A.; Castelli, M.; Piobbico, D.; Pieroni, S.; Servillo, G. HOPS and P53: Thick as Thieves in Life and Death. Cell Cycle 2020, 19, 2996–3003. [Google Scholar] [CrossRef] [PubMed]
- Xu, J. Preparation, Culture, and Immortalization of Mouse Embryonic Fibroblasts. Curr. Protoc. Mol. Biol. 2005, 28, 28.1.1–28.1.8. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferracchiato, S.; Di-Iacovo, N.; Scopetti, D.; Piobbico, D.; Castelli, M.; Pieroni, S.; Gargaro, M.; Manni, G.; Brancorsini, S.; Della-Fazia, M.A.; et al. Hops/Tmub1 Heterozygous Mouse Shows Haploinsufficiency Effect in Influencing p53-Mediated Apoptosis. Int. J. Mol. Sci. 2021, 22, 7186. https://doi.org/10.3390/ijms22137186
Ferracchiato S, Di-Iacovo N, Scopetti D, Piobbico D, Castelli M, Pieroni S, Gargaro M, Manni G, Brancorsini S, Della-Fazia MA, et al. Hops/Tmub1 Heterozygous Mouse Shows Haploinsufficiency Effect in Influencing p53-Mediated Apoptosis. International Journal of Molecular Sciences. 2021; 22(13):7186. https://doi.org/10.3390/ijms22137186
Chicago/Turabian StyleFerracchiato, Simona, Nicola Di-Iacovo, Damiano Scopetti, Danilo Piobbico, Marilena Castelli, Stefania Pieroni, Marco Gargaro, Giorgia Manni, Stefano Brancorsini, Maria Agnese Della-Fazia, and et al. 2021. "Hops/Tmub1 Heterozygous Mouse Shows Haploinsufficiency Effect in Influencing p53-Mediated Apoptosis" International Journal of Molecular Sciences 22, no. 13: 7186. https://doi.org/10.3390/ijms22137186
APA StyleFerracchiato, S., Di-Iacovo, N., Scopetti, D., Piobbico, D., Castelli, M., Pieroni, S., Gargaro, M., Manni, G., Brancorsini, S., Della-Fazia, M. A., & Servillo, G. (2021). Hops/Tmub1 Heterozygous Mouse Shows Haploinsufficiency Effect in Influencing p53-Mediated Apoptosis. International Journal of Molecular Sciences, 22(13), 7186. https://doi.org/10.3390/ijms22137186