
Biomarkers for Comorbidities Modulate the Activity of T-Cells in COPD


Abstract
:1. Introduction
2. Results
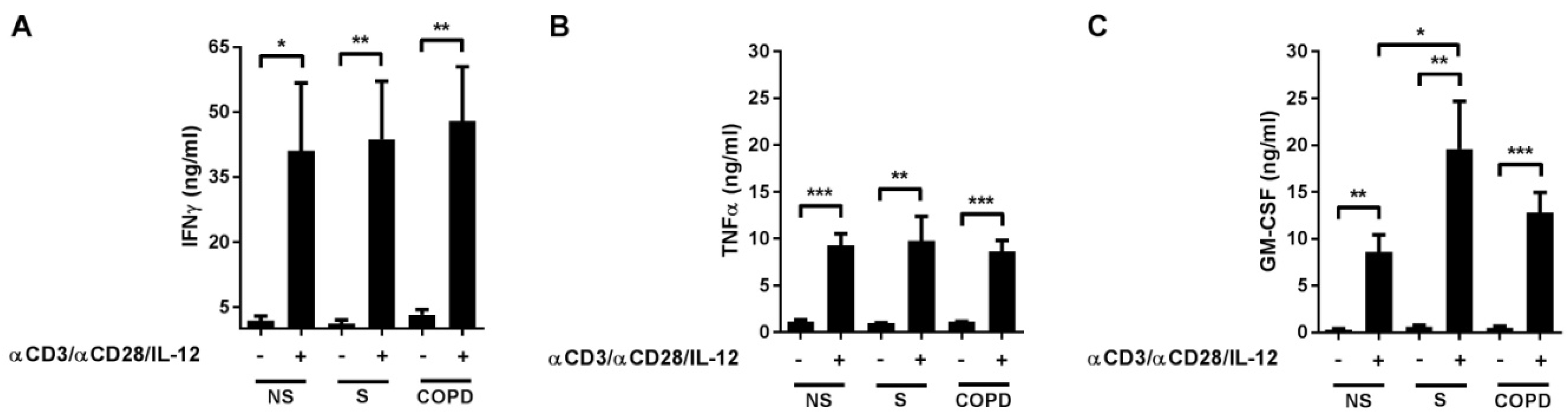
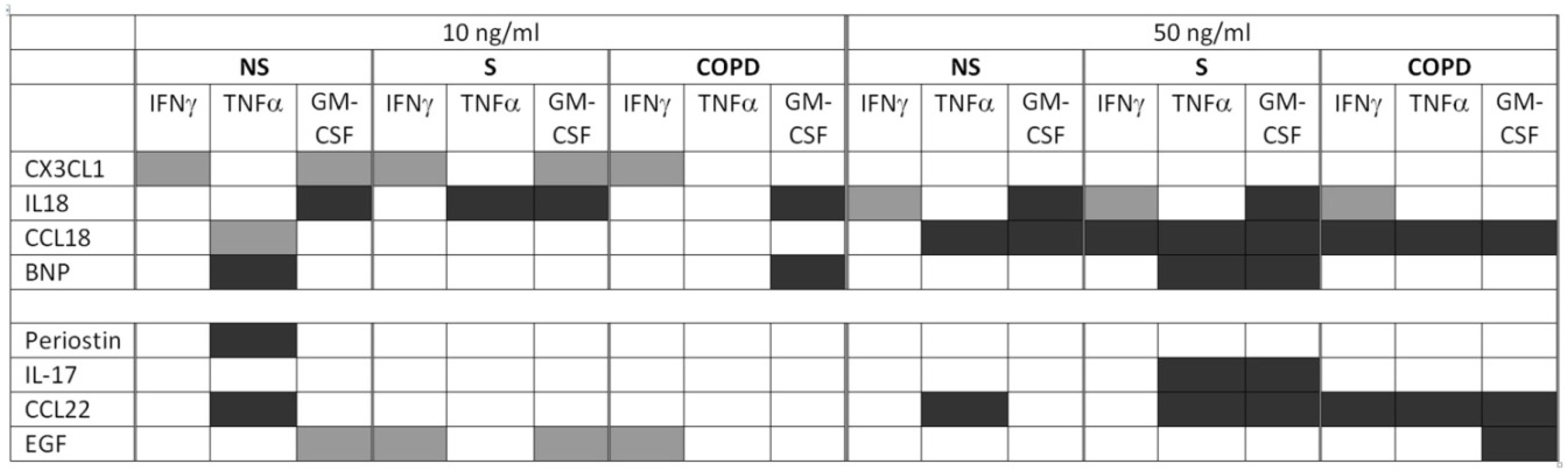
2.1. Cytokine Release in Response to T-Cell Activation
2.2. CX3CL1 Increased IFNγ, TNFα, and GM-CSF Release of T-Cells
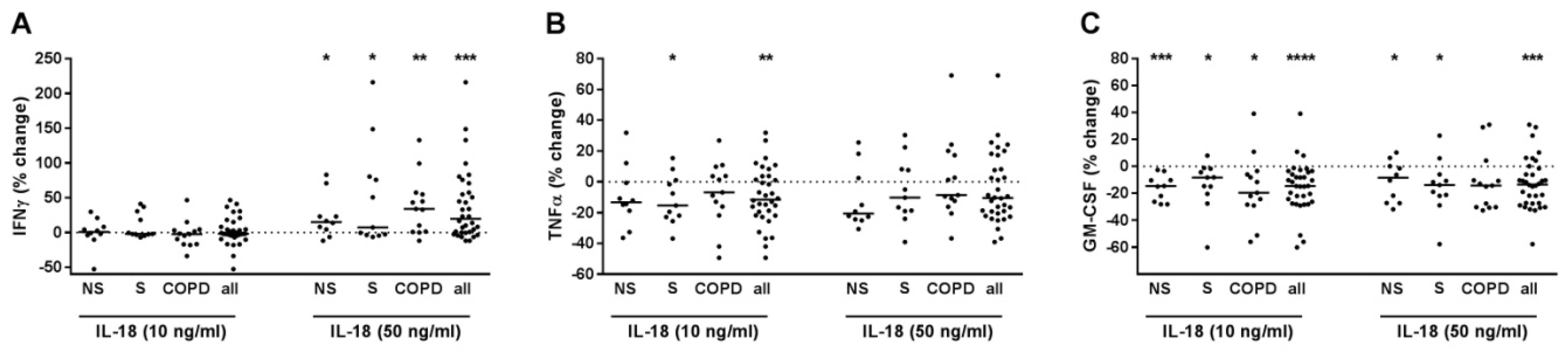
2.3. IL-18 Increased IFNγ but Reduced TNFα and GM-CSF Release of T-Cells
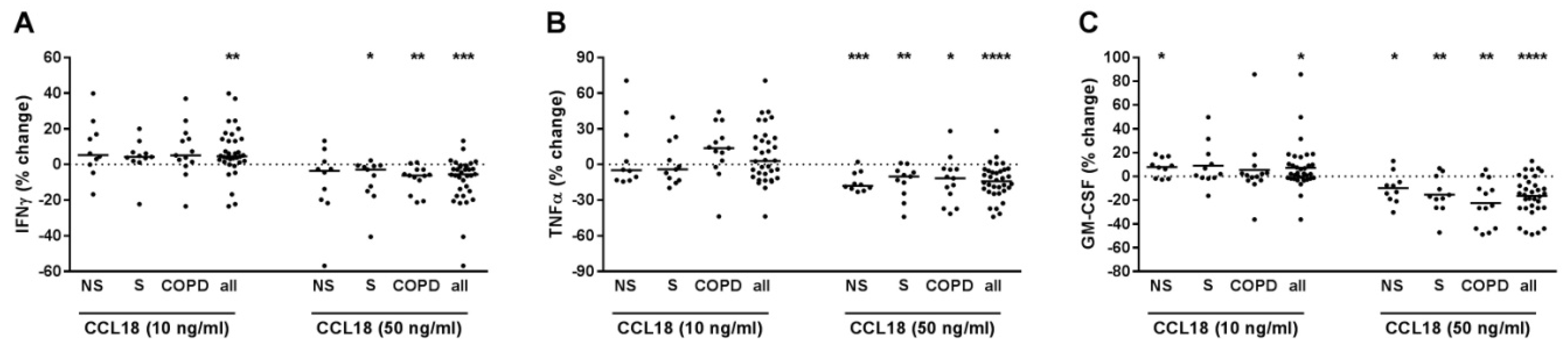
2.4. CCL18 Concentration-Dependently Increased or Decreased the Cytokine Release of T-Cells
2.5. BNP Reduced TNFα and GM-CSF Release of T-Cells
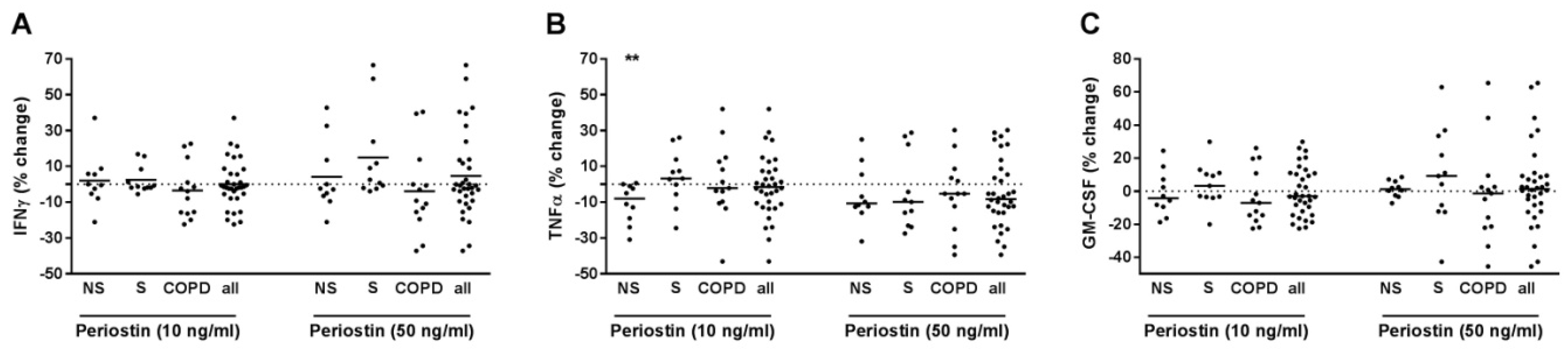
2.6. Periostin Did Not Modulate IFNγ, TNFα or GM-CSF Release of T-Cells from Current Smokers without Respiratory Symptoms and from COPD Subjects
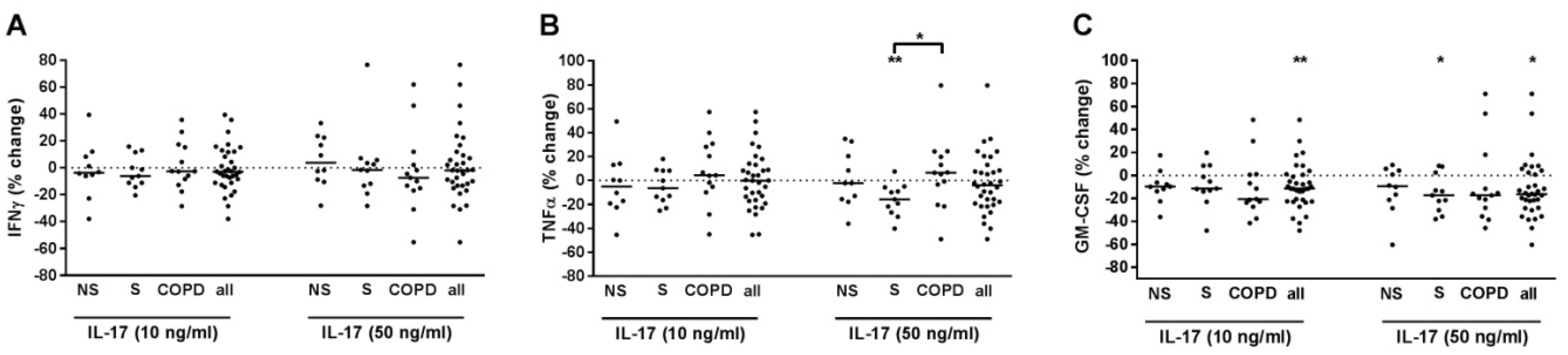
2.7. IL-17 Suppressed GM-CSF Release of T-Cells
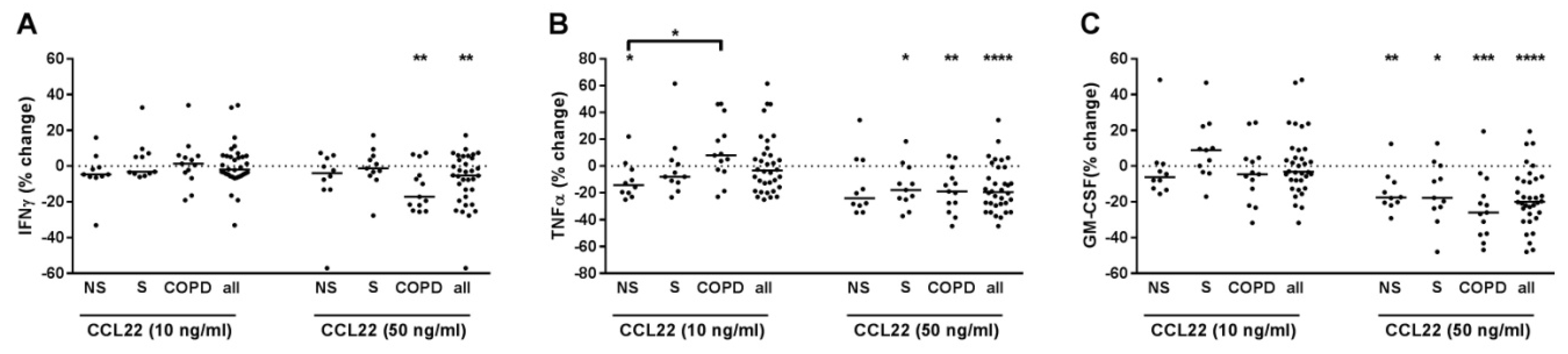
2.8. CCL22 Suppressed IFNγ, TNFα and GM-CSF Release of T-Cells
2.9. EGF Modulated IFNγ and GM-CSF Release of T-Cells
3. Discussion
4. Material and Methods
4.1. Study Subjects
4.2. Isolation of Peripheral Blood Mononuclear Cells (PBMCs)
4.3. Enzyme-Linked Immunosorbent Assay for IFNγ, TNFα and GM-CSF
4.4. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pelgrim, C.E.; Peterson, J.D.; Gosker, H.R.; Schols, A.M.; van Helvoort, A.; Garssen, J.; Folkerts, G.; Kraneveld, A.D. Psychological co-morbidities in COPD: Targeting systemic inflammation, a benefit for both? Eur. J. Pharmacol. 2019, 842, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Westerik, J.A.M.; Metting, E.I.; Van Boven, J.F.M.; Tiersma, W.; Kocks, J.W.H.; Schermer, T.R. Associations between chronic comorbidity and exacerbation risk in primary care patients with COPD. Respir. Res. 2017, 18, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangelov, K.; Sethi, S. Role of Infections. Clin. Chest Med. 2014, 35, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Paats, M.; Bergen, I.; Hoogsteden, H.; van der Eerden, M.; Hendriks, R. Systemic CD4+ and CD8+ T-cell cytokine profiles correlate with GOLD stage in stable COPD. Eur. Respir. J. 2011, 40, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, M.E.P.; Higgs, B.; Brohawn, B.P.; Pilataxi, B.F.; Guo, X.; Kuziora, M.; Bowler, R.P.; White, W.I. CD4+ T-Cell Profiles and Peripheral Blood Ex-Vivo Responses to T-Cell Directed Stimulation Delineate COPD Phenotypes. Chronic Obstr. Pulm. Dis. J. COPD Found. 2015, 2, 268–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef]
