
Myocardial Tissue Characterization in Heart Failure with Preserved Ejection Fraction: From Histopathology and Cardiac Magnetic Resonance Findings to Therapeutic Targets

 , ,
, , 
 ,
,
Abstract
:1. Introduction
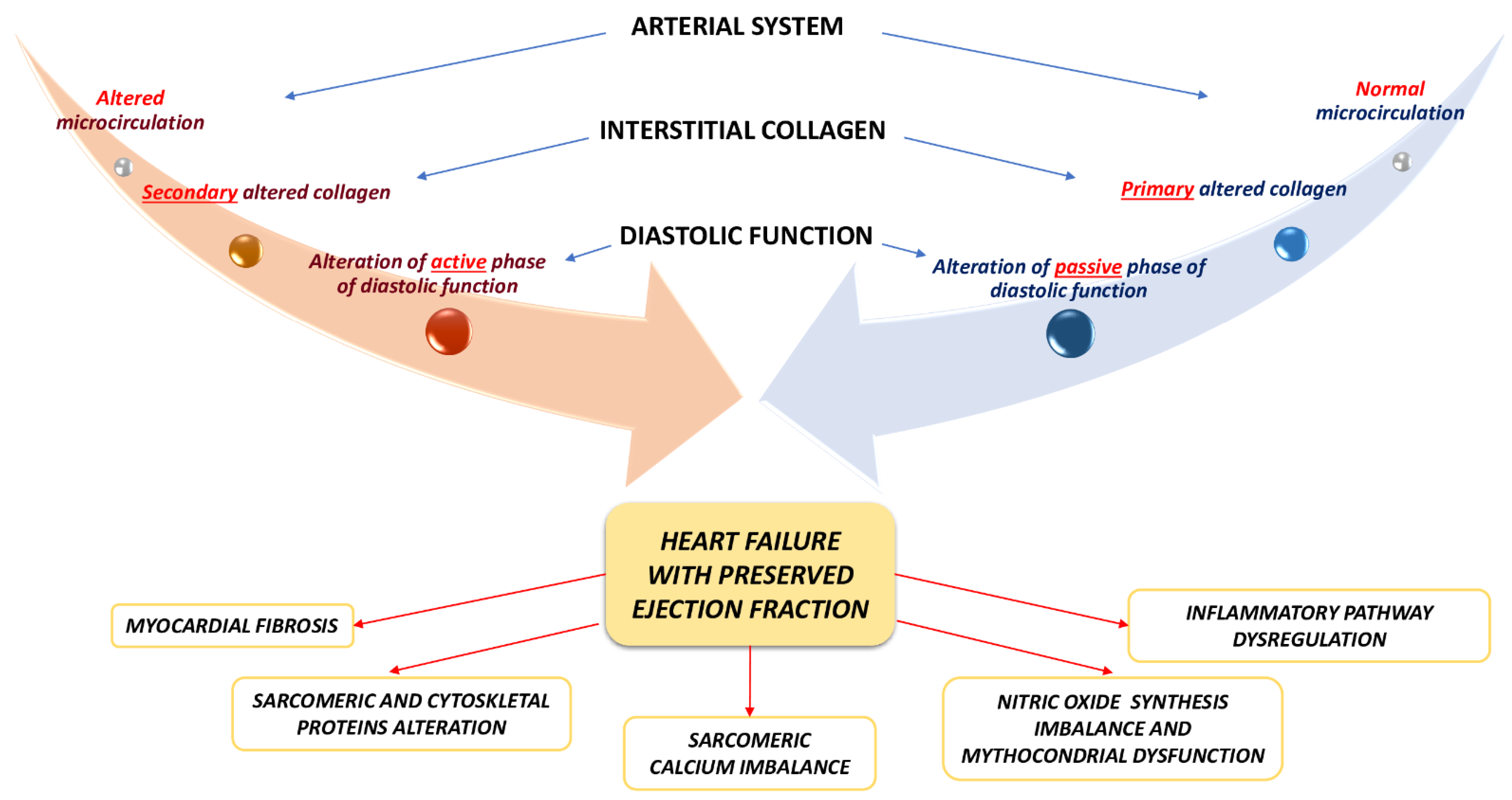
2. The Pathophysiology of Heart Failure with Preserved Ejection Fraction
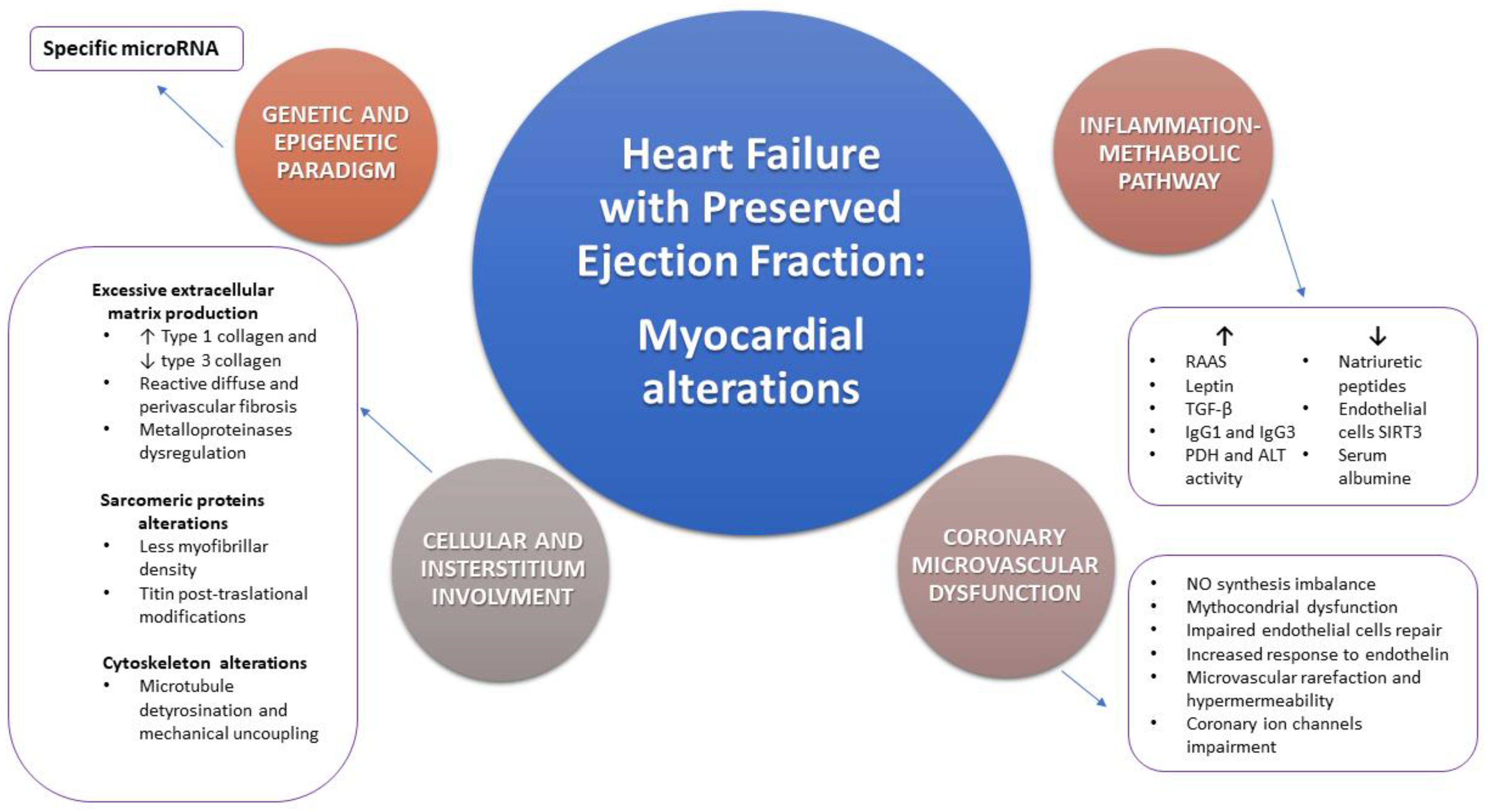
2.1. Heart Failure with Preserved Ejection Fraction and Myocardial Morphological Alterations: Cellular and Interstitial Involvement
2.2. Heart Failure with Preserved Ejection Fraction and Coronary Microvascular Dysfunction: Morphological and Functional Aspects
2.3. Heart Failure with Preserved Ejection Fraction and the Inflammation-Metabolic Pathway
3. Precision Medicine and Personalized Approach to Heart Failure with Preserved Ejection Fraction: The Genetic and Epigenetic Paradigm
4. Role of Myocardial Tissue Characterization and Pathophysiological Mechanisms for the Identification of New Therapeutic Targets
4.1. The Renin-Angiotensin-Aldosterone System and Neprilysin Pathway
4.2. The Oxidative Stress and the Nitric Oxide Pathway
4.3. Role of Inflammation, Fibrosis and Calcium Handling as Therapeutic Targets
4.4. The Heart Rate and Volemic Status Regulation as Therapeutic Targets
5. Conclusions
Funding
Conflicts of Interest
References
- Van Riet, E.E.S.; Hoes, A.W.; Limburg, A.; Landman, M.A.J.; Van der Hoeven, H.; Rutten, F.H. Prevalence of unrecognized heart failure in older persons with shortness of breath on exertion. Eur. J. Heart Fail. 2014, 16, 772–777. [Google Scholar] [CrossRef]
- Van Riet, E.E.S.; Hoes, A.W.; Wagenaar, K.P.; Limburg, A.; Landman, M.A.J.; Rutten, F.H. Epidemiology of heart failure: The prevalence of heart failure and ventricular dysfunction in older adults over time. A systematic review. Eur. J. Heart Fail. 2016, 18, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Mureddu, G.F.; Agabiti, N.; Rizzello, V.; Forastiere, F.; Latini, R.; Cesaroni, G.; Masson, S.; Cacciatore, G.; Colivicchi, F.; Uguccioni, M.; et al. Prevalence of preclinical and clinical heart failure in the elderly. A population-based study in Central Italy. Eur. J. Heart Fail. 2012, 14, 718–729. [Google Scholar] [CrossRef]
- Ponikowski, P.; Voors, A.A.; Anker, S.; Bueno, H.; Cleland, G.F.J.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [CrossRef] [PubMed]
- Gerber, Y.; Weston, S.A.; Redfield, M.M.; Chamberlain, A.M.; Manemann, S.M.; Jiang, R.; Killian, J.M.; Roger, V.L. A contemporary appraisal of the heart failure epidemic in Olmsted County, Minnesota, 2000 to 2010. JAMA Intern Med. 2015, 175, 996–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meta-analysis Global Group in Chronic Heart Failure (MAGGIC). The survival of patients with heart failure with preserved or reduced left ventricular ejection fraction: An individual patient data meta-analysis. Eur. Heart J. 2012, 33, 1750–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borlaug, B.A. The pathophysiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 2014, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Pieske, B.; Tschöpe, C.; De Boer, R.A.; Fraser, A.G.; Anker, S.D.; Donal, E.; Edelmann, F.; Fu, M.; Guazzi, M.; Lam, C.S.P.; et al. How to diagnose heart failure with preserved ejection fraction: The HFA-PEFF diagnostic algorithm: A consensus recommendation from the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur. Heart J. 2019, 40, 3297–3317. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, M.A.; Shah, A.M.; Borlaug, B.A. Heart Failure with Preserved Ejection Fraction In Perspective. Circ. Res. 2019, 124, 1598–1617. [Google Scholar] [CrossRef]
- Dunlay, S.M.; Roger, V.L.; Redfield, M.M. Epidemiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 2017, 14, 591–602. [Google Scholar] [CrossRef]
- Vaduganathan, M.; Patel, R.B.; Michel, A.; Shah, S.J.; Senni, M.; Gheorghiade, M.; Butler, J. Mode of Death in Heart Failure With Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2017, 69, 556–569. [Google Scholar] [CrossRef] [PubMed]
- Maggioni, A.P.; Dahlström, U.; Filippatos, G.; Chioncel, O.; Leiro, M.C.; Drozdz, J.; Fruhwald, F.; Gullestad, L.; Logeart, D.; Fabbri, G.; et al. EURObservational Research Programme: Regional differences and 1-year follow-up results of the Heart Failure Pilot Survey (ESC-HF Pilot). Eur. J. Heart Fail. 2013, 15, 808–817. [Google Scholar] [CrossRef] [Green Version]
- Fonarow, G.C.; Gattis Stough, W.; Abraham, W.T.; Albert, N.M.; Gheorghiade, M.; Greenberg, B.H.; O’Connor, C.M.; Sun, J.-L.; Clyde, W.; Yancy, C.W.; et al. Characteristics, treatments, and outcomes of patients with preserved systolic function hospitalized for heart failure: A report from the OPTIMIZE-HF registry. J. Am. Coll. Cardiol. 2007, 50, 768–777. [Google Scholar] [CrossRef] [Green Version]
- Obokata, M.; Reddy, Y.N.V.; Borlaug, B.A. Diastolic dysfunction and heart failure with preserved ejection fraction: Understanding mechanisms by using noninvasive methods. J. Am. Coll. Cardiol. Imaging 2020, 13, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Paulus, W.J.; Tschope, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tschope, C.; Paulus, W.J. Is echocardiographic evaluation of diastolic function useful in determining clinical care? Doppler echocardiography yields dubious estimates of left ventricular diastolic pressures. Circulation 2009, 120, 810–820. [Google Scholar] [CrossRef]
- Westermann, D.; Kasner, M.; Steendijk, P.; Spillmann, F.; Riad, A.; Weitmann, K.; Hoffmann, W.; Poller, W.; Pauschinger, M.; Schultheiss, H.-P.; et al. Role of left ventricular stiffness in heart failure with normal ejection fraction. Circulation 2008, 117, 2051–2060. [Google Scholar] [CrossRef] [PubMed]
- Treibel, T.A.; White, S.K.; Moon, J.C. Myocardial Tissue Characterization: Histological and Pathophysiological Correlation. Curr. Cardiovasc. Imaging Rep. 2014, 7, 9254. [Google Scholar] [CrossRef] [Green Version]
- Moon, J.C.; Messroghli, D.R.; Kellman, P.; Piechnik, S.K.; Robson, M.D.; Ugander, M.; Gatehouse, P.D.; Arai, A.E.; Friedrich, M.G.; Neubauer, S.; et al. Myocardial T1 mapping and extracellular volume quantification: A Society for Cardiovascular Magnetic Resonance (SCMR) and CMR Working Group of the European Society of Cardiology consensus statement. J. Cardiovasc. Magn. Reson. 2013, 15, 92. [Google Scholar] [CrossRef] [Green Version]
- Flett, A.S.; Hayward, M.P.; Ashworth, M.T.; Hansen, M.S.; Taylor, A.M.; Elliott, P.M.; McGregor, C.; Moon, J.C. Equilibrium contrast cardiovascular magnetic resonance for the measurement of diffuse myocardial fibrosis: Preliminary validation in humans. Circulation 2010, 122, 138–144. [Google Scholar] [CrossRef]
- Pan, J.A.; Kerwin, M.J.; Salerno, M. Native T1 Mapping, Extracellular Volume Mapping, and Late Gadolinium Enhancement in Cardiac Amyloidosis: A Meta-Analysis. JACC Cardiovasc. Imaging 2020, 13, 1299–1310. [Google Scholar] [CrossRef]
