
Novelty of Sphingolipids in the Central Nervous System Physiology and Disease: Focusing on the Sphingolipid Hypothesis of Neuroinflammation and Neurodegeneration

Abstract
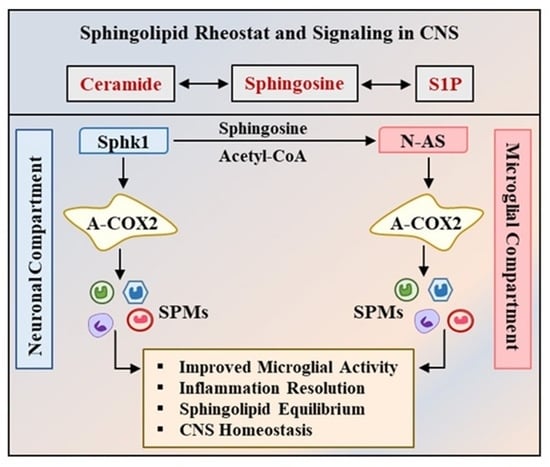
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Functional Significance of Bioactive Lipids in Health
Endogenous Regulators of Bioactive Lipid Synthesis: Enzymes and Pathways
3. Compartmentalization of Sphingolipids in the CNS as Drivers of Physiology and Pathology
3.1. Deregulation of Neuronal Sphk1–S1P Signaling in AD
3.2. Sphk1 and COX2 Together Act as a Bridge between CNS Homeostasis and AD Pathology
3.3. N-acetyl Sphingosine: Direct Modulator of COX2 Acetylation in the CNS
3.4. Sphingolipids in the Pathobiology of Parkinson’s Disease
3.5. Sphingolipids and Other Neurodegenerative Disorders
4. Conclusions
Therapeutic Implications and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shevchenko, A.; Simons, K. Lipidomics: Coming to grips with lipid diversity. Nat. Rev. Mol. Cell Biol. 2010, 11, 593–598. [Google Scholar] [CrossRef]
- Bari, M.; Bisogno, T. Bioactive Lipids in Health and Disease. Biomolecules 2020, 10, 1698. [Google Scholar] [CrossRef]
- Dennis, E.A.; Norris, P.C. Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef] [Green Version]
- Cai, T.; Yang, F. Phospholipid and Phospholipidomics in Health and Diseases. In Lipidomics in Health & Disease: Methods & Application; Wang, X., Wu, D., Shen, H., Eds.; Springer: Singapore, 2018; pp. 177–202. [Google Scholar]
- Kleuser, B. The Enigma of Sphingolipids in Health and Disease. Int. J. Mol. Sci. 2018, 19, 3126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Marzo, V.; Bifulco, M.; De Petrocellis, L. The endocannabinoid system and its therapeutic exploitation. Nat. Rev. Drug Discov. 2004, 3, 771–784. [Google Scholar] [CrossRef]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, G.; Wang, J.; Rasul, A.; Anwar, H.; Imran, A.; Qasim, M.; Zafar, S.; Kamran, S.K.S.; Razzaq, A.; Aziz, N.; et al. Role of cholesterol and sphingolipids in brain development and neurological diseases. Lipids Health Dis. 2019, 18, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, T. Lipid mediators in health and disease: Enzymes and receptors as therapeutic targets for the regulation of immunity and inflammation. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 123–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruegel, M.; Ceglarek, U.; Thiery, J. Eicosanoids: Essential mediators in health and disease/Eicosanoide: Bedeutende Faktoren in der Homöostase und ihre Bedeutung in der Pathogenese multipler Erkrankungen. Lab. Med. 2009, 33, 333–339. [Google Scholar] [CrossRef]
- Imig, J.D. Eicosanoid blood vessel regulation in physiological and pathological states. Clin. Sci. 2020, 134, 2707–2727. [Google Scholar] [CrossRef] [PubMed]
- Tassoni, D.; Kaur, G.; Weisinger, R.S.; Sinclair, A.J. The role of eicosanoids in the brain. Asia Pac. J. Clin. Nutr. 2008, 17 (Suppl. 1), 220–228. [Google Scholar] [PubMed]
- Peiris, H.N.; Vaswani, K.; Almughlliq, F.; Koh, Y.Q.; Mitchell, M.D. Review: Eicosanoids in preterm labor and delivery: Potential roles of exosomes in eicosanoid functions. Placenta 2017, 54, 95–103. [Google Scholar] [CrossRef]
- Cullis, P.R.; Kruijff, B.D.; Hope, M.J.; Nayar, R.; Schmid, S.L. Phospholipids and membrane transport. Can. J. Biochem. 1980, 58, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Bohdanowicz, M.; Grinstein, S. Role of phospholipids in endocytosis, phagocytosis, and macropinocytosis. Physiol. Rev. 2013, 93, 69–106. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, V.B.; Rossjohn, J.; Wakelam, M.J. Phospholipid signaling in innate immune cells. J. Clin. Investig. 2018, 128, 2670–2679. [Google Scholar] [CrossRef]