- McKendry, R.T.; Spalluto, C.M.; Burke, H.; Nicholas, B.; Cellura, M.D.; Al-Shamkhani, A.; Staples, K.; Wilkinson, T.M.A. Dysregulation of Antiviral Function of CD8+T Cells in the Chronic Obstructive Pulmonary Disease Lung. Role of the PD-1–PD-L1 Axis. Am. J. Respir. Crit. Care Med. 2016, 193, 642–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knobloch, J.; Schild, K.; Jungck, D.; Urban, K.; Müller, K.; Schweda, E.K.H.; Rupp, J.; Koch, A. The T-Helper Cell Type 1 Immune Response to Gram-Negative Bacterial Infections Is Impaired in COPD. Am. J. Respir. Crit. Care Med. 2011, 183, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Curtis, J.L. At the Checkpoint: Lung CD8+T Cells, Respiratory Viruses, and Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2016, 193, 600–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, P.J. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2016, 138, 16–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Gadgil, A.S.; Givelber, R.; George, M.P.; Stoner, M.W.; Sciurba, F.; Duncan, S.R. Peripheral T Cell Functions Correlate with the Severity of Chronic Obstructive Pulmonary Disease. J. Immunol. 2009, 182, 3270–3277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gayle, A.; Dickinson, S.; Poole, C.; Pang, M.; Fauconnot, O.; Quint, J.K. Incidence of type II diabetes in chronic obstructive pulmonary disease: A nested case–control study. NPJ Prim. Care Respir. Med. 2019, 29, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trinkmann, F.; Saur, J.; Borggrefe, M.; Akin, I. Cardiovascular Comorbidities in Chronic Obstructive Pulmonary Disease (COPD)—Current Considerations for Clinical Practice. J. Clin. Med. 2019, 8, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, R.; Hinkle, C.C.; Ferguson, J.; Mehta, N.N.; Li, M.; Qu, L.; Lu, Y.; Putt, M.E.; Ahima, R.S.; Reilly, M.P. Fractalkine Is a Novel Human Adipochemokine Associated With Type 2 Diabetes. Diabetes 2011, 60, 1512–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaharieva, E.; Kamenov, Z.; Velikova, T.; Tsakova, A.; El-Darawish, Y.; Okamura, H. Interleukin-18 serum level is elevated in type 2 diabetes and latent autoimmune diabetes. Endocr. Connect. 2018, 7, 179–185. [Google Scholar] [CrossRef] [Green Version]
