
Identification of Dihydromyricetin and Metabolites in Serum and Brain Associated with Acute Anti-Ethanol Intoxicating Effects in Mice

 , , ,
, , ,  , and
, and
Abstract
:1. Introduction
2. Results
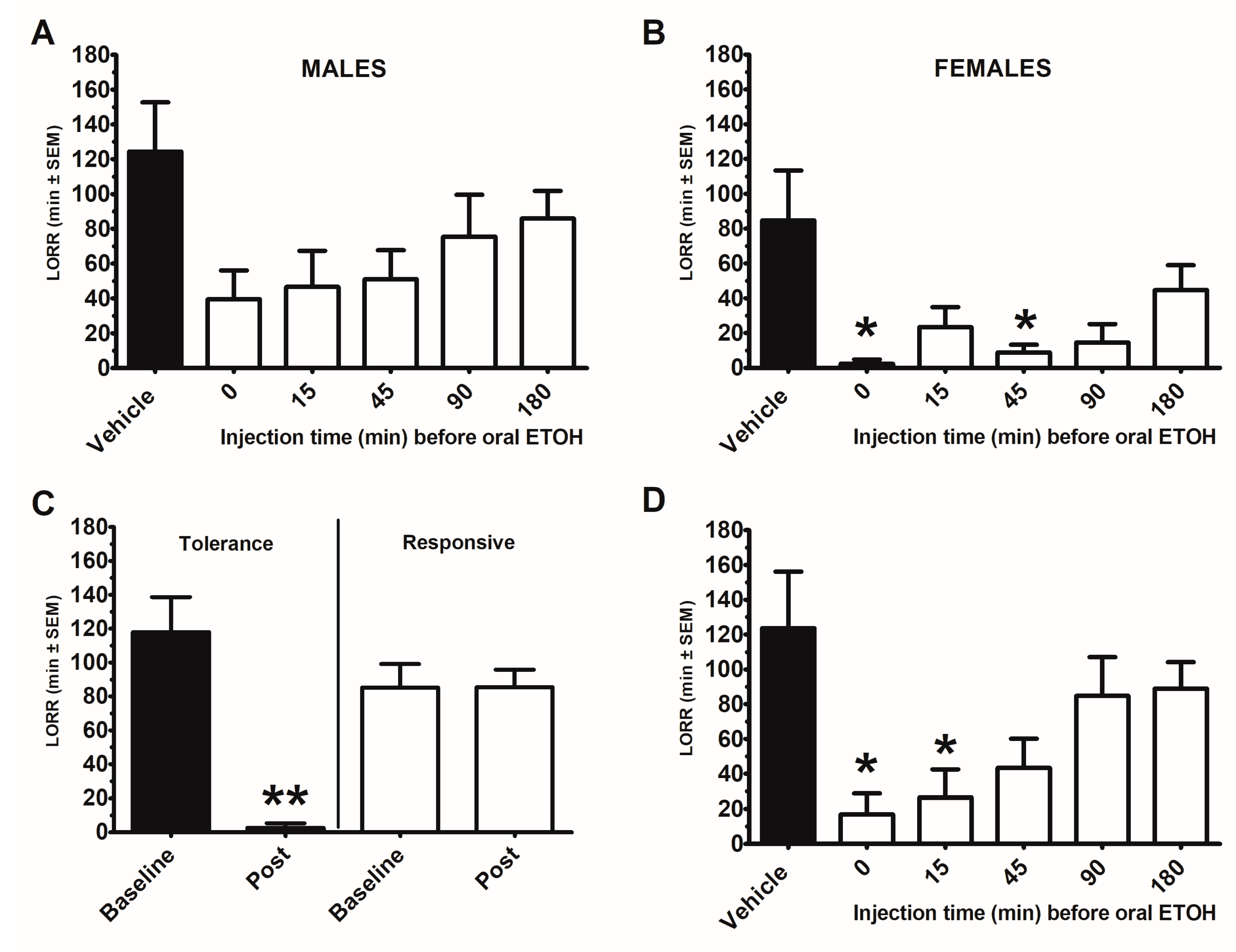
2.1. Ethanol-Induced Loss of Righting Reflex