- Diao, K.Y.; Yang, Z.G.; Xu, H.Y.; Liu, X.; Zhang, Q.; Shi, K.; Jiang, L.; Xie, L.J.; Wen, L.Y.; Guo, Y.K. Histologic validation of myocardial fibrosis measured by T1 mapping: A systematic review and meta-analysis. J. Cardiovasc. Magn. Reson. 2016, 18, 92. [Google Scholar] [CrossRef] [Green Version]
- Bull, S.; White, S.K.; Piechnik, S.K.; Flett, A.S.; Ferreira, V.M.; Loudon, M.; Francis, J.M.; Karamitsos, T.D.; Prendergast, B.D.; Robson, M.D.; et al. Human non-contrast T1 values and correlation with histology in diffuse fibrosis. Heart 2013, 99, 932–937. [Google Scholar] [CrossRef] [Green Version]
- Quarta, G.; Gori, M.; Iorio, A.; D’Elia, E.; Moon, J.C.; Iacovoni, A.; Burocchi, S.; Schelbert, E.B.; Brambilla, P.; Sironi, S.; et al. Cardiac magnetic resonance in heart failure with preserved ejection fraction: Myocyte, interstitium, microvascular, and metabolic abnormalities. Eur. J. Heart Fail. 2020, 22, 1065–1075. [Google Scholar] [CrossRef]
- Lam, C.S.; Roger, V.L.; Rodeheffer, R.J.; Bursi, F.; Borlaug, B.A.; Ommen, S.R.; Kass, D.A.; Redfield, M.M. Cardiac structure and ventricular-vascular function in persons with heart failure and preserved ejection fraction from Olmsted County, Minnesota. Circulation 2007, 115, 1982–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maestrini, V.; Treibel, T.A.; White, S.K.; Fontana, M.; Moon, J.C. T1 Mapping for Characterization of Intracellular and Extracellular Myocardial Diseases in Heart Failure. Curr. Cardiovasc. Imaging Rep. 2014, 7, 9287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sado, D.M.; Maestrini, V.; Piechnik, S.K.; Banypersad, S.M.; White, S.K.; Flett, A.S.; Robson, M.D.; Neubauer, S.; Ariti, C.; Arai, A.; et al. Noncontrast myocardial T1 mapping using cardiovascular magnetic resonance for iron overload. J. Magn. Reson. Imaging 2015, 41, 1505–1511. [Google Scholar] [CrossRef] [PubMed]
- Karamitsos, T.D.; Piechnik, S.K.; Banypersad, S.M.; Fontana, M.; Ntusi, N.B.; Ferreira, V.M.; Whelan, C.J.; Myerson, S.G.; Robson, M.D.; Hawkins, P.N.; et al. Noncontrast T1 mapping for the diagnosis of cardiac amyloidosis. JACC Cardiovasc. Imaging 2013, 6, 488–497. [Google Scholar] [CrossRef] [Green Version]
- Severino, P.; Maestrini, V.; Mariani, M.V.; Birtolo, L.I.; Scarpati, R.; Mancone, M.; Fedele, F. Structural and myocardial dysfunction in heart failure beyond ejection fraction. Heart Fail. Rev. 2020, 25, 9–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedele, F.; Mancone, M.; Adamo, F.; Severino, P. Heart Failure With Preserved, Mid-Range, and Reduced Ejection Fraction: The Misleading Definition of the New Guidelines. Cardiol. Rev. 2017, 25, 4–5. [Google Scholar] [CrossRef] [Green Version]
- Severino, P.; Mariani, M.V.; Fedele, F. Futility in cardiology: The need for a change in perspectives. Eur. J. Heart Fail. 2019, 21, 1483–1484. [Google Scholar] [CrossRef] [PubMed]
- Fedele, F.; Severino, P.; Calcagno, S.; Mancone, M. Heart failure: TNM-like classification. J. Am. Coll. Cardiol. 2014, 63, 1959–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Severino, P.; Mather, P.J.; Pucci, M.; D’Amato, A.; Mariani, M.V.; Infusino, F.; Birtolo, L.I.; Maestrini, V.; Mancone, M.; Fedele, F. Advanced Heart Failure and End-Stage Heart Failure: Does a Difference Exist. Diagnostics (Basel) 2019, 9, 170. [Google Scholar] [CrossRef] [Green Version]
- Severino, P.; D’Amato, A.; Saglietto, A.; D’Ascenzo, F.; Marini, C.; Schiavone, M.; Ghionzoli, N.; Pirrotta, F.; Troiano, F.; Cannillo, M.; et al. Reduction in heart failure hospitalization rate during coronavirus disease 19 pandemic outbreak. ESC Heart Fail. 2020, 7, 4182–4188. [Google Scholar] [CrossRef]
- Hayashi, T.; Yamada, S.; Iwano, H.; Nakabachi, M.; Sakakibara, M.; Okada, K.; Murai, D.; Nishino, H.; Kusunose, K.; Watanabe, K.; et al. Left ventricular global strain for estimating relaxation and filling pressure—A multicenter study. Circ. J. 2016, 80, 1163–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, H.; Ishida, M.; Makino, W.; Goto, Y.; Ichikawa, Y.; Kitagawa, K.; Omori, T.; Dohi, K.; Ito, M.; Sakuma, H. Cardiovascular magnetic resonance feature tracking for characterization of patients with heart failure with preserved ejection fraction: Correlation of global longitudinal strain with invasive diastolic functional indices. J. Cardiovasc. Magn. Reson. 2020, 22, 42. [Google Scholar] [CrossRef] [PubMed]
- De Boer, R.A.; De Keulenaer, G.; Bauersachs, J.; Brutsaert, D.; Cleland, J.G.; Diez, J.; Du, X.J.; Ford, P.; Heinzel, F.R.; Lipson, K.E.; et al. Towards better definition, quantification and treatment of fibrosis in heart failure. A scientific roadmap by the Committee of Translational Research of the Heart Failure Association (HFA) of the European Society of Cardiology. Eur. J. Heart Fail. 2019, 21, 272–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, V.S.; Yanek, L.R.; Vaishnav, J.; Ying, W.; Vaidya, D.; Lee, Y.Z.J.; Riley, S.J.; Subramanya, V.; Brown, E.E.; Hopkins, C.D.; et al. Endomyocardial Biopsy Characterization of Heart Failure with Preserved Ejection Fraction and Prevalence of Cardiac Amyloidosis. JACC Heart Fail. 2020, 8, 712–724. [Google Scholar] [CrossRef]
- Russell, S.B.; Smith, J.C.; Huang, M.; Trupin, J.S.; Williams, S.M. Pleiotropic Effects of Immune Responses Explain Variation in the Prevalence of Fibroproliferative Diseases. PLoS Genet 2015, 11, e1005568. [Google Scholar] [CrossRef] [Green Version]
- Hellwege, J.N.; Torstenson, E.S.; Russell, S.B.; Edwards, T.L.; Velez Edwards, D.R. Evidence of selection as a cause for racial disparities in fibroproliferative disease. PLoS ONE 2017, 12, e0182791. [Google Scholar] [CrossRef] [Green Version]
- Lavalle, C.; Mariani, M.V.; Piro, A.; Straito, M.; Severino, P.; Della Rocca, D.G.; Forleo, G.B.; Romero, J.; Di Biase, L.; Fedele, F. Electrocardiographic features, mapping and ablation of idiopathic outflow tract ventricular arrhythmias. J. Interv. Card. Electrophysiol. 2020, 57, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Piro, A.; Magnocavallo, M.; Della Rocca, D.G.; Neccia, M.; Manzi, G.; Mariani, M.V.; Straito, M.; Bernardini, A.; Severino, P.; Iannucci, G.; et al. Management of cardiac implantable electronic device follow-up in COVID-19 pandemic: Lessons learned during Italian lockdown. J. Cardiovasc. Electrophysiol. 2020, 31, 2814–2823. [Google Scholar] [CrossRef] [PubMed]
- Cuijpers, I.; Simmonds, S.J.; Van Bilsen, M.; Czarnowska, E.; González Miqueo, A.; Heymans, S.; Kuhn, A.R.; Mulder, P.; Ratajska, A.; Jones, E.A.V.; et al. Microvascular and lymphatic dysfunction in HFpEF and its associated comorbidities. Basic Res. Cardiol. 2020, 115, 39. [Google Scholar] [CrossRef] [PubMed]