- Blankenberg, S.; Rupprecht, H.J.; Poirier, O.; Bickel, C.; Smieja, M.; Hafner, G.; Meyer, J.; Cambien, F.; Tiret, L. Plasma Concentrations and Genetic Variation of Matrix Metalloproteinase 9 and Prognosis of Patients With Cardiovascular Disease. Circulation 2003, 107, 1579–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damås, J.K.; Boullier, A.; Waehre, T.; Smith, C.; Sandberg, W.J.; Green, S.; Aukrust, P.; Quehenberger, O. Expression of Fractalkine (CX3CL1) and its Receptor, CX3CR1, Is Elevated in Coronary Artery Disease and Is Reduced During Statin Therapy. Arter. Thromb. Vasc. Biol. 2005, 25, 2567–2572. [Google Scholar] [CrossRef] [Green Version]
- Versteylen, M.O.; Manca, M.; Joosen, I.A.; Schmidt, D.E.; Das, M.; Hofstra, L.; Crijns, H.J.; Biessen, E.A.; Kietselaer, B.L. CC chemokine ligands in patients presenting with stable chest pain: Association with atherosclerosis and future cardiovascular events. Neth. Hear. J. 2016, 24, 722–729. [Google Scholar] [CrossRef] [Green Version]
- Kraaijeveld, A.; de Jager, S.; de Jager, W.; Prakken, B.; McColl, S.; Haspels, I.; Putter, H.; van Berkel, T.; Nagelkerken, L.; Jukema, J.; et al. CC Chemokine Ligand-5 (CCL5/RANTES) and CC Chemokine Ligand-18 (CCL18/PARC) Are Specific Markers of Refractory Unstable Angina Pectoris and Are Transiently Raised During Severe Ischemic Symptoms. Circulation 2007, 116, 1931–1941. [Google Scholar] [CrossRef] [Green Version]
- Stolla, M.; Pelisek, J.; Von Brühl, M.-L.; Schäfer, A.; Barocke, V.; Heider, P.; Lorenz, M.; Tirniceriu, A.; Steinhart, A.; Bauersachs, J.; et al. Fractalkine Is Expressed in Early and Advanced Atherosclerotic Lesions and Supports Monocyte Recruitment via CX3CR1. PLoS ONE 2012, 7, e43572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velagaleti, R.S.; Vasan, R.S. Heart Failure in the Twenty-First Century: Is it a Coronary Artery Disease or Hypertension Problem? Cardiol. Clin. 2007, 25, 487–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maisel, A.S.; Krishnaswamy, P.; Nowak, R.M.; Mccord, J.; Hollander, J.E.; Duc, P.; Omland, T.; Storrow, A.B.; Abraham, W.T.; Wu, A.H.; et al. Rapid Measurement of B-Type Natriuretic Peptide in the Emergency Diagnosis of Heart Failure. N. Engl. J. Med. 2002, 347, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Hao, W.; Li, M.; Zhang, C.; Zhang, Y.; Du, W. Increased levels of inflammatory biomarker CX3CL1 in patients with chronic obstructive pulmonary disease. Cytokine 2020, 126, 154881. [Google Scholar] [CrossRef] [PubMed]