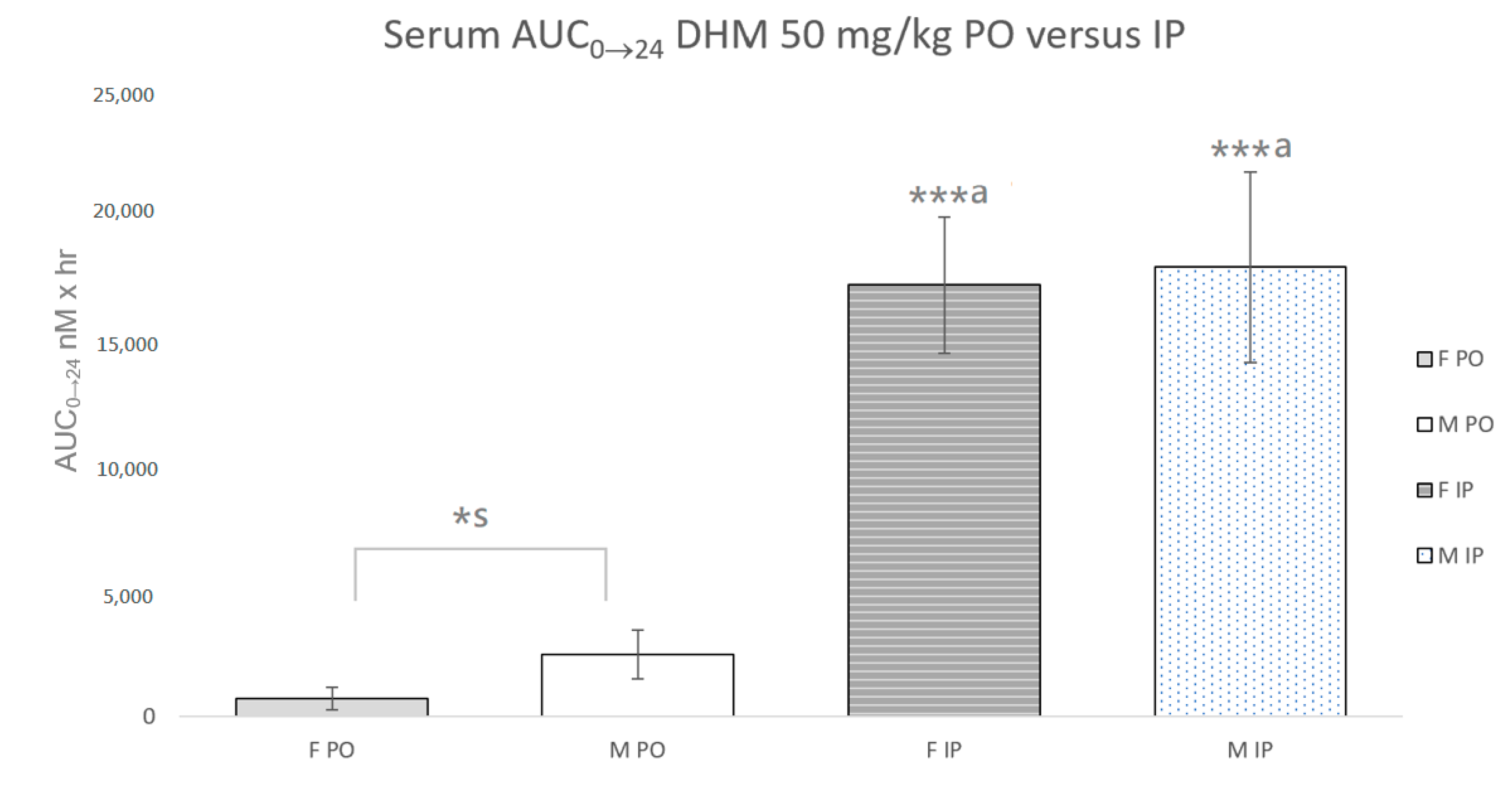
2.2. Quantification of DHM in Serum and Brain Samples
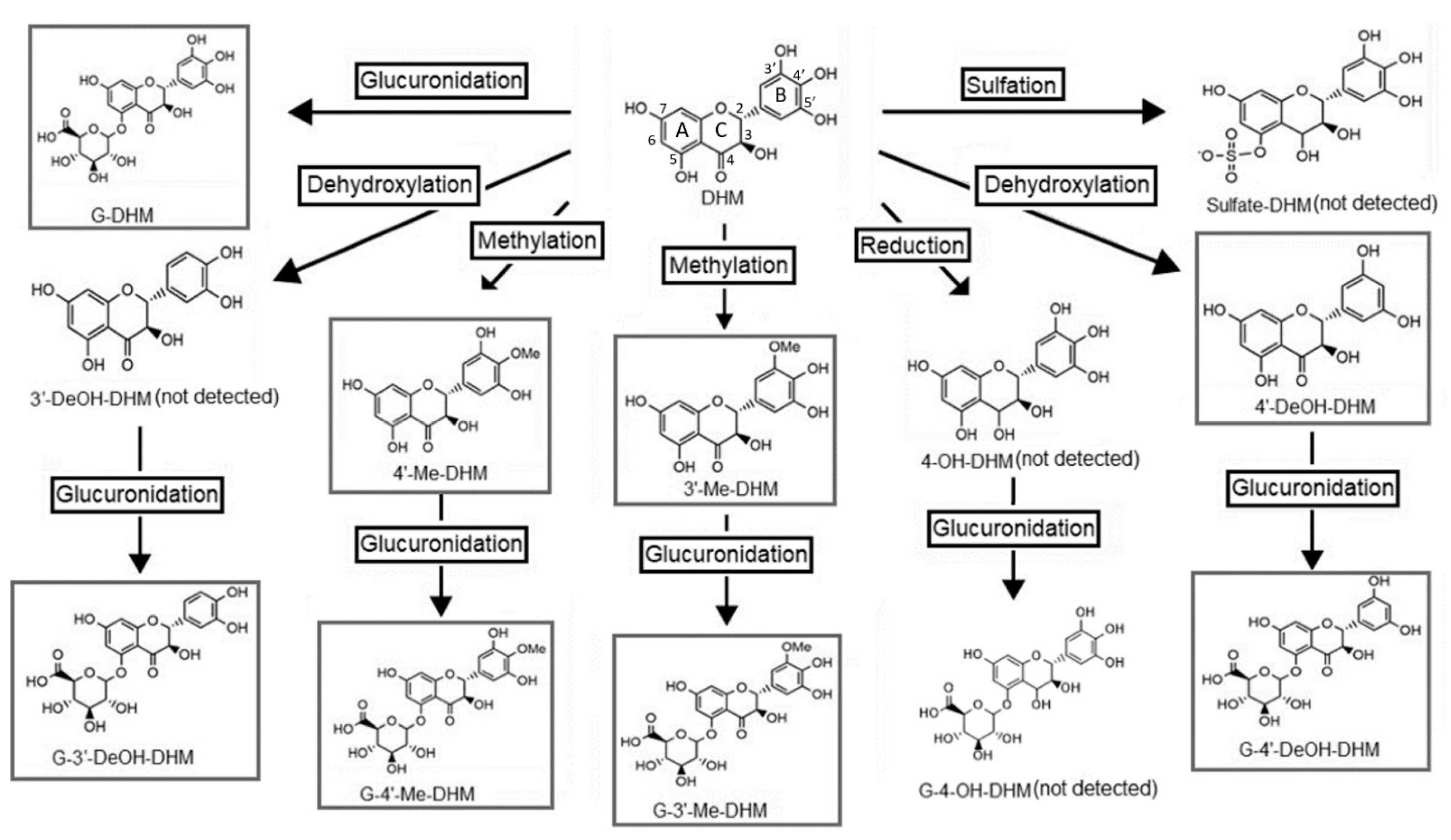
2.3. Identification of Dihydromyricetin Metabolites
2.4. Intrinsic Activity in α5β3γ2 GABAA Receptors Expressed in Xenopus Oocytes