- García-Morales, V.; Montero, F.; González-Forero, D.; Rodríguez-Bey, G.; Gómez-Pérez, L.; Medialdea-Wandossell, M.J.; Domínguez-Vías, G.; García-Verdugo, J.M.; Moreno-López, B. Membrane-derived phospholipids control synaptic neurotransmission and plasticity. PLoS Biol. 2015, 13, e1002153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand-Yavin, A.; Yavin, E. Brain Oxidative Stress from a Phospholipid Perspective. In Handbook of Neurochemistry and Molecular Neurobiology: Neural Lipids; Lajtha, A., Tettamanti, G., Goracci, G., Eds.; Springer: Boston, MA, USA, 2009; pp. 603–630. [Google Scholar]
- Pitson, S.M. Regulation of sphingosine kinase and sphingolipid signaling. Trends Biochem. Sci. 2011, 36, 97–107. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef]
- Buckley, C.D.; Gilroy, D.W.; Serhan, C.N. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity 2014, 40, 315–327. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekharan, J.A.; Sharma-Walia, N. Lipoxins: Nature’s way to resolve inflammation. J. Inflamm. Res. 2015, 8, 181–192. [Google Scholar]
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Investig. 2018, 128, 2657–2669. [Google Scholar] [CrossRef]
- Hansen, T.V.; Vik, A.; Serhan, C.N. The Protectin Family of Specialized Pro-resolving Mediators: Potent Immunoresolvents Enabling Innovative Approaches to Target Obesity and Diabetes. Front. Pharmacol. 2018, 9, 1582. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Wan, M.; Huang, W. Maresins: Specialized Proresolving Lipid Mediators and Their Potential Role in Inflammatory-Related Diseases. Mediat. Inflamm. 2018, 2018, 2380319. [Google Scholar] [CrossRef] [Green Version]
- Christie, W.W.; Harwood, J.L. Oxidation of polyunsaturated fatty acids to produce lipid mediators. Essays Biochem. 2020, 64, 401–421. [Google Scholar] [PubMed]
- Smith, W.L.; Murphy, R.C. CHAPTER 12—The Eicosanoids: Cyclooxygenase, Lipoxygenase, and Epoxygenase Pathways. In Biochemistry of Lipids, Lipoproteins and Membranes, 5th ed.; Vance, D.E., Vance, J.E., Eds.; Elsevier: San Diego, CA, USA, 2008; pp. 331–362. [Google Scholar]
- Rouzer, C.A.; Marnett, L.J. Cyclooxygenases: Structural and functional insights. J. Lipid Res. 2009, 50, S29–S34. [Google Scholar] [CrossRef] [Green Version]
- Gibellini, F.; Smith, T.K. The Kennedy pathway—De novo synthesis of phosphatidylethanolamine and phosphatidylcholine. IUBMB Life 2010, 62, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Calzada, E.; Onguka, O.; Claypool, S.M. Chapter Two—Phosphatidylethanolamine Metabolism in Health and Disease. In International Review of Cell and Molecular Biology; Jeon, K.W., Ed.; Academic Press: Cambridge, MA, USA, 2016; Volume 321, pp. 29–88. [Google Scholar]
- Bankaitis, V.A.; Grabon, A. Phosphatidylinositol synthase and diacylglycerol platforms bust a move. Dev. Cell 2011, 21, 810–812. [Google Scholar] [CrossRef] [Green Version]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An overview of sphingolipid metabolism: From synthesis to breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar]
- Chatterjee, A.; Komshian, S.; Sansbury, B.E.; Wu, B.; Mottola, G.; Chen, M.; Spite, M.; Conte, M.S. Biosynthesis of proresolving lipid mediators by vascular cells and tissues. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 3393–3402. [Google Scholar] [CrossRef] [Green Version]
- Morteau, O. Prostaglandins and inflammation: The cyclooxygenase controversy. Arch. Immunol. Ther. Exp. 2000, 48, 473–480. [Google Scholar]