- Imaoka, H.; Hoshino, T.; Takei, S.; Kinoshita, T.; Okamoto, M.; Kawayama, T.; Kato, S.; Iwasaki, H.; Watanabe, K.; Aizawa, H. Interleukin-18 production and pulmonary function in COPD. Eur. Respir. J. 2008, 31, 287–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sin, D.D.; Miller, B.E.; Duvoix, A.; Man, S.F.P.; Zhang, X.; Silverman, E.K.; Connett, J.E.; Anthonisen, N.A.; Wise, R.; Tashkin, D.; et al. Serum PARC/CCL-18 Concentrations and Health Outcomes in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2011, 183, 1187–1192. [Google Scholar] [CrossRef] [Green Version]
- Gagnat, A.A.; Gjerdevik, M.; Gallefoss, F.; Coxson, H.O.; Gulsvik, A.; Bakke, P. Incidence of non-pulmonary cancer and lung cancer by amount of emphysema and airway wall thickness: A community-based cohort. Eur. Respir. J. 2017, 49, 1601162. [Google Scholar] [CrossRef] [Green Version]
- Maselli, D.J.; Hanania, N.A. Asthma COPD overlap: Impact of associated comorbidities. Pulm. Pharmacol. Ther. 2018, 52, 27–31. [Google Scholar] [CrossRef]
- Izuhara, K.; Conway, S.J.; Moore, B.; Matsumoto, H.; Holweg, C.T.J.; Matthews, J.G.; Arron, J.R. Roles of Periostin in Respiratory Disorders. Am. J. Respir. Crit. Care Med. 2016, 193, 949–956. [Google Scholar] [CrossRef]
- González-González, L.; Alonso, J. Periostin: A Matricellular Protein with Multiple Functions in Cancer Development and Progression. Front. Oncol. 2018, 8, 225. [Google Scholar] [CrossRef]
- Hong, L.-Z.; Wei, X.-W.; Chen, J.-F.; Shi, Y. Overexpression of periostin predicts poor prognosis in non-small cell lung cancer. Oncol. Lett. 2013, 6, 1595–1603. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.-H.; Wang, W.; Lin, Y.; Qian, L.-H.; Zhang, X.-W.; Wang, Q.-B.; Yu, L.-K. Diagnostic and prognostic value of serum periostin in patients with non-small cell lung cancer. Oncotarget 2016, 8, 18746–18753. [Google Scholar] [CrossRef] [PubMed]
- Agache, I.; Ciobanu, C.; Agache, C.; Anghel, M. Increased serum IL-17 is an independent risk factor for severe asthma. Respir. Med. 2010, 104, 1131–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Pociask, D.A.; McAleer, J.P.; Chan, Y.R.; Alcorn, J.F.; Kreindler, J.L.; Keyser, M.R.; Shapiro, S.D.; Houghton, A.M.; Kolls, J.K.; et al. IL-17RA Is Required for CCL2 Expression, Macrophage Recruitment, and Emphysema in Response to Cigarette Smoke. PLoS ONE 2011, 6, e20333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jafarzadeh, A.; Fooladseresht, H.; Minaee, K.; Bazrafshani, M.R.; Khosravimashizi, A.; Nemati, M.; Mohammadizadeh, M.; Mohammadi, M.M.; Ghaderi, A. Higher circulating levels of chemokine