- Michels da Silva, D.; Langer, H.; Graf, T. Inflammatory and molecular pathways in heart failure-ischemia, HFpEF and transthyretin cardiac amyloidosis. Int. J. Mol. Sci. 2019, 20, 2322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, A.; Schelbert, E.B.; Díez, J.; Butler, J. Myocardial Interstitial Fibrosis in Heart Failure: Biological and Translational Perspectives. J. Am. Coll. Cardiol. 2018, 71, 1696–1706. [Google Scholar] [CrossRef]
- Su, M.Y.; Lin, L.Y.; Tseng, Y.H.; Chang, C.C.; Wu, C.K.; Lin, J.L.; Tseng, W.Y. CMR-verified diffuse myocardial fibrosis is associated with diastolic dysfunction in HFpEF. JACC Cardiovasc. Imaging 2014, 7, 991–997. [Google Scholar] [CrossRef] [Green Version]
- Kanagala, P.; Cheng, A.S.H.; Singh, A.; Khan, J.N.; Gulsin, G.S.; Patel, P.; Gupta, P.; Arnold, J.R.; Squire, I.B.; Ng, L.L.; et al. Relationship between focal and diffuse fibrosis assessed by CMR and clinical outcomes in heart failure with preserved ejection fraction. J. Am. Coll. Cardiol. Imaging 2019, 11 Pt 2, 2291–2301. [Google Scholar] [CrossRef]
- Franssen, C.; González Miqueo, A. The role of titin and extracellular matrixremodeling in heart failure with preserved ejection fraction. Neth Heart J. 2016, 24, 259–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasner, M.; Westermann, D.; Lopez, B.; Gaub, R.; Escher, F.; Kuhl, U.; Schultheiss, H.P.; Tschope, C. Diastolic tissue Doppler indexes correlate with the degree of collagen expression and cross-linking in heart failure and normal ejection fraction. J. Am. Coll. Cardiol. 2011, 57, 977–985. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, S.F.; Hussain, S.; Mirzoyev, S.A.; Edwards, W.D.; Maleszewski, J.J.; Redfield, M.M. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation 2015, 131, 550–559. [Google Scholar] [CrossRef] [Green Version]
- Zile, M.R.; Baicu, C.F.; Ikonomidis, J.S.; Stroud, R.E.; Nietert, P.J.; Bradshaw, A.D.; Slater, R.; Palmer, B.M.; Van Buren, P.; Meyer, M.; et al. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: Contributions of collagen and titin. Circulation 2015, 131, 1247–1259. [Google Scholar] [CrossRef]
- Rodriguez, C.; Martinez-Gonzalez, J. The role of Lysyl oxidase enzymes in cardiac function and remodeling. Cells 2019, 8, 1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolajević Starčević, J.; Janić, M.; Šabovič, M. Molecular Mechanisms Responsible for Diastolic Dysfunction in Diabetes Mellitus Patients. Int. J. Mol. Sci. 2019, 20, 1197. [Google Scholar] [CrossRef] [Green Version]
- Nakamori, S.; Dohi, K.; Ishida, M.; Goto, Y.; Imanaka-Yoshida, K.; Omori, T.; Goto, I.; Kumagai, N.; Fujimoto, N.; Ichikawa, Y.; et al. Native T1 Mapping and Extracellular Volume Mapping for the Assessment of Diffuse Myocardial Fibrosis in Dilated Cardiomyopathy. JACC Cardiovasc. Imaging 2018, 11, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Rommel, K.P.; von Roeder, M.; Latuscynski, K.; Oberueck, C.; Blazek, S.; Fengler, K.; Besler, C.; Sandri, M.; Lücke, L.; Gutberlet, M.; et al. Extracellular volume fraction for characterization of patients with heart failure and preserved ejection fraction. J. Am. Coll. Cardiol. 2016, 67, 1815–1825. [Google Scholar] [CrossRef]
- Ruberg, F.L.; Berk, J.L. Transthyretin (TTR) cardiac amyloidosis. Circulation 2012, 126, 1286–1300. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Pavia, P.; Rapezzi, C.; Adler, Y.; Arad, M.; Basso, C.; Brucato, A.; Burazor, I.; Caforio, A.L.P.; Damy, T.; Eriksson, U.; et al. Diagnosis and treatment of cardiac amyloidosis: A position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2021, 42, 1554–1568. [Google Scholar] [CrossRef]
- Hawkins, P.N.; Ando, Y.; Dispenzeri, A.; Gonzalez-Duarte, A.; Adams, D.; Suhr, O.B. Evolving landscape in the management of transthyretin amyloidosis. Ann. Med. 2015, 47, 625–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treibel, T.A.; Fontana, M.; Gilbertson, J.A.; Castelletti, S.; White, S.K.; Scully, P.R.; Roberts, N.; Hutt, D.F.; Rowczenio, D.M.; Whelan, C.J.; et al. Occult Transthyretin Cardiac Amyloid in Severe Calcific Aortic Stenosis: Prevalence and Prognosis in Patients Undergoing Surgical Aortic Valve Replacement. Circ. Cardiovasc. Imaging 2016, 9, e005066. [Google Scholar] [CrossRef] [Green Version]
- Maceira, A.M.; Joshi, J.; Prasad, S.K.; Moon, J.C.; Perugini, E.; Harding, I.; Sheppard, M.N.; Poole-Wilson, P.A.; Hawkins, P.N.; Pennell, D.J. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation 2005, 111, 186–193. [Google Scholar] [CrossRef] [Green Version]
- Dungu, J.N.; Valencia, O.; Pinney, J.H.; Gibbs, S.D.; Rowczenio, D.; Gilbertson, J.A.; Lachmann, H.J.; Wechalekar, A.; Gillmore, J.D.; Whelan, C.J.; et al. CMR-based differentiation of AL and ATTR cardiac amyloidosis. JACC Cardiovasc. Imaging 2014, 7, 133–142. [Google Scholar] [CrossRef] [Green Version]
- Mishra, S.; Kass, D.A. Cellular and molecular pathobiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 2021, 18, 400–423. [Google Scholar] [CrossRef]
- Van Heerebeek, L.; Franssen, C.P.; Hamdani, N.; Verheugt, F.W.; Somsen, G.A.; Paulus, W.J. Molecular and cellular basis for diastolic dysfunction. Curr. Heart Fail. Rep. 2012, 9, 293–302. [Google Scholar] [CrossRef]
- Caporizzo, M.A.; Chen, C.Y.; Bedi, K.; Margulies, K.B.; Prosser, B.L. Microtubules Increase Diastolic Stiffness in Failing Human Cardiomyocytes and Myocardium. Circulation 2020, 141, 902–915. [Google Scholar] [CrossRef] [PubMed]
- Benech, J.C.; Benech, N.; Zambrana, A.I.; Rauschert, I.; Bervejillo, V.; Oddone, N.; Damián, J.P. Diabetes increases stiffness of live cardiomyocytes measured by atomic force microscopy nanoindentation. Am. J. Physiol. Cell Physiol. 2014, 307, C910–C919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmonds, S.J.; Cuijpers, I.; Heymans, S.; Jones, E.A.V. Cellular and Molecular Differences between HFpEF and HFrEF: A Step Ahead in an Improved Pathological Understanding. Cells 2020, 9, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouad, F.M.; Slominski, J.M.; Tarazi, R.C. Left ventricular diastolic function in hypertension: Relation to left ventricular mass and systolic function. J. Am. Coll. Cardiol. 1984, 3, 1500–1506. [Google Scholar] [CrossRef] [Green Version]
- Zile, M.R.; Baicu, C.F.; Gaasch, W.H. Diastolic heart failure—abnormalities in active relaxation and passive stiffness of the left ventricle. N. Engl. J. Med. 2004, 350, 1953–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alyodo, D.; Anderson, R.W.; Parrish, D.G.; Dai, X.Z.; Bache, R.J. Alteration of myocardial blood flow associated with experimental canine left ventricular hypertrophy secondary to valvular aortic stenosis. Circ. Res. 1986, 58, 47–57. [Google Scholar] [CrossRef] [Green Version]
- Anversa, P.; Rocci, R.; Olivetti, G. Quantitative structural analysis of the myocardium during physiologic growth and induced cardiac hypertrophy: A review. J. Am. Coll. Cardiol. 1986, 7, 1140–1149. [Google Scholar] [CrossRef] [Green Version]