- Lee, J.Y.; Jin, H.K.; Bae, J.S. Sphingolipids in neuroinflammation: A potential target for diagnosis and therapy. BMB Rep. 2020, 53, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Russo, R.; Cristiano, C.; Avagliano, C.; De Caro, C.; La Rana, G.; Raso, G.M.; Canani, R.B.; Meli, R.; Calignano, A. Gut-brain Axis: Role of Lipids in the Regulation of Inflammation, Pain and CNS Diseases. Curr. Med. Chem. 2018, 25, 3930–3952. [Google Scholar] [CrossRef]
- Wheeler, D.; Knapp, E.; Bandaru, V.V.; Wang, Y.; Knorr, D.; Poirier, C.; Mattson, M.P.; Geiger, J.D.; Haughey, N.J. Tumor necrosis factor-alpha-induced neutral sphingomyelinase-2 modulates synaptic plasticity by controlling the membrane insertion of NMDA receptors. J. Neurochem. 2009, 109, 1237–1249. [Google Scholar] [CrossRef] [Green Version]
- Tabatadze, N.; Savonenko, A.; Song, H.; Bandaru, V.V.; Chu, M.; Haughey, N.J. Inhibition of neutral sphingomyelinase-2 perturbs brain sphingolipid balance and spatial memory in mice. J. Neurosci. Res. 2010, 88, 2940–2951. [Google Scholar] [CrossRef] [Green Version]
- Maceyka, M.; Spiegel, S. Sphingolipid metabolites in inflammatory disease. Nature 2014, 510, 58–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mencarelli, C.; Martinez-Martinez, P. Ceramide function in the brain: When a slight tilt is enough. Cell. Mol. Life Sci. 2013, 70, 181–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoica, B.A.; Movsesyan, V.A.; Lea, P.M.; Faden, A.I. Ceramide-induced neuronal apoptosis is associated with dephosphorylation of Akt, BAD, FKHR, GSK-3beta, and induction of the mitochondrial-dependent intrinsic caspase pathway. Mol. Cell Neurosci. 2003, 22, 365–382. [Google Scholar] [CrossRef]
- Movsesyan, V.A.; Yakovlev, A.G.; Dabaghyan, E.A.; Stoica, B.A.; Faden, A.I. Ceramide induces neuronal apoptosis through the caspase-9/caspase-3 pathway. Biochem. Biophys. Res. Commun. 2002, 299, 201–207. [Google Scholar] [CrossRef]
- de Wit, N.M.; den Hoedt, S.; Martinez-Martinez, P.; Rozemuller, A.J.; Mulder, M.T.; de Vries, H.E. Astrocytic ceramide as possible indicator of neuroinflammation. J. Neuroinflammation 2019, 16, 48. [Google Scholar] [CrossRef]
- Filippov, V.; Song, M.A.; Zhang, K.; Vinters, H.V.; Tung, S.; Kirsch, W.M.; Yang, J.; Duerksen-Hughes, P.J. Increased ceramide in brains with Alzheimer’s and other neurodegenerative diseases. J. Alzheimer’s Dis. 2012, 29, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Jazvinšćak Jembrek, M.; Hof, P.R.; Šimić, G. Ceramides in Alzheimer’s Disease: Key Mediators of Neuronal Apoptosis Induced by Oxidative Stress and Aβ Accumulation. Oxidative Med. Cell. Longev. 2015, 2015, 346783. [Google Scholar] [CrossRef] [Green Version]
- Katsel, P.; Li, C.; Haroutunian, V. Gene expression alterations in the sphingolipid metabolism pathways during progression of dementia and Alzheimer’s disease: A shift toward ceramide accumulation at the earliest recognizable stages of Alzheimer’s disease? Neurochem. Res. 2007, 32, 845–856. [Google Scholar] [CrossRef]
- Healy, L.M.; Antel, J.P. Sphingosine-1-Phosphate Receptors in the Central Nervous and Immune Systems. Curr. Drug Targets 2016, 17, 1841–1850. [Google Scholar] [CrossRef] [PubMed]
- Yanagida, K.; Liu, C.H.; Faraco, G.; Galvani, S.; Smith, H.K.; Burg, N.; Anrather, J.; Sanchez, T.; Iadecola, C.; Hla, T. Size-selective opening of the blood-brain barrier by targeting endothelial sphingosine 1-phosphate receptor 1. Proc. Natl. Acad. Sci. USA 2017, 114, 4531–4536. [Google Scholar] [CrossRef] [Green Version]