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animal Studies
4.3. Bioanalysis of Serum and Brain Samples
4.4. Instrumentation and Conditions
4.5. Identification of DHM and Metabolites in Serum and Brain Samples
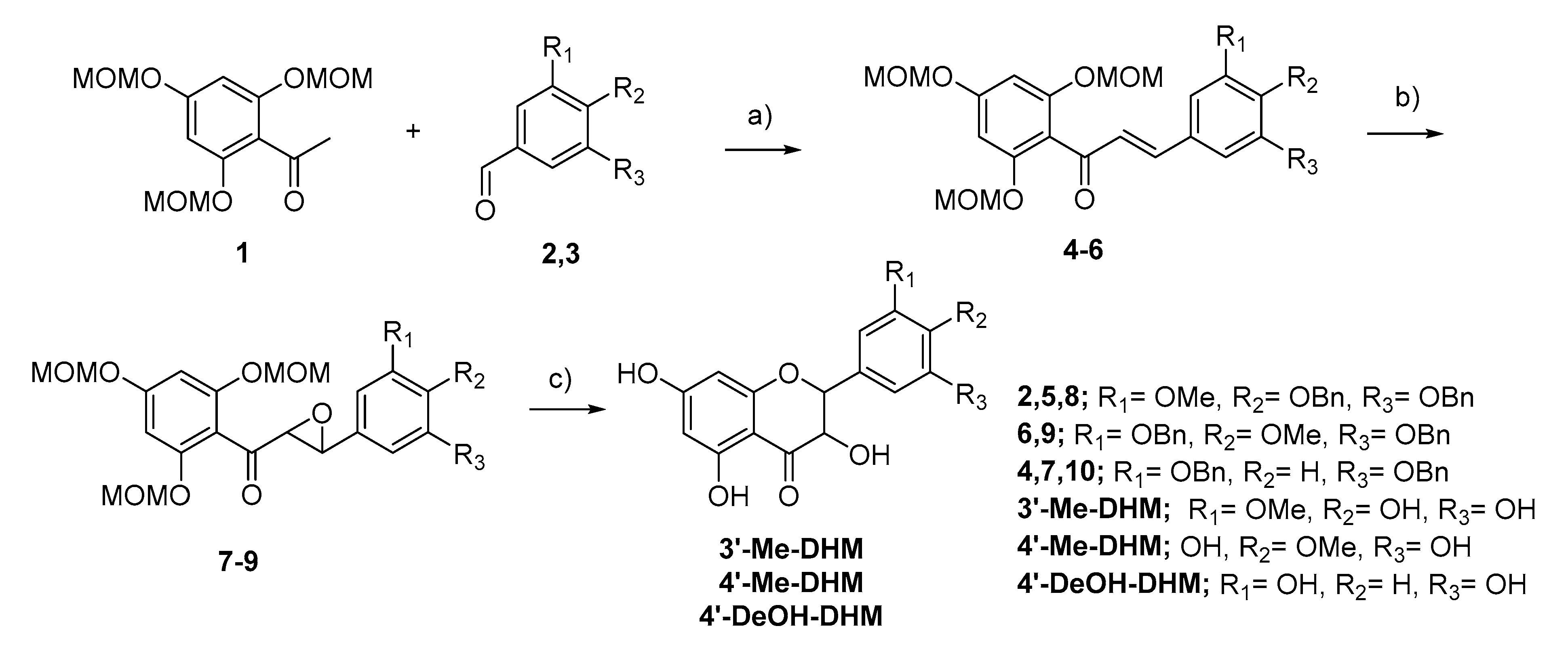
4.6. Synthesis and Identification of DHM Metabolites
4.7. Xenopus Oocytes Preparation
4.8. Electrophysiology Studies
4.9. Pharmacokinetic and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. Alcohol: Fact Sheet. 2018. Available online: https://www.who.int/en/news-room/fact-sheets/detail/alcohol (accessed on 9 June 2020).