- Omori, T.; Nakamori, S.; Fujimoto, N.; Ishida, M.; Kitagawa, K.; Ichikawa, Y.; Kumagai, N.; Kurita, T.; Imanaka-Yoshida, K.; Hiroe, M.; et al. Myocardial Native T1 Predicts Load-Independent Left Ventricular Chamber Stiffness in Patients With HFpEF. JACC Cardiovasc. Imaging 2020, 13, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- White, S.K.; Sado, D.M.; Fontana, M.; Banypersad, S.M.; Maestrini, V.; Flett, A.S.; Piechnik, S.K.; Robson, M.D.; Hausenloy, D.J.; Sheikh, A.M.; et al. T1 mapping for myocardial extracellular volume measurement by CMR: Bolus only versus primed infusion technique. JACC Cardiovasc. Imaging 2013, 6, 955e62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everett, R.J.; Stirrat, C.G.; Semple, S.I.; Newby, D.E.; Dweck, M.R.; Mirsadraee, S. Assessment of myocardial fibrosis with T1 mapping MRI. Clin. Radiol. 2016, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Poyhonen, P.; Kivisto, S.; Holmstrom, M.; Hanninen, H. Quantifying late gadolinium enhancement on CMR provides additional prognostic information in early risk stratification of nonischemic cardiomyopathy: A cohort study. BMC Cardiovasc. Disord. 2014, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, S.; Saito, N.; Kirigaya, H.; Gyotoku, D.; Iinuma, N.; Kusakawa, Y.; Iguchi, K.; Nakachi, T.; Fukui, K.; Futaki, M.; et al. Prognostic significance of quantitative assessment of focal myocardial fibrosis in patients with heart failure with preserved ejection fraction. Int. J. Cardiol. 2015, 191, 314–319. [Google Scholar] [CrossRef]
- Mascherbauer, J.; Marzluf, B.A.; Tufaro, C.; Pfaffenberger, S.; Graf, A.; Wexberg, P.; Panzenböck, A.; Jakowitsch, J.; Bangert, C.; Laimer, D.; et al. Cardiac magnetic resonance postcontrast T1 time is associated with outcome in patients with heart failure and preserved ejection fraction. Circ. Cardiovasc. Imaging 2013, 6, 1056–1065. [Google Scholar] [CrossRef] [Green Version]
- Assadi, H.; Jones, R.; Swift, A.J.; Al-Mohammad, A.; Garg, P. Cardiac MRI for the prognostication of heart failure with preserved ejection fraction: A systematic review and meta-analysis. Magn. Reson. Imaging 2021, 76, 116–122. [Google Scholar] [CrossRef]
- Schelbert, E.B.; Fridman, Y.; Wong, T.C.; Abu Daya, H.; Piehler, K.M.; Kadakkal, A.; Miller, C.A.; Ugander, M.; Maanja, M.; Kellman, P.; et al. Temporal relation between myocardial fibrosis and heart failure with preserved ejection fraction: Association with baseline disease severity and subsequent outcome. JAMA Cardiol. 2017, 2, 995–1006. [Google Scholar] [CrossRef]
- Roy, C.; Slimani, A.; de Meester, C.; Amzulescu, M.; Pasquet, A.; Vancraeynest, D.; Beauloye, C.; Vanoverschelde, J.-L.; Gerber, B.L.; Pouleur, A.C. Associations and prognostic significance of diffuse myocardial fibrosis by cardiovascular magnetic resonance in heart failure with preserved ejection fraction. J. Cardiovasc. Magn. Reson. 2018, 20, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duca, F.; Kammerlander, A.A.; Zotter-Tufaro, C.; Aschauer, S.; Schwaiger, M.L.; Marzluf, B.A.; Bonderman, D.; Mascherbauer, J. Interstitial Fibrosis, Functional Status, and Outcomes in Heart Failure with Preserved Ejection Fraction Insights From a Prospective Cardiac Magnetic Resonance Imaging Study. Circ. Cardiovasc. Imaging 2016, 9, e005277. [Google Scholar] [CrossRef] [Green Version]
- Kunadian, V.; Chieffo, A.; Camici, P.G.; Berry, C.; Escaned, J.; Maas, A.H.E.M.; Prescott, E.; Karam, N.; Appelman, Y.; Fraccaro, C.; et al. An EAPCI Expert Consensus Document on Ischaemia with Non-Obstructive Coronary Arteries in Collaboration with European Society of Cardiology Working Group on Coronary Pathophysiology & Microcirculation Endorsed by Coronary Vasomotor Disorders International Study Group. EuroIntervention 2021, 16, 1049–1069. [Google Scholar] [CrossRef]
- Padro, T.; Manfrini, O.; Bugiardini, R.; Canty, J.; Cenko, E.; De Luca, G.; Duncker, D.J.; Eringa, E.C.; Koller, A.; Tousoulis, D.; et al. ESC Working Group on Coronary Pathophysiology and Microcirculation position paper on ‘coronary microvascular dysfunction in cardiovascular disease’. Cardiovasc. Res. 2020, 116, 741–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knuuti, J.; Wijns, W.; Saraste, A.; Capodanno, D.; Barbato, E.; Funck-Brentano, C.; Eva Prescott, E.; Storey, R.F.; Deaton, C.; Cuisset, T.; et al. 2019 ESC Guidelines for the diagnosis and management of chronic coronary syndromes: The Task Force for the diagnosis and management of chronic coronary syndromes of the European Society of Cardiology (ESC). Eur. Heart J. 2020, 41, 407–477. [Google Scholar] [CrossRef] [PubMed]
- Alexander, Y.; Osto, E.; Schmidt-Trucksäss, A.; Shechter, M.; Trifunovic, D.; Duncker, D.J.; Aboyans, V.; Bäck, M.; Badimon, L.; Cosentino, F.; et al. Endothelial function in cardiovascular medicine: A consensus paper of the European Society of Cardiology Working Groups on Atherosclerosis and Vascular Biology, Aorta and Peripheral Vascular Diseases, Coronary Pathophysiology and Microcirculation, and Thrombosis. Cardiovasc. Res. 2021, 117, 29–42. [Google Scholar] [CrossRef] [Green Version]
- Severino, P.; D’Amato, A.; Pucci, M.; Infusino, F.; Adamo, F.; Birtolo, L.I.; Netti, L.; Montefusco, G.; Chimenti, C.; Lavalle, C.; et al. Ischemic Heart Disease Pathophysiology Paradigms Overview: From Plaque Activation to Microvascular Dysfunction. Int. J. Mol. Sci. 2020, 21, 8118. [Google Scholar] [CrossRef]
- Camici, P.G.; Tschöpe, C.; Di Carli, M.F.; Rimoldi, O.; Van Linthout, S. Coronary microvascular dysfunction in hypertrophy and heart failure. Cardiovasc. Res. 2020, 116, 806–816. [Google Scholar] [CrossRef] [PubMed]
- Chimenti, C.; Russo, A.; Pieroni, M.; Calabrese, F.; Verardo, R.; Thiene, G.; Russo, M.A.; Maseri, A.; Frustaci, A. Intramyocyte detection of Epstein-Barr virus genome by laser capture microdissection in patients with inflammatory cardiomyopathy. Circulation 2004, 110, 3534–3539. [Google Scholar] [CrossRef] [Green Version]
- Basoli, A.; Cametti, C.; Satriani, F.G.; Mariani, P.; Severino, P. Hemocompatibility of stent materials: Alterations in electrical parameters of erythrocyte membranes. Vasc Health Risk Manag. 2012, 8, 197–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alecrin, I.N.; Aldrighi, J.M.; Caldas, M.A.; Gebara, O.C.; Lopes, N.H.; Ramires, J.A.F. Acute and chronic effects of oestradiol on left ventricular diastolic function in hypertensive postmenopausal women with left ventricular diastolic dysfunction. Heart 2004, 90, 777–781. [Google Scholar] [CrossRef] [Green Version]
- Severino, P.; D’Amato, A.; Netti, L.; Pucci, M.; Infusino, F.; Maestrini, V.; Mancone, M.; Fedele, F. Myocardial Ischemia and Diabetes Mellitus: Role of Oxidative Stress in the Connection between Cardiac Metabolism and Coronary Blood Flow. J. Diabetes Res. 2019, 2019, 9489826. [Google Scholar] [CrossRef]