- Kanno, T.; Nishizaki, T.; Proia, R.L.; Kajimoto, T.; Jahangeer, S.; Okada, T.; Nakamura, S. Regulation of synaptic strength by sphingosine 1-phosphate in the hippocampus. Neuroscience 2010, 171, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Riganti, L.; Antonucci, F.; Gabrielli, M. Sphingosine-1-Phosphate (S1P) Impacts Presynaptic Functions by Regulating Synapsin I Localization in the Presynaptic Compartment. J. Neurosci. 2016, 36, 4624–4634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, T.; Kajimoto, T.; Jahangeer, S.; Nakamura, S.-I. Sphingosine kinase/sphingosine 1-phosphate signalling in central nervous system. Cell. Signal. 2009, 21, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Fischer, I.; Alliod, C.; Martinier, N.; Newcombe, J.; Brana, C.; Pouly, S. Sphingosine kinase 1 and sphingosine 1-phosphate receptor 3 are functionally upregulated on astrocytes under pro-inflammatory conditions. PLoS ONE 2011, 6, e23905. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.P.; Mizugishi, K.; Bektas, M.; Sandhoff, R.; Proia, R.L. Sphingosine kinase 1/S1P receptor signaling axis controls glial proliferation in mice with Sandhoff disease. Hum. Mol. Genet. 2008, 17, 2257–2264. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Han, S.H.; Park, M.H.; Baek, B.; Song, I.S.; Choi, M.K.; Takuwa, Y.; Ryu, H.; Kim, S.H.; He, X.; et al. Neuronal SphK1 acetylates COX2 and contributes to pathogenesis in a model of Alzheimer’s Disease. Nat. Commun. 2018, 9, 1479. [Google Scholar] [CrossRef]
- Couttas, T.A.; Kain, N.; Daniels, B.; Lim, X.Y.; Shepherd, C.; Kril, J.; Pickford, R.; Li, H.; Garner, B.; Don, A.S. Loss of the neuroprotective factor Sphingosine 1-phosphate early in Alzheimer’s disease pathogenesis. Acta Neuropathol. Commun. 2014, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Ceccom, J.; Loukh, N.; Lauwers-Cances, V.; Touriol, C.; Nicaise, Y.; Gentil, C.; Uro-Coste, E.; Pitson, S.; Maurage, C.A.; Duyckaerts, C.; et al. Reduced sphingosine kinase-1 and enhanced sphingosine 1-phosphate lyase expression demonstrate deregulated sphingosine 1-phosphate signaling in Alzheimer’s disease. Acta Neuropathol. Commun. 2014, 2, 12. [Google Scholar] [CrossRef]
- Gassowska, M.; Cieslik, M.; Wilkaniec, A.; Strosznajder, J.B. Sphingosine kinases/sphingosine-1-phosphate and death Signalling in APP-transfected cells. Neurochem. Res. 2014, 39, 645–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.P.; Hu, Z.; Sieburth, D. Recruitment of sphingosine kinase to presynaptic terminals by a conserved muscarinic signaling pathway promotes neurotransmitter release. Genes Dev. 2012, 26, 1070–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toman, R.E.; Payne, S.G.; Watterson, K.R.; Maceyka, M.; Lee, N.H.; Milstien, S.; Bigbee, J.W.; Spiegel, S. Differential transactivation of sphingosine-1-phosphate receptors modulates NGF-induced neurite extension. J. Cell Biol. 2004, 166, 381–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minghetti, L. Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J. Neuropathol. Exp. Neurol. 2004, 63, 901–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.P.; Dong, L.; Yuan, C.; Noon, K.R.; Smith, W.L. Asymmetric acetylation of the cyclooxygenase-2 homodimer by aspirin and its effects on the oxygenation of arachidonic, eicosapentaenoic, and docosahexaenoic acids. Mol. Pharmacol. 2010, 77, 979–986. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Ronowska, A.; Szutowicz, A.; Bielarczyk, H.; Gul-Hinc, S.; Klimaszewska-Łata, J.; Dyś, A.; Zyśk, M.; Jankowska-Kulawy, A. The Regulatory Effects of Acetyl-CoA Distribution in the Healthy and Diseased Brain. Front. Cell. Neurosci. 2018, 12, 169. [Google Scholar] [CrossRef] [Green Version]