- Litten, R.Z.; Egli, M.; Heilig, M.; Cui, C.; Fertig, J.B.; Ryan, M.L.; Falk, D.E.; Moss, H.; Huebner, R.; Noronha, A. Medications development to treat alcohol dependence: A vision for the next decade. Addict. Biol. 2012, 17, 513–527. [Google Scholar] [CrossRef] [Green Version]
- Litten, R.Z.; Wilford, B.B.; Falk, D.E.; Ryan, M.L.; Fertig, J.B. Potential medications for the treatment of alcohol use disorder: An evaluation of clinical efficacy and safety. Subst. Abus. 2016, 37, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Tanum, L.; Solli, K.K.; Benth, J.Š.; Opheim, A.; Sharma-Haase, K.; Krajci, P.; Kunøe, N. Effectiveness of injectable extended-release naltrexone vs daily buprenorphine-naloxone for opioid dependence: A randomized clinical noninferiority trial. JAMA Psychiatry 2017, 74, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, B.P.; Holtyn, A.F.; Subramaniam, S.; Tompkins, D.A.; Oga, E.A.; Bigelow, G.E.; Silverman, K.J.A. Extended-release injectable naltrexone for opioid use disorder: A systematic review. Addiction 2018, 113, 1188–1209. [Google Scholar] [CrossRef]
- Clark, A.K.; Wilder, C.M.; Winstanley, E.L. A systematic review of community opioid overdose prevention and naloxone distribution programs. J. Addict. Med. 2014, 8, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.E.; Witt, R.L. Respiratory effects of ethyl alcohol intoxication. JAMA 1972, 222, 486. [Google Scholar] [CrossRef]
- Wilson, K.C.; Saukkonen, J.J. Acute respiratory failure from abused substances. J. Intensive Care Med. 2004, 19, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Kerensky, T.; Walley, A.Y. Opioid overdose prevention and naloxone rescue kits: What we know and what we don’t know. Addict. Sci. Clin. Pract. 2017, 12, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Enoch, M.-A. The role of GABAA receptors in the development of alcoholism. Pharmacol. Biochem. Behav. 2008, 90, 95–104. [Google Scholar] [CrossRef] [Green Version]
- Olsen, R.W.; Liang, J. Role of GABA A receptors in alcohol use disorders suggested by chronic intermittent ethanol (CIE) rodent model. Mol. Brain 2017, 10, 45. [Google Scholar] [CrossRef]
- Liang, J.; Olsen, R.W. Alcohol use disorders and current pharmacological therapies: The role of GABA A receptors. Acta Pharmacol. Sin. 2014, 35, 981–993. [Google Scholar] [CrossRef] [Green Version]
- Olsen, R.W.; Hanchar, H.J.; Meera, P.; Wallner, M. GABAA receptor subtypes: The “one glass of wine” receptors. Alcohol 2007, 41, 201–209. [Google Scholar] [CrossRef] [Green Version]
- Koob, G.F. A role for GABA mechanisms in the motivational effects of alcohol. Biochem. Pharmacol. 2004, 68, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Wick, J.Y. The history of benzodiazepines. Consult Pharm. 2013, 28, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Lindemeyer, A.K.; Liang, J.; Marty, V.N.; Meyer, E.M.; Suryanarayanan, A.; Olsen, R.W.; Spigelman, I. Ethanol-Induced Plasticity of GABAA Receptors in the Basolateral Amygdala. Neurochem. Res. 2014, 39, 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- Naito, A.; Muchhala, K.H.; Asatryan, L.; Trudell, J.R.; Homanics, G.E.; Perkins, D.I.; Davies, D.L.; Alkana, R.L. Glycine and GABAA ultra-sensitive ethanol receptors as novel tools for alcohol and brain research. Mol. Pharmacol. 2014, 86, 635–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindemeyer, A.K.; Shen, Y.; Yazdani, F.; Shao, X.M.; Spigelman, I.; Davies, D.L.; Olsen, R.W.; Liang, J. α2 Subunit–Containing GABAA Receptor Subtypes Are Upregulated and Contribute to Alcohol-Induced Functional Plasticity in the Rat Hippocampus. Mol. Pharmacol. 2017, 92, 101–112. [Google Scholar]