- Severino, P.; D’Amato, A.; Netti, L.; Pucci, M.; De Marchis, M.; Palmirotta, R.; Volterrani, M.; Mancone, M.; Fedele, F. Diabetes Mellitus and Ischemic Heart Disease: The Role of Ion Channels. Int. J. Mol. Sci. 2018, 19, 802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Severino, P.; D’Amato, A.; Pucci, M.; Infusino, F.; Birtolo, L.I.; Mariani, M.V.; Lavalle, C.; Maestrini, V.; Mancone, M.; Fedele, F. Ischemic Heart Disease and Heart Failure: Role of Coronary Ion Channels. Int. J. Mol. Sci. 2020, 21, 3167. [Google Scholar] [CrossRef]
- Mitacchione, G.; Schiavone, M.; Curnis, A.; Arca, M.; Antinori, S.; Gasperetti, A.; Mascioli, G.; Severino, P.; Sabato, F.; Caracciolo, M.M.; et al. Impact of prior statin use on clinical outcomes in COVID-19 patients: Data from tertiary referral hospitals during COVID-19 pandemic in Italy. J. Clin. Lipidol. 2021, 15, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Van de Wouw, J.; Sorop, O.; Van Drie, R.W.A.; Van Duin, R.W.B.; Nguyen, I.T.N.; Joles, J.A.; Verhaar, M.C.; Merkus, D.; Duncker, D.J. Perturbations in myocardial perfusion and oxygen balance in swine with multiple risk factors: A novel model of ischemia and no obstructive coronary artery disease. Basic Res. Cardiol. 2020, 115, 21. [Google Scholar] [CrossRef] [PubMed]
- Campbell, D.J.; Somaratne, J.B.; Prior, D.L.; Yii, M.; Kenny, J.F.; Newcomb, A.E.; Kelly, D.J.; Black, M.J. Obesity is associated with lower coronary microvascular density. PLoS ONE 2013, 8, e81798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron, A.D.; Tarshoby, M.; Hook, G.; Lazaridis, E.N.; Cronin, J.; Johnson, A.; Steinberg, H.O. Interaction between insulin sensitivity and muscle perfusion on glucose uptake in human skeletal muscle: Evidence for capillary recruitment. Diabetes 2000, 49, 768–774. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Quesada, C.; Cavalera, M.; Biernacka, A.; Kong, P.; Lee, D.W.; Saxena, A.; Frunza, O.; Dobaczewski, M.; Shinde, A.; Frangogiannis, N.G. Thrombospondin-1 induction in the diabetic myocardium stabilizes the cardiac matrix in addition to promoting vascular rarefaction through angiopoietin-2 upregulation. Circ. Res. 2013, 113, 1331–1344. [Google Scholar] [CrossRef] [Green Version]
- Alex, L.; Russo, I.; Holoborodko, V.; Frangogiannis, N.G. Characterization of a mouse model of obesity-related fibrotic cardiomyopathy that recapitulates features of human heart failure with preserved ejection fraction. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H934–H949. [Google Scholar] [CrossRef] [Green Version]
- Jacob, M.; Paul, O.; Mehringer, L.; Chappell, D.; Rehm, M.; Welsch, U.; Kaczmarek, I.; Conzen, P.; Becker, B.F. Albumin augmentation improves condition of guinea pig hearts after 4 h of cold ischemia. Transplantation 2009, 87, 956–965. [Google Scholar] [CrossRef]
- Welsh, L. Vascular permeability—The essentials. Upsala J. Med. Sci. 2015, 120, 135–143. [Google Scholar] [CrossRef] [Green Version]
- Van Dijk, C.G.M.; Oosterhuis, N.R.; Xu, Y.J.; Brandt, M.; Paulus, W.J.; van Heerebeek, L.; Duncker, D.J.; Verhaar, M.C.; Fontoura, D.; Lourenço, A.P.; et al. Distinct Endothelial Cell Responses in the Heart and Kidney Microvasculature Characterize the Progression of Heart Failure With Preserved Ejection Fraction in the Obese ZSF1 Rat with Cardiorenal Metabolic Syndrome. Circ. Heart Fail. 2016, 9, e002760. [Google Scholar] [CrossRef]
- Zeng, H.; He, X.; Tuo, Q.H.; Liao, D.F.; Zhang, G.Q.; Chen, J.X. LPS causes pericyte loss and microvascular dysfunction via disruption of Sirt3/angiopoietins/Tie-2 and HIF-2α/Notch3 pathways. Sci. Rep. 2016, 6, 20931. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; Chen, J.X. Sirtuin 3, Endothelial Metabolic Reprogramming, and Heart Failure With Preserved Ejection Fraction. J. Cardiovasc. Pharmacol. 2019, 74, 315–323. [Google Scholar] [CrossRef]
- Fedele, F.; Severino, P.; Bruno, N.; Stio, R.; Caira, C.; D’Ambrosi, A.; Brasolin, B.; Ohanyan, V.; Mancone, M. Role of ion channels in coronary microcirculation: A review of the literature. Future Cardiol. 2013, 9, 897–905. [Google Scholar] [CrossRef]
- Fedele, F.; Mancone, M.; Chilian, W.M.; Severino, P.; Canali, E.; Logan, S.; De Marchis, M.L.; Volterrani, M.; Palmirotta, R.; Guadagni, F. Role of genetic polymorphisms of ion channels in the pathophysiology of coronary microvascular dysfunction and ischemic heart disease. Basic Res. Cardiol. 2013, 108, 387. [Google Scholar] [CrossRef] [Green Version]
- Severino, P.; D’Amato, A.; Netti, L.; Pucci, M.; Mariani, M.V.; Cimino, S.; Birtolo, L.I.; Infusino, F.; De Orchi, P.; Palmirotta, R.; et al. Susceptibility to ischaemic heart disease: Focusing on genetic variants for ATP-sensitive potassium channel beyond traditional risk factors. Eur. J. Prev. Cardiol. 2020, 2047487320926780. [Google Scholar] [CrossRef] [PubMed]
- Goodwill, A.G.; Dick, G.M.; Kiel, A.M.; Tune, J.D. Regulation of Coronary Blood Flow. Compr. Physiol. 2017, 7, 321–382. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, L.L.; Schertel, E.R.; Pratt, J.W.; McClure, D.E.; Ying, A.J.; Heck, C.F.; Myerowitz, P.D. Impairment of left ventricular function by acute cardiac lymphatic obstruction. Cardiovasc. Res. 1997, 33, 164–171. [Google Scholar] [CrossRef] [Green Version]
- Reddy, Y.N.V.; Obokata, M.; Wiley, B.; Koepp, K.E.; Jorgenson, C.C.; Egbe, A.; Melenovsky, V.; Carter, R.E.; Borlaug, B.A. The haemodynamic basis of lung congestion during exercise in heart failure with preserved ejection fraction. Eur. Heart J. 2019, 40, 3721–3730. [Google Scholar] [CrossRef] [PubMed]
- Zawieja, S.D.; Gasheva, O.; Zawieja, D.C.; Muthuchamy, M. Blunted flow-mediated responses and diminished nitric oxide synthase expression in lymphatic thoracic ducts of a rat model of metabolic syndrome. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H385–H393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zawieja, S.D.; Wang, W.; Chakraborty, S.; Zawieja, D.C.; Muthuchamy, M. Macrophage alterations within the mesenteric lymphatic tissue are associated with impairment of lymphatic pump in metabolic syndrome. Microcirculation 2016, 23, 558–570. [Google Scholar] [CrossRef] [Green Version]
- Nitti, M.D.; Hespe, G.E.; Kataru, R.P.; GarcíaNores, G.D.; Savetsky, I.L.; Torrisi, J.S.; Gardenier, J.C.; Dannenberg, A.J.; Mehrara, B.J. Obesity-induced lymphatic dysfunction is reversible with weight loss. J. Physiol. 2016, 594, 7073–7087. [Google Scholar] [CrossRef] [PubMed]