CCL22 in patients with breast cancer: Evaluation of the influences of tumor stage and chemokine gene polymorphism. Tumor Biol. 2014, 36, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Prieto, S.; De Chiara, L.; Rodríguez-Girondo, M.; Vázquez-Iglesias, L.; Rodríguez-Berrocal, F.J.; Fernandez-Villar, A.; Botana-Rial, M.I.; De La Cadena, M.P. Highly Sensitive Marker Panel for Guidance in Lung Cancer Rapid Diagnostic Units. Sci. Rep. 2017, 7, srep41151. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Hochwald, S.; Deng, S.; Zhu, Y.; Tan, C.; Zhong, Q.; Zhou, Y.; Zhao, H.; Huang, H. Evaluation of EGF, EGFR, and E-cadherin as potential biomarkers for gastrointestinal cancers. Front. Lab. Med. 2017, 1, 135–140. [Google Scholar] [CrossRef]
- Carpaij, O.A.; Muntinghe, F.O.W.; Wagenaar, M.B.; Habing, J.W.; Timens, W.; Kerstjens, H.A.M.; Nawijn, M.; Kunz, L.I.Z.; Hiemstra, P.S.; Tew, G.W.; et al. Serum periostin does not reflect type 2-driven inflammation in COPD. Respir. Res. 2018, 19, 112. [Google Scholar] [CrossRef] [Green Version]
- Kim, V.; Cornwell, W.D.; Oros, M.; Durra, H.; Criner, G.J.; Rogers, T.J. Plasma Chemokine signature correlates with lung goblet cell hyperplasia in smokers with and without chronic obstructive pulmonary disease. BMC Pulm. Med. 2015, 15, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loza, M.J.; Watt, R.; Baribaud, F.; Barnathan, E.S.; Rennard, S.I. Systemic inflammatory profile and response to anti-tumor necrosis factor therapy in chronic obstructive pulmonary disease. Respir. Res. 2012, 13, 12. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Chen, X.; Liu, J.; Zhou, D.B.; Kuang, X.; Xiao, J.; Yu, Q.; Lu, X.; Li, W.; Xie, B.; et al. Serum IL-1β and IL-17 levels in patients with COPD: Associations with clinical parameters. Int. J. Chronic Obstr. Pulm. Dis. 2017, 12, 1247–1254. [Google Scholar] [CrossRef] [Green Version]
- Chung, K.F.; Caramori, G.; Adcock, I.M.; Di Stefano, A. Cytokine inhibition in the treatment of COPD. Int. J. Chronic Obstr. Pulm. Dis. 2014, 9, 397–412. [Google Scholar] [CrossRef] [Green Version]
- Vlahos, R.; Bozinovski, S.; Chan, S.P.J.; Ivanov, S.; Lindén, A.; Hamilton, J.A.; Anderson, G.P. Neutralizing Granulocyte/Macrophage Colony–Stimulating Factor Inhibits Cigarette Smoke–induced Lung Inflammation. Am. J. Respir. Crit. Care Med. 2010, 182, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Vlahos, R.; Bozinovski, S.; Hamilton, J.A.; Anderson, G. Therapeutic potential of treating chronic obstructive pulmonary disease (COPD) by neutralising granulocyte macrophage-colony stimulating factor (GM-CSF). Pharmacol. Ther. 2006, 112, 106–115. [Google Scholar] [CrossRef]