- Amato, L.; Minozzi, S.; Vecchi, S.; Davoli, M. Benzodiazepines for alcohol withdrawal. Cochrane Database Syst. Rev. 2010, 3. [Google Scholar] [CrossRef]
- Khan, U.; Wilson, A. Alcohol Withdrawal, in Surgical Critical Care Therapy; Springer: New York, NY, USA, 2018; pp. 53–60. [Google Scholar]
- Li, H.; Li, Q.; Liu, Z.; Yang, K.; Chen, Z.; Cheng, Q.; Wu, L. The versatile effects of dihydromyricetin in health. Evid. Based Complement. Alternat. Med. 2017, 2017, 1053617. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Lindemeyer, A.K.; Gonzalez, C.; Shao, X.M.; Spigelman, I.; Olsen, R.W.; Liang, J. Dihydromyricetin as a novel anti-alcohol intoxication medication. J. Neurosci. 2012, 32, 390–401. [Google Scholar] [CrossRef] [Green Version]
- Silva, J.; Yu, X.; Qi, L.; Davies, D.L.; Liang, J.J.N.P.C. Antialcohol Effects of Dihydromyricetin in Combination with Other Flavonoids. Nat. Prod. Commun. 2020, 15, 1934578X20946250. [Google Scholar] [CrossRef]
- Liang, J.; Shen, Y.; Shao, X.M.; Scott, M.B.; Ly, E.; Wong, S.; Nguyen, A.; Tan, K.; Kwon, B.; Olsen, R.W.J.N.r. Dihydromyricetin prevents fetal alcohol exposure-induced behavioral and physiological deficits: The roles of GABA A receptors in adolescence. Neurochem. Res. 2014, 39, 1147–1161. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Yin, X.; Wang, X.; Li, X. Determination of dihydromyricetin in rat plasma by LC-MS/MS and its application to a pharmacokinetic study. Pharm. Biol. 2017, 55, 657–662. [Google Scholar] [CrossRef] [Green Version]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- Hitchcock, S.A.; Pennington, L.D. Structure–brain exposure relationships. J. Med. Chem. 2006, 49, 7559–7583. [Google Scholar] [CrossRef]
- Liu, L.; Sun, S.; Rui, H.; Li, X. In vitro inhibitory effects of dihydromyricetin on human liver cytochrome P450 enzymes. Pharm. Biol. 2017, 55, 1868–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, J.; Liu, J.; Chen, T.-M.; Lan, Q.; Zhang, Q.-Y.; Liu, B.; Dai, D.; Zhang, W.-D.; Hu, L.-P.; Zhu, R.-Z. Dihydromyricetin alleviates carbon tetrachloride-induced acute liver injury via JNK-dependent mechanism in mice. World J. Gastroenterol. 2015, 21, 5473. [Google Scholar] [CrossRef] [PubMed]
- Williamson, G.; Kay, C.D.; Crozier, A.; Safety, F. The bioavailability, transport, and bioactivity of dietary flavonoids: A review from a historical perspective. Compr. Rev. Food Sci. Food Saf. 2018, 17, 1054–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, L.; Tong, Q.; Dong, W.; Yang, G.; Hou, X.; Xiong, W.; Shi, C.; Fang, J.; Wang, W. Tissue Distribution, excretion, and metabolic profile of dihydromyricetin, a flavonoid from vine tea (Ampelopsis grossedentata) after oral administration in rats. J. Agric. Food Chem. 2017, 65, 4597–4604. [Google Scholar] [CrossRef]