- Scallan, J.P.; Hill, M.A.; Davis, M.J. Lymphatic vascular integrity is disrupted in type 2 diabetes due to impaired nitric oxide signalling. Cardiovasc. Res. 2015, 107, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.H.; Zhou, X.; Ji, W.J.; Zeng, S.; Dong, Y.; Tian, L.; Bi, Y.; Guo, Z.Z.; Gao, F.; Chen, H.; et al. Overexpression of VEGF-C attenuates chronic high salt intake-induced left ventricular maladaptive remodeling in spontaneously hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H598–H609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez Gelston, C.A.; Balasubbramanian, D.; Abouelkheir, G.R.; Lopez, A.H.; Hudson, K.R.; Johnson, E.R.; Muthuchamy, M.; Mitchell, B.M.; Rutkowski, J.M. Enhancing Renal Lymphatic Expansion Prevents Hypertension in Mice. Circ. Res. 2018, 122, 1094–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, S.; Saito, N.; Kirigaya, H.; Gyotoku, D.; Iinuma, N.; Kusakawa, Y.; Iguchi, K.; Nakachi, T.; Fukui, K.; Futaki, M.; et al. Impairment of coronary flow reserve evaluated by phase contrast cine-magnetic resonance imaging in patients with heart failure with preserved ejection fraction. J. Am. Heart Assoc. 2016, 5, e002649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löffler, A.I.; Pan, J.A.; Balfour, P.C., Jr.; Shaw, P.W.; Yang, Y.; Nasir, M.; Auger, D.A.; Epstein, F.H.; Kramer, C.M.; Gan, L.M.; et al. Frequency of Coronary Microvascular Dysfunction and Diffuse Myocardial Fibrosis (Measured by Cardiovascular Magnetic Resonance) in Patients With Heart Failure and Preserved Left Ventricular Ejection Fraction. Am. J. Cardiol. 2019, 124, 1584–1589. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.J.; Lam, C.S.P.; Svedlund, S.; Saraste, A.; Hage, C.; Tan, R.S.; Beussink-Nelson, L.; Fermer, M.L.; Broberg, M.A.; Gan, L.M.; et al. Prevalence and correlates of coronary microvascular dysfunction in heart failure with preserved ejection fraction: PROMIS-HFpEF. Eur. Heart J. 2018, 39, 3439–3450. [Google Scholar] [CrossRef]
- Packer, M.; Lam, C.S.P.; Lund, L.H.; Maurer, M.S.; Borlaug, B.A. Characterization of the inflammatory-metabolic phenotype of heart failure with a preserved ejection fraction: A hypothesis to explain influence of sex on the evolution and potential treatment of the disease. Eur. J. Heart Fail. 2020, 22, 1551–1567. [Google Scholar] [CrossRef]
- Severino, P.; Netti, L.; Mariani, M.V.; Maraone, A.; D’Amato, A.; Scarpati, R.; Infusino, F.; Pucci, M.; Lavalle, C.; Maestrini, V.; et al. Prevention of Cardiovascular Disease: Screening for Magnesium Deficiency. Cardiol. Res. Pr. 2019, 2019, 4874921. [Google Scholar] [CrossRef] [Green Version]
- Obokata, M.; Reddy, Y.N.V.; Pislaru, S.V.; Melenovsky, V.; Borlaug, B.A. Evidence supporting the existence of a distinct obese phenotype of heart failure with preserved ejection fraction. Circulation 2017, 136, 6–19. [Google Scholar] [CrossRef]
- Foussier, C.; Barral, P.A.; Jerosh-Herold, M.; Gariboldi, V.; Rapacchi, S.; Gallon, A.; Bartoli, A.; Bentatou, Z.; Guye, M.; Bernard, M.; et al. Quantification of diffuse myocardial fibrosis using CMR extracellular volume fraction and serum biomarkers of collagen turnover with histologic quantification as standard of reference. Diagn. Interv. Imaging. 2021, 102, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Venteclef, N.; Guglielmi, V.; Balse, E.; Gaborit, B.; Cotillard, A.; Atassi, F.; Amour, J.; Leprince, P.; Dutour, A.; Clément, K.; et al. Human epicardial adipose tissue induces fibrosis of the atrial myocardium through the secretion of adipo-fibrokines. Eur. Heart J. 2015, 36, 795–805. [Google Scholar] [CrossRef] [Green Version]
- Severino, P.; Mariani, M.V.; Maraone, A.; Piro, A.; Ceccacci, A.; Tarsitani, L.; Maestrini, V.; Mancone, M.; Lavalle, C.; Pasquini, M.; et al. Triggers for Atrial Fibrillation: The Role of Anxiety. Cardiol. Res. Pr. 2019, 2019, 1208505. [Google Scholar] [CrossRef]
- Magnocavallo, M.; Bellasi, A.; Mariani, M.V.; Fusaro, M.; Ravera, M.; Paoletti, E.; Di Iorio, B.; Barbera, V.; Della Rocca, D.G.; Palumbo, R.; et al. Thromboembolic and Bleeding Risk in Atrial Fibrillation Patients with Chronic Kidney Disease: Role of Anticoagulation Therapy. J. Clin. Med. 2020, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Mariani, M.V.; Magnocavallo, M.; Straito, M.; Piro, A.; Severino, P.; Iannucci, G.; Chimenti, C.; Mancone, M.; Rocca, D.G.D.; Forleo, G.B.; et al. Direct oral anticoagulants versus vitamin K antagonists in patients with atrial fibrillation and cancer a meta-analysis. J. Thromb. Thrombolysis 2021, 51, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.K.; Lee, J.K.; Hsu, J.C.; Su, M.M.; Wu, Y.F.; Lin, T.T.; Lan, C.W.; Hwang, J.J.; Lin, L.Y. Myocardial adipose deposition and the development of heart failure with preserved ejection fraction. Eur. J. Heart Fail. 2020, 22, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Koepp, K.E.; Obokata, M.; Reddy, Y.N.V.; Olson, T.P.; Borlaug, B.A. Hemodynamic and functional impact of epicardial adipose tissue in heart failure with preserved ejection fraction. JACC Heart Fail. 2020, 8, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Gronemeyer, S.A.; Steen, R.G.; Kauffman, W.M.; Reddick, W.E.; Glass, J.O. Fast adipose tissue (FAT) assessment by MRI. Magn. Reson. Imaging 2000, 18, 815–818. [Google Scholar] [CrossRef]
- Wu, C.K.; Tsai, H.Y.; Su, M.M.; Wu, Y.F.; Hwang, J.J.; Lin, J.L.; Lin, L.Y.; Chen, J.J. Evolutional change in epicardial fat and its correlation with myocardial diffuse fibrosis in heart failure patients. J. Clin. Lipidol. 2017, 11, 1421–1431. [Google Scholar] [CrossRef] [PubMed]
- Mahmod, M.; Pal, N.; Rayner, J.; Holloway, C.; Raman, B.; Dass, S.; Levelt, E.; Ariga, R.; Ferreira, V.; Banerjee, R.; et al. The interplay between metabolic alterations, diastolic strain rate and exercise capacity in mild heart failure with preserved ejection fraction: A cardiovascular magnetic resonance study. J. Cardiovasc. Magn. Reson. 2018, 20, 88. [Google Scholar] [CrossRef]
- Pu, Q.; Larouche, I.; Schiffrin, E.L. Effect of dual angiotensin converting enzyme/neutral endopeptidase inhibition, angiotensin converting enzyme inhibition, or AT1 antagonism on coronary microvasculature in spontaneously hypertensive rats. Am. J. Hypertens. 2003, 11 Pt 1, 931–937. [Google Scholar] [CrossRef] [Green Version]
- Tamura, N.; Ogawa, Y.; Chusho, H.; Nakamura, K.; Nakao, K.; Suda, M.; Kasahara, M.; Hashimoto, R.; Katsuura, G.; Mukoyama, M.; et al. Cardiac fibrosis in mice lacking brain natriuretic peptide. Proc. Natl. Acad. Sci. USA 2000, 97, 4239–4244. [Google Scholar] [CrossRef] [Green Version]
- Santos-Alvarez, J.; Goberna, R.; Sánchez-Margalet, V. Human leptin stimulates proliferation and activation of human circulating monocytes. Cell Immunol. 1999, 194, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Saucillo, D.C.; Gerriets, V.A.; Sheng, J.; Rathmell, J.C.; Maciver, N.J. Leptin metabobolically licenses T cells for activation to link nutrition and immunity. J. Immunol. 2014, 192, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Moudgil, R.; Samra, G.; Ko, K.A.; Vu, H.T.; Thomas, T.N.; Luo, W.; Chang, J.; Reddy, A.K.; Fujiwara, K.; Abe, J.I. Topoisomerase 2B Decrease Results in Diastolic Dysfunction via p53 and Akt: A Novel Pathway. Front. Cardiovasc. Med. 2020, 7, 594123. [Google Scholar] [CrossRef] [PubMed]