- Yasuda, K.; Nakanishi, K.; Tsutsui, H. Interleukin-18 in Health and Disease. Int. J. Mol. Sci. 2019, 20, 649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bischoff, L.; Alvarez, S.; Dai, D.L.; Soukhatcheva, G.; Orban, P.C.; Verchere, C.B. Cellular Mechanisms of CCL22-Mediated Attenuation of Autoimmune Diabetes. J. Immunol. 2015, 194, 3054–3064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vellaichamy, E.; Kaur, K.; Pandey, K.N. Enhanced activation of pro-inflammatory cytokines in mice lacking natriuretic peptide receptor-A. Peptides 2007, 28, 893–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glezeva, N.; Collier, P.; Voon, V.; Ledwidge, M.; McDonald, K.; Watson, C.; Baugh, J. Attenuation of Monocyte Chemotaxis—A Novel Anti-inflammatory Mechanism of Action for the Cardio-protective Hormone B-Type Natriuretic Peptide. J. Cardiovasc. Transl. Res. 2013, 6, 545–557. [Google Scholar] [CrossRef]
- Jones, B.A.; Beamer, M.; Ahmed, S. Fractalkine/CX3CL1: A Potential New Target for Inflammatory Diseases. Mol. Interv. 2010, 10, 263–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gracie, J.A.; Robertson, S.E.; McInnes, I. Interleukin-18. J. Leukoc. Biol. 2003, 73, 213–224. [Google Scholar] [CrossRef]
- Xu, D.; Chan, W.L.; Leung, B.P.; Hunter, D.; Schulz, K.; Carter, R.W.; McInnes, I.; Robinson, J.H.; Liew, F.Y. Selective Expression and Functions of Interleukin 18 Receptor on T Helper (Th) Type 1 but not Th2 Cells. J. Exp. Med. 1998, 188, 1485–1492. [Google Scholar] [CrossRef] [Green Version]
- Soler, D.; Chapman, T.R.; Poisson, L.R.; Wang, L.; Cote-Sierra, J.; Ryan, M.; McDonald, A.; Badola, S.; Fedyk, E.; Coyle, A.J.; et al. CCR8 expression identifies CD4 memory T cells enriched for FOXP3+ regulatory and Th2 effector lymphocytes. J. Immunol. 2006, 177, 6940–6951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Endo, R.I.; Nemerow, G.R. Upregulation of integrins alpha v beta 3 and alpha v beta 5 on human monocytes and T lymphocytes facilitates adenovirus-mediated gene delivery. J. Virol. 1995, 69, 2257–2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchiya, K.; Jo, T.; Takeda, N.; Al Heialy, S.; Siddiqui, S.; Shalaby, K.H.; Risse, P.-A.; Maghni, K.; Martin, J.G. EGF receptor activation during allergic sensitization affects IL-6-induced T-cell influx to airways in a rat model of asthma. Eur. J. Immunol. 2010, 40, 1590–1602. [Google Scholar] [CrossRef]