- Medeiros, R.; Kitazawa, M.; Passos, G.F.; Baglietto-Vargas, D.; Cheng, D.; Cribbs, D.H.; LaFerla, F.M. Aspirin-triggered lipoxin A4 stimulates alternative activation of microglia and reduces Alzheimer disease-like pathology in mice. Am. J. Pathol. 2013, 182, 1780–1789. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.X.; Tan, B.C.; Zhou, W.; Wei, T.; Lai, W.H.; Tan, J.W.; Dong, J.H. Resolvin D1, an endogenous lipid mediator for inactivation of inflammation-related signaling pathways in microglial cells, prevents lipopolysaccharide-induced inflammatory responses. CNS Ther. 2013, 19, 235–243. [Google Scholar] [CrossRef]
- Lee, J.Y.; Han, S.H.; Park, M.H.; Song, I.S.; Choi, M.K.; Yu, E. N-AS-triggered SPMs are direct regulators of microglia in a model of Alzheimer’s disease. Nat. Commun. 2020, 11, 2358. [Google Scholar] [CrossRef]
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Abbott, S.K.; Li, H.; Muñoz, S.S.; Knoch, B.; Batterham, M.; Murphy, K.E.; Halliday, G.M.; Garner, B. Altered ceramide acyl chain length and ceramide synthase gene expression in Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 518–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mielke, M.M.; Maetzler, W.; Haughey, N.J.; Bandaru, V.V.; Savica, R.; Deuschle, C.; Gasser, T.; Hauser, A.K.; Gräber-Sultan, S.; Schleicher, E.; et al. Plasma ceramide and glucosylceramide metabolism is altered in sporadic Parkinson’s disease and associated with cognitive impairment: A pilot study. PLoS ONE 2013, 8, e73094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arkadir, D.; Dinur, T.; Mullin, S.; Mehta, A.; Baris, H.N.; Alcalay, R.N.; Zimran, A. Trio approach reveals higher risk of PD in carriers of severe vs. mild GBA mutations. Blood Cells Mol. Dis. 2018, 68, 115–116. [Google Scholar] [CrossRef] [PubMed]
- Pchelina, S.; Emelyanov, A.; Baydakova, G.; Andoskin, P.; Senkevich, K.; Nikolaev, M.; Miliukhina, I.; Yakimovskii, A.; Timofeeva, A.; Fedotova, E.; et al. Oligomeric α-synuclein and glucocerebrosidase activity levels in GBA-associated Parkinson’s disease. Neurosci. Lett. 2017, 636, 70–76. [Google Scholar] [CrossRef]
- Badawy, S.M.M.; Okada, T.; Kajimoto, T.; Hirase, M.; Matovelo, S.A.; Nakamura, S.; Yoshida, D.; Ijuin, T.; Nakamura, S.I. Extracellular α-synuclein drives sphingosine 1-phosphate receptor subtype 1 out of lipid rafts, leading to impaired inhibitory G-protein signaling. J. Biol. Chem. 2018, 293, 8208–8216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyszko, J.A.; Strosznajder, J.B. The key role of sphingosine kinases in the molecular mechanism of neuronal cell survival and death in an experimental model of Parkinson’s disease. Folia Neuropathol. 2014, 52, 260–269. [Google Scholar] [CrossRef]
- Motyl, J.; Przykaza, Ł.; Boguszewski, P.M.; Kosson, P.; Strosznajder, J.B. Pramipexole and Fingolimod exert neuroprotection in a mouse model of Parkinson’s disease by activation of sphingosine kinase 1 and Akt kinase. Neuropharmacology 2018, 135, 139–150. [Google Scholar] [CrossRef]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Primers 2015, 1, 15005. [Google Scholar] [CrossRef]
- Di Pardo, A.; Basit, A.; Armirotti, A.; Amico, E.; Castaldo, S.; Pepe, G.; Marracino, F.; Buttari, F.; Digilio, A.F.; Maglione, V. De novo Synthesis of Sphingolipids Is Defective in Experimental Models of Huntington’s Disease. Front. Neurosci. 2017, 11, 698. [Google Scholar] [CrossRef]
- Di Pardo, A.; Amico, E.; Basit, A. Defective Sphingosine-1-phosphate metabolism is a druggable target in Huntington’s disease. Sci. Rep. 2017, 7, 5280. [Google Scholar] [CrossRef] [Green Version]