- Van den Hoogen, P.; De Jager, S.C.A.; Huibers, M.M.H.; Schoneveld, A.H.; Puspitasari, Y.M.; Valstar, G.B.; Oerlemans, M.I.F.J.; De Weger, R.A.; Doevendans, P.A.; Den Ruijter, H.M.; et al. Increased circulating IgG levels, myocardial immune cells and IgG deposits support a role for an immune response in pre- and end-stage heart failure. J. Cell. Mol. Med. 2019, 23, 7505–7516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prenner, S.B.; Pillutla, R.; Yenigalla, S.; Gaddam, S.; Lee, J.; Obeid, M.J.; Ans, A.H.; Jehangir, Q.; Kim, J.; Zamani, P.; et al. Serum Albumin Is a Marker of Myocardial Fibrosis, Adverse Pulsatile Aortic Hemodynamics, and Prognosis in Heart Failure With Preserved Ejection Fraction. J. Am. Heart Assoc. 2020, 9, e014716. [Google Scholar] [CrossRef]
- Charles, C.J.; Lee, P.; Li, R.R.; Yeung, T.; Ibraham Mazlan, S.M.; Tay, Z.W.; Abdurrachim, D.; Teo, X.Q.; Wang, W.H.; De Kleijn, D.P.V.; et al. A porcine model of heart failure with preserved ejection fraction: Magnetic resonance imaging and metabolic energetics. ESC Heart Fail. 2020, 7, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Sankaralingam, S.; Lopaschuk, G.D. Cardiac energy metabolic alterations in pressure overload–induced left and right heart failure (2013 Grover Conference Series). Pulm. Circ. 2015, 5, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Lydell, C.P.; Chan, C.P.; Wambolt, R.B.; Sambandam, N.; Parsons, H.; Bondy, G.P.; Rodrigues, B.; Popov, K.M.; Harris, R.A.; Brownsey, R.W.; et al. Pyruvate dehydrogenase and the regulation of glucose oxidation in hypertrophied rat hearts. Cardiovasc. Res. 2002, 53, 841–851. [Google Scholar] [CrossRef] [Green Version]
- Doenst, T.; Pytel, G.; Schrepper, A.; Amorim, P.; Färber, G.; Shingu, Y.; Mohr, F.W.; Schwarzer, M. Decreased rates of substrate oxidation ex vivo predict the onset of heart failure and contractile dysfunction in rats with pressure overload. Cardiovasc. Res. 2010, 86, 461–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kundu, B.K.; Zhong, M.; Sen, S.; Davogustto, G.; Keller, S.R.; Taegtmeyer, H. Remodeling of glucose metabolism precedes pressure overload-induced left ventricular hypertrophy: Review of a hypothesis. Cardiology 2015, 130, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Sampath, S.; Parimal, A.S.; Huang, W.; Manigbas, E.; Gsell, W.; Chang, M.M.L.; Qiu, A.; Jacobsen, K.; Evelhoch, J.L.; Chin, C.L. Quantification of regional myocardial mean intracellular water lifetime: A nonhuman primate study in myocardial stress. NMR Biomed. 2020, 33, e4248. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Poirier-Quinot, M.; Springer, C.S., Jr.; Balschi, J.A. Active trans-plasma membrane water cycling in yeast is revealed by NMR. Biophys. J. 2011, 101, 2833–2842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinman, B.; Lachin, J.M.; Inzucchi, S.E. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 374, 1094. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.J. Precision Medicine for Heart Failure with Preserved Ejection Fraction: An Overview. J. Cardiovasc. Transl. Res. 2017, 10, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Hahn, V.S.; Knutsdottir, H.; Luo, X.; Bedi, K.; Margulies, K.B.; Haldar, S.M.; Stolina, M.; Yin, J.; Khakoo, A.Y.; Vaishnav, J.; et al. Myocardial Gene Expression Signatures in Human Heart Failure with Preserved Ejection Fraction. Circulation 2021, 143, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.T.; Wong, L.L.; Liew, O.W.; Richards, A.M. Heart Failure with Reduced Ejection Fraction (HFrEF) and Preserved Ejection Fraction (HFpEF): The Diagnostic Value of Circulating MicroRNAs. Cells 2019, 8, 1651. [Google Scholar] [CrossRef] [Green Version]
- Zile, M.R.; Jhund, P.S.; Baicu, C.F.; Claggett, B.L.; Pieske, B.; Voors, A.A.; Prescott, M.F.; Shi, V.; Lefkowitz, M.; McMurray, J.J.; et al. Prospective Comparison of ARNI with ARB on Management of Heart Failure with Preserved Ejection Fraction (PARAMOUNT) Investigators. Plasma Biomarkers Reflecting Profibrotic Processes in Heart Failure with a Preserved Ejection Fraction: Data from the Prospective Comparison of ARNI with ARB on Management of Heart Failure with Preserved Ejection Fraction Study. Circ. Heart Fail. 2016, 9, e002551. [Google Scholar] [CrossRef] [Green Version]
- Vlachos, I.S.; Zagganas, K.; Paraskevopoulou, M.D.; Georgakilas, G.; Karagkouni, D.; Vergoulis, T.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-miRPath v3.0: Deciphering microRNA function with experimental support. Nucleic Acids Res. 2015, 43, W460–W466. [Google Scholar] [CrossRef]
- Zhou, Y.; Shiok, T.C.; Richards, A.M.; Wang, P. MicroRNA-101a suppresses fibrotic programming in isolated cardiac fibroblasts and in vivo fibrosis following trans-aortic constriction. J. Mol. Cell. Cardiol. 2018, 121, 266–276. [Google Scholar] [CrossRef]
- Rech, M.; Barandiarán Aizpurua, A.; Van Empel, V.; Van Bilsen, M.; Schroen, B. Pathophysiological understanding of HFpEF: MicroRNAs as part of the puzzle. Cardiovasc. Res. 2018, 114, 782–793. [Google Scholar] [CrossRef]
- Essandoh, K.; Li, Y.; Huo, J.; Fan, G.C. MiRNA-Mediated Macrophage Polarization and its Potential Role in the Regulation of Inflammatory Response. Shock 2016, 46, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Chen, S.; Gordon, A.D.; Chakrabarti, S. miR-146a mediates inflammatory changes and fibrosis in the heart in diabetes. J. Mol. Cell. Cardiol. 2017, 105, 70–76. [Google Scholar] [CrossRef]
- Zampetaki, A.; Kiechl, S.; Drozdov, I.; Willeit, P.; Mayr, U.; Prokopi, M.; Mayr, A.; Weger, S.; Oberhollenzer, F.; Bonora, E.; et al. Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circ. Res. 2010, 107, 810–817. [Google Scholar] [CrossRef]
- Widmer, R.J.; Chung, W.Y.; Herrmann, J.; Jordan, K.L.; Lerman, L.O.; Lerman, A. The association between circulating microRNA levels and coronary endothelial function. PLoS ONE 2014, 9, e109650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dellago, H.; Bobbili, M.R.; Grillari, J. MicroRNA-17-5p: At the Crossroads of Cancer and Aging—A Mini-Review. Gerontology 2017, 63, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Gurha, P.; Abreu-Goodger, C.; Wang, T.; Ramirez, O.M.; Drumond, A.L.; Van Dongen, S.; Chen, Y.; Bartonicek, N.; Enright, A.J.; Lee, B.; et al. Targeted deletion of microRNA-22 promotes stress-induced cardiac dilation and contractile dysfunction. Circulation 2012, 125, 2751–2761. [Google Scholar] [CrossRef] [PubMed]