- Nanki, T.; Lipsky, P.E. Lack of correlation between chemokine receptor and Th1/Th2 cytokine expression by individual memory T cells. Int. Immunol. 2000, 12, 1659–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purandare, A.; Somerville, J. Antagonists of CCR4 as Immunomodulatory Agents. Curr. Top. Med. Chem. 2006, 6, 1335–1344. [Google Scholar] [CrossRef] [PubMed]
- Stolberg, V.R.; Martin, B.; Mancuso, P.; Olszewski, M.A.; Freeman, C.M.; Curtis, J.L.; Chensue, S.W. Role of CC Chemokine Receptor 4 in Natural Killer Cell Activation during Acute Cigarette Smoke Exposure. Am. J. Pathol. 2014, 184, 454–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zrioual, S.; Toh, M.-L.; Tournadre, A.; Zhou, Y.; Cazalis, M.-A.; Pachot, A.; Miossec, V.; Miossec, P. IL-17RA and IL-17RC Receptors Are Essential for IL-17A-Induced ELR+ CXC Chemokine Expression in Synoviocytes and Are Overexpressed in Rheumatoid Blood. J. Immunol. 2007, 180, 655–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, M.; Todd, I.; Fairclough, L.C. The role of CD8 + T lymphocytes in chronic obstructive pulmonary disease: A systematic review. Inflamm. Res. 2021, 70, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Sun, Y. Role of Regulatory T Cells in Disturbed Immune Homeostasis in Patients with Chronic Obstructive Pulmonary Disease. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Barnes, P.J. The cytokine network in asthma and chronic obstructive pulmonary disease. J. Clin. Investig. 2008, 118, 3546–3556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knobloch, J.; Panek, S.; Yanik, S.D.; Jameel, K.J.; Bendella, Z.; Jungck, D.; Bürger, P.; Bülthoff, E.; Struck, B.; Giannakis, N.; et al. The monocyte-dependent immune response to bacteria is suppressed in smoking-induced COPD. J. Mol. Med. 2019, 97, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Knobloch, J.; Yakin, Y.; Körber, S.; Grensemann, B.; Bendella, Z.; Boyaci, N.; Gallert, W.-J.; Yanik, S.D.; Jungck, D.; Koch, A.; et al. Simvastatin requires activation in accessory cells to modulate T-cell responses in asthma and COPD. Eur. J. Pharmacol. 