- Zhang, Y.; Que, S.; Yang, X.; Wang, B.; Qiao, L.; Zhao, Y. Isolation and identification of metabolites from dihydromyricetin. Magn. Reson. Chem. 2007, 45, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Xiang, D.; Fan, L.; Hou, X.-l.; Xiong, W.; Shi, C.-y.; Wang, W.-q.; Fang, J.-g. Uptake and Transport Mechanism of Dihydromyricetin Across Human Intestinal Caco-2 Cells. J. Food Sci. 2018, 83, 1941–1947. [Google Scholar] [CrossRef]
- Xiang, D.; Wang, C.-g.; Wang, W.-q.; Shi, C.-y.; Xiong, W.; Wang, M.-d.; Fang, J.-g. Gastrointestinal stability of dihydromyricetin, myricetin, and myricitrin: An in vitro investigation. Int. J. Food Sci. Nutr. 2017, 68, 704–711. [Google Scholar] [CrossRef]
- Huang, Y.; Zhao, J.; Jian, W.; Wang, G. Effects of verapamil on the pharmacokinetics of dihydromyricetin in rats and its potential mechanism. Xenobiotica 2018, 48, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.; Carry, E.; Xue, C.; Zhang, J.; Liang, J.; Roberge, J.Y.; Davies, D.L.J.M. A Novel Dual Drug Approach That Combines Ivermectin and Dihydromyricetin (DHM) to Reduce Alcohol Drinking and Preference in Mice. Molecules 2021, 26, 1791. [Google Scholar] [CrossRef] [PubMed]
- Wong, I.L.; Wang, B.C.; Yuan, J.; Duan, L.X.; Liu, Z.; Liu, T.; Li, X.M.; Hu, X.; Zhang, X.Y.; Jiang, T. Potent and Nontoxic Chemosensitizer of P-Glycoprotein-Mediated Multidrug Resistance in Cancer: Synthesis and Evaluation of Methylated Epigallocatechin, Gallocatechin, and Dihydromyricetin Derivatives. J. Med. Chem. 2015, 58, 4529–4549. [Google Scholar] [CrossRef]
- Meibohm, B.; Beierle, I.; Derendorf, H. How important are gender differences in pharmacokinetics? Clin. Pharmacokinet. 2002, 41, 329–342. [Google Scholar] [CrossRef]
- Fröhlich, M.; Albermann, N.; Sauer, A.; Walter-Sack, I.; Haefeli, W.E.; Weiss, J. In vitro and ex vivo evidence for modulation of P-glycoprotein activity by progestins. Biochem. Pharmacol. 2004, 68, 2409–2416. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Walle, T. Methylated flavonoids have greatly improved intestinal absorption and metabolic stability. Drug Metab. Dispos. 2006, 34, 1786–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Liu, T.; Gu, J.; Luo, X.; Ji, R.; Cao, Y.; Xue, H.; Wong, J.T.-F.; Wong, B.L.; Pei, G. 3D-QSAR model of flavonoids binding at benzodiazepine site in GABAA receptors. J. Med. Chem. 2001, 44, 1883–1891. [Google Scholar] [CrossRef]
- Duchowicz, P.R.; Vitale, M.G.; Castro, E.A.; Autino, J.C.; Romanelli, G.P.; Bennardi, D.O. QSAR modeling of the interaction of flavonoids with GABA (A) receptor. Eur. J. Med. Chem. 2008, 43, 1593–1602. [Google Scholar] [CrossRef]
- Hanrahan, J.R.; Chebib, M.; Johnston, G.A. Flavonoid modulation of GABAA receptors. Br. J. Pharmacol. 2011, 163, 234–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deeb, O.; Shaik, B.; Agrawal, V.K. Exploring QSARs of the interaction of flavonoids with GABA (A) receptor using MLR, ANN and SVM techniques. J. Enzym. Inhib. Med. Chem. 2014, 29, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.; Yu, X.; Moradian, R.; Folk, C.; Spatz, M.H.; Kim, P.; Bhatti, A.A.; Davies, D.L.; Liang, J. Dihydromyricetin Protects the Liver via Changes in Lipid Metabolism and Enhanced Ethanol Metabolism. Alcohol. Clin. Exp. Res. 2020, 44, 1046–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, E.J.; Kwon, H.C.; Sohn, Y.C.; Nam, C.W.; Park, H.B.; Kim, C.Y.; Yang, H.O. Four flavonoids from Echinosophora koreensis and their effects on alcohol metabolizing enzymes. Arch. Pharm. Res. 2009, 32, 851–855. [Google Scholar] [CrossRef] [PubMed]