- Di Pardo, A.; Amico, E.; Favellato, M.; Castrataro, R.; Fucile, S.; Squitieri, F.; Maglione, V. FTY720 (fingolimod) is a neuroprotective and disease-modifying agent in cellular and mouse models of Huntington disease. Hum. Mol. Genet. 2014, 23, 2251–2265. [Google Scholar] [CrossRef] [Green Version]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Dodge, J.C.; Treleaven, C.M.; Pacheco, J.; Cooper, S.; Bao, C.; Abraham, M.; Cromwell, M.; Sardi, S.P.; Chuang, W.L.; Sidman, R.L.; et al. Glycosphingolipids are modulators of disease pathogenesis in amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2015, 112, 8100–8105. [Google Scholar] [CrossRef] [Green Version]
- Henriques, A.; Croixmarie, V.; Bouscary, A.; Mosbach, A.; Keime, C.; Boursier-Neyret, C.; Walter, B.; Spedding, M.; Loeffler, J.P. Sphingolipid Metabolism Is Dysregulated at Transcriptomic and Metabolic Levels in the Spinal Cord of an Animal Model of Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2017, 10, 433. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Corcia, P.; Fergani, A.; Gonzalez De Aguilar, J.L.; Bonnefont-Rousselot, D.; Bittar, R.; Seilhean, D.; Hauw, J.J.; Lacomblez, L.; Loeffler, J.P.; et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 2008, 70, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Larrauri, A.; Presa, N.; Dominguez-Herrera, A.; Ouro, A.; Trueba, M.; Gomez-Muñoz, A. Role of bioactive sphingolipids in physiology and pathology. Essays Biochem. 2020, 64, 579–589. [Google Scholar] [PubMed]
- Alessenko, A.V.; Albi, E. Exploring Sphingolipid Implications in Neurodegeneration. Front. Neurol. 2020, 11, 437. [Google Scholar] [CrossRef]
- Ryan, J.; Storey, E.; Murray, A.M.; Woods, R.L.; Wolfe, R.; Reid, C.M.; Nelson, M.R.; Chong, T.T.J.; Williamson, J.D.; Ward, S.A.; et al. Randomized placebo-controlled trial of the effects of aspirin on dementia and cognitive decline. Neurology 2020, 95, e320–e331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grodzicki, W.; Dziendzikowska, K. The Role of Selected Bioactive Compounds in the Prevention of Alzheimer’s Disease. Antioxidants 2020, 9, 229. [Google Scholar] [CrossRef] [Green Version]
- John, T.; Samuel, B.; Abolaji, O.; Folashade, O.; Oyetooke, A.; Oluwatosin, F. Functional foods and bioactive compounds: Roles in the prevention, treatment and management of neurodegenerative diseases. GSC Biol. Pharm. Sci. 2020, 11, 297–313. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ayub, M.; Jin, H.-K.; Bae, J.-s. Novelty of Sphingolipids in the Central Nervous System Physiology and Disease: Focusing on the Sphingolipid Hypothesis of Neuroinflammation and Neurodegeneration. Int. J. Mol. Sci. 2021, 22, 7353. https://doi.org/10.3390/ijms22147353
Ayub M, Jin H-K, Bae J-s. Novelty of Sphingolipids in the Central Nervous System Physiology and Disease: Focusing on the Sphingolipid Hypothesis of Neuroinflammation and Neurodegeneration. International Journal of Molecular Sciences. 2021; 22(14):7353. https://doi.org/10.3390/ijms22147353
Chicago/Turabian StyleAyub, Maria, Hee-Kyung Jin, and Jae-sung Bae. 2021. "Novelty of Sphingolipids in the Central Nervous System Physiology and Disease: Focusing on the Sphingolipid Hypothesis of Neuroinflammation and Neurodegeneration" International Journal of Molecular Sciences 22, no. 14: 7353. https://doi.org/10.3390/ijms22147353
APA StyleAyub, M., Jin, H. -K., & Bae, J. -s. (2021). Novelty of Sphingolipids in the Central Nervous System Physiology and Disease: Focusing on the Sphingolipid Hypothesis of Neuroinflammation and Neurodegeneration. International Journal of Molecular Sciences, 22(14), 7353. https://doi.org/10.3390/ijms22147353