2016, 788, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.; Raidl, M.; Lux, M.; Müller, K.; Büning, H.; Humme, S.; Erdmann, E. IL-12-induced T-bet expression and IFNγ release in lymphocytes from asthmatics—Role of MAPkinases ERK-1/-2, p38MAPK and effect of dexamethasone. Respir. Med. 2007, 101, 1321–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mat, Z.; Grensemann, B.; Yakin, Y.; Knobloch, J.; Koch, A. Effect of lipoteichoic acid on IL-2 and IL-5 release from T lymphocytes in asthma and COPD. Int. Immunopharmacol. 2012, 13, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Knobloch, J.; Hag, H.; Jungck, D.; Urban, K.; Koch, A. Resveratrol Impairs the Release of Steroid-resistant Cytokines from Bacterial Endotoxin-Exposed Alveolar Macrophages in Chronic Obstructive Pulmonary Disease. Basic Clin. Pharmacol. Toxicol. 2011, 109, 138–143. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | NS | S | COPD |
|---|---|---|---|
| N | 10 | 11 | 13 |
| Age, y | 56.6 ± 3.6 | 56.7 ± 2.6 | 61.9 ± 2.3 |
| Gender, m:f | 2:8 | 4:7 | 7:6 |
| FEV1, % pred. | 104.8 ± 4.7 | 105.5 ± 4.3 | 45.9 ± 2.9 *** |
| FEV1/FVC, % | 79.4 ± 1.8 | 79.3 ± 1.3 | 45 ± 3.1 *** |
| FVC, % pred. | 109.1 ± 5.5 | 108.9 ± 4.2 | 84.5 ± 4.3 ** |
| Pack-Years | 0 | 50.7 ± 10.8 | 53.7 ± 7.5 |
| Monocytes, %WBC | 6.3 ± 0.5 | 9.2 ± 0.7 ## | 4.5 ± 0.5 §§ |
| Lymphocytes, %WBC | 37.5 ± 4.7 | 34.3 ± 3.5 | 27.7 ± 2.1 |
| Neutrophils, %WBC | 55.2 ± 4.9 | 56.5 ± 4.0 | 67.3 ± 2.6 |
| Eosinophils, %WBC | 1.0 ± 0.3 | 0.9 ± 0.4 | 0.5 ± 0.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jamal Jameel, K.; Gallert, W.-J.; Yanik, S.D.; Panek, S.; Kronsbein, J.; Jungck, D.; Koch, A.; Knobloch, J. Biomarkers for Comorbidities Modulate the Activity of T-Cells in COPD. Int. J. Mol. Sci. 2021, 22, 7187. https://doi.org/10.3390/ijms22137187
Jamal Jameel K, Gallert W-J, Yanik SD, Panek S, Kronsbein J, Jungck D, Koch A, Knobloch J. Biomarkers for Comorbidities Modulate the Activity of T-Cells in COPD. International Journal of Molecular Sciences. 2021; 22(13):7187. https://doi.org/10.3390/ijms22137187
Chicago/Turabian StyleJamal Jameel, Kaschin, Willem-Jakob Gallert, Sarah D. Yanik, Susanne Panek, Juliane Kronsbein, David Jungck, Andrea Koch, and Jürgen Knobloch. 2021. "Biomarkers for Comorbidities Modulate the Activity of T-Cells in COPD" International Journal of Molecular Sciences 22, no. 13: 7187. https://doi.org/10.3390/ijms22137187
APA StyleJamal Jameel, K., Gallert, W. -J., Yanik, S. D., Panek, S., Kronsbein, J., Jungck, D., Koch, A., & Knobloch, J. (2021). Biomarkers for Comorbidities Modulate the Activity of T-Cells in COPD. International Journal of Molecular Sciences, 22(13), 7187. https://doi.org/10.3390/ijms22137187