- Skotnicova, A.; Boubinova, G.; Boštíková, Z.; Dušková, Š.; šulc, M.; Kutinova-Canova, N.; Mraz, J.; Hodek, P.J.P.R. Does Dihydromyricetin Impact on Alcohol Metabolism. Physiol. Res. 2020, 69 (Suppl. 4), S573–S581. [Google Scholar] [CrossRef]
- Ponomarev, I.; Crabbe, J.C. A novel method to assess initial sensitivity and acute functional tolerance to hypnotic effects of ethanol. J. Pharmacol. Exp. Ther. 2002, 302, 257–263. [Google Scholar] [CrossRef] [Green Version]
- Cayla, N.S.; Dagne, B.A.; Wu, Y.; Lu, Y.; Rodriguez, L.; Davies, D.L.; Gross, E.R.; Heifets, B.D.; Davies, M.F.; MacIver, M.B. A newly developed anesthetic based on a unique chemical core. Proc. Natl. Acad. Sci. USA 2019, 116, 15706–15715. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| F PO | M PO | F IP | M IP | ||
|---|---|---|---|---|---|
| Serum | T1/2 (h) | 1.83 | 1.91 | 0.76 | 1.59 |
| Cmax (nM) | 623 ± 159 | 1885 ± 590 | 23,777 ± 1998 | 38,251 ± 7441 | |
| AUC0→24 (nM × h) | 728 ± 461 | 2508 ± 988 | 17,360 ± 2734 | 18,079 ± 3840 | |
| Tmax (min) | 15 | 15 | 15 | 15 | |
| Brain | Concentration (nmol/g) 15 min post DHM IP | 50.9 ± 2.6 | 423 ± 66 | 864 ± 220 | 672 ± 85 |
| DHM | 4′-Me-DHM | 3′-Me-DHM | 4-DeOH-DHM | |
|---|---|---|---|---|
| GABAAR Potentiation | +43.2 ± 5.6 ** | Not Active | −12.6 ± 1.5 * | +5.3 ± 2.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carry, E.; Kshatriya, D.; Silva, J.; Davies, D.L.; Yuan, B.; Wu, Q.; Patel, H.; Park, E.R.; Gilleran, J.; Hao, L.; et al. Identification of Dihydromyricetin and Metabolites in Serum and Brain Associated with Acute Anti-Ethanol Intoxicating Effects in Mice. Int. J. Mol. Sci. 2021, 22, 7460. https://doi.org/10.3390/ijms22147460
Carry E, Kshatriya D, Silva J, Davies DL, Yuan B, Wu Q, Patel H, Park ER, Gilleran J, Hao L, et al. Identification of Dihydromyricetin and Metabolites in Serum and Brain Associated with Acute Anti-Ethanol Intoxicating Effects in Mice. International Journal of Molecular Sciences. 2021; 22(14):7460. https://doi.org/10.3390/ijms22147460
Chicago/Turabian StyleCarry, Eileen, Dushyant Kshatriya, Joshua Silva, Daryl L. Davies, Bo Yuan, Qingli Wu, Harna Patel, Elizabeth R. Park, John Gilleran, Lihong Hao, and et al. 2021. "Identification of Dihydromyricetin and Metabolites in Serum and Brain Associated with Acute Anti-Ethanol Intoxicating Effects in Mice" International Journal of Molecular Sciences 22, no. 14: 7460. https://doi.org/10.3390/ijms22147460
APA StyleCarry, E., Kshatriya, D., Silva, J., Davies, D. L., Yuan, B., Wu, Q., Patel, H., Park, E. R., Gilleran, J., Hao, L., Roberge, J., Bello, N. T., & Simon, J. E. (2021). Identification of Dihydromyricetin and Metabolites in Serum and Brain Associated with Acute Anti-Ethanol Intoxicating Effects in Mice. International Journal of Molecular Sciences, 22(14), 7460. https://doi.org/10.3390/ijms22147460