- Jazbutyte, V.; Fiedler, J.; Kneitz, S.; Galuppo, P.; Just, A.; Holzmann, A.; Bauersachs, J.; Thum, T. MicroRNA22 increases senescence and activates cardiac fibroblasts in the aging heart. Age (Dordr) 2013, 35, 747–762. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, S.; He, A.; Kong, S.W.; Lu, J.; Bejar, R.; Bodyak, N.; Lee, K.H.; Ma, Q.; Kang, P.M.; Golub, T.R.; et al. MicroRNA-1 negatively regulates expression of the hypertrophy-associated calmodulin and Mef2a genes. Mol. Cell. Biol. 2009, 29, 2193–2204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florijn, B.W.; Valstar, G.B.; Duijs, J.M.G.J.; Menken, R.; Cramer, M.J.; Teske, A.J.; Ghossein-Doha, C.; Rutten, F.H.; Spaanderman, M.E.A.; den Ruijter, H.M.; et al. Sex-specific microRNAs in women with diabetes and left ventricular diastolic dysfunction or HFpEF associate with microvascular injury. Sci. Rep. 2020, 10, 13945. [Google Scholar] [CrossRef] [PubMed]
- Kaschina, E.; Unger, T. Angiotensin AT1/AT2 receptors: Regulation, signalling and function. Blood Press 2003, 12, 70–88. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Ostergren, J.; Swedberg, K.; Granger, C.B.; Held, P.; Michelson, E.L.; Olofsson, B.; Yusuf, S.; Pfeffer, M.A. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function taking angiotensin-converting-enzyme inhibitors: The CHARM-Added trial. Lancet 2003, 362, 767–771. [Google Scholar] [CrossRef]
- CONSENSUS Trial Study Group. Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). N. Engl. J. Med. 1987, 316, 1429–1435. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, S.; Pitt, B.; Davis, C.E.; Hood, W.B.; Cohn, J.N. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N. Engl. J. Med. 1991, 325, 293–302. [Google Scholar] [CrossRef]
- Cleland, J.G.; Tendera, M.; Adamus, J.; Freemantle, N.; Polonski, L.; Taylor, J. The perindopril in elderly people with chronic heart failure (PEP-CHF) study. Eur. Heart J. 2006, 27, 2338–2345. [Google Scholar] [CrossRef] [Green Version]
- Yusuf, S.; Pfeffer, M.A.; Swedberg, K.; Granger, C.B.; Held, P.; McMurray, J.J.; Michelson, E.L.; Olofsson, B.; Ostergren, J. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: The CHARM-preserved trial. Lancet 2003, 362, 777–781. [Google Scholar] [CrossRef]
- Beldhuis, I.E.; Streng, K.W.; Ter Maaten, J.M.; Voors, A.A.; Van der Meer, P.; Rossignol, P.; McMurray, J.J.; Damman, K. Renin-Angiotensin System Inhibition, Worsening Renal Function, and Outcome in Heart Failure Patients With Reduced and Preserved Ejection Fraction: A Meta-Analysis of Published Study Data. Circ. Heart Fail. 2017, 10, e003588. [Google Scholar] [CrossRef]
- Yip, G.W.; Wang, M.; Wang, T.; Chan, S.; Fung, J.W.; Yeung, L.; Yip, T.; Lau, S.T.; Lau, C.P.; Tang, M.O.; et al. The Hong Kong diastolic heart failure study: A randomised controlled trial of diuretics, irbesartan and ramipril on quality of life, exercise capacity, left ventricular global and regional function in heart failure with a normal ejection fraction. Heart 2008, 94, 573–580. [Google Scholar] [CrossRef]
- Zi, M.; Carmichael, N.; Lye, M. The effect of quinapril on functional status of elderly patients with diastolic heart failure. Cardiovasc. Drugs 2003, 17, 133–139. [Google Scholar] [CrossRef]
- CIBIS Investigators and Committees. A randomized trial of beta-blockade in heart failure. Card. Insufficiency Bisoprolol Study (CIBIS) Circ. 1994, 90, 1765–1773. [Google Scholar] [CrossRef] [Green Version]
- Zannad, F.; McMurray, J.J.; Krum, H.; Van Veldhuisen, D.J.; Swedberg, K.; Shi, H.; Vincent, J.; Pocock, S.J.; Pitt, B. EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N. Engl. J. Med. 2011, 364, 11–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massie, B.M.; Carson, P.E.; McMurray, J.J.; Komajda, M.; McKelvie, R.; Zile, M.R.; Anderson, S.; Donovan, M.; Iverson, E.; Staiger, C.; et al. I-PRESERVE Investigators. Irbesartan in patients with heart failure and preserved ejection fraction. N. Engl. J. Med. 2008, 359, 2456–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lund, L.H.; Claggett, B.; Liu, J.; Lam, C.S.; Jhund, P.S.; Rosano, G.M.; Swedberg, K.; Yusuf, S.; Granger, C.B.; Pfeffer, M.A.; et al. Heart failure with mid-range ejection fraction in CHARM: Characteristics, outcomes and effect of candesartan across the entire ejection fraction spectrum. Eur. J. Heart Fail. 2018, 20, 1230–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, S.D.; Janardhanan, R.; Verma, A.; Bourgoun, M.; Daley, W.L.; Purkayastha, D.; Lacourciere, Y.; Hippler, S.E.; Fields, H.; Naqvi, T.Z.; et al. Effect of angiotensin receptor blockade and antihypertensive drugs on diastolic function in patients with hypertension and diastolic dysfunction: A randomised trial. Lancet 2007, 369, 2079–2087. [Google Scholar] [CrossRef]
- Tschöpe, C.; Birner, C.; Böhm, M.; Bruder, O.; Frantz, S.; Luchner, A.; Maier, L.; Stork, S.; Kherad, B.; Laufs, U. Heart failure with preserved ejection fraction: Current management and future strategies: Expert opinion on the behalf of the Nucleus of the “Heart Failure Working Group” of the German Society of Cardiology (DKG). Clin. Res. Cardiol. 2018, 107, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.D.; McMurray, J.J.V.; Anand, I.S.; Ge, J.; Lam, C.S.P.; Maggioni, A.P.; Martinez, F.; Packer, M.; Pfeffer, M.A.; Pieske, B.; et al. Angiotensin-neprilysin inhibition in heart failure with preserved ejection fraction. N. Engl. J. Med. 2019, 381, 1609–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuno, T.; Ueyama, H.; Fujisaki, T.; Briasouli, A.; Takagi, H.; Briasoulis, A. Meta-Analysis Evaluating the Effects of Renin-Angiotensin-Aldosterone System Blockade on Outcomes of Heart Failure with Preserved Ejection Fraction. Am. J. Cardiol. 2020, 125, 1187–1193. [Google Scholar] [CrossRef]
- Brown, N.J. Aldosterone and end-organ damage. Curr. Opin. Nephrol Hypertens. 2005, 14, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Edelmann, F.; Wachter, R.; Schmidt, A.G.; Kraigher-Krainer, E.; Colantonio, C.; Kamke, W.; Duvinage, A.; Stahrenberg, R.; Durstewitz, K.; Löffler, M.; et al. Effect of spironolactone on diastolic function and exercise capacity in patients with heart failure with preserved ejection fraction: The Aldo-DHF randomized controlled trial. JAMA 2013, 309, 781–791. [Google Scholar] [CrossRef] [Green Version]
- Pitt, B.; Pfeffer, M.A.; Assmann, S.F.; Boineau, R.; Anand, I.S.; Claggett, B.; Clausell, N.; Desai, A.S.; Diaz, R.; Fleg, J.L.; et al. Spironolactone for heart failure with preserved ejection fraction. N. Engl. J. Med. 2014, 370, 1383–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuchi, C.; Tritto, I.; Carluccio, E.; Mattei, C.; Cattadori, G.; Ambrosio, G. Role of endothelial dysfunction in heart failure. Heart Fail. Rev. 2020, 25, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Redfield, M.M.; Chen, H.H.; Borlaug, B.A.; Semigran, M.J.; Lee, K.L.; Lewis, G.; LeWinter, M.M.; Rouleau, J.L.; Bull, D.A.; Mann, D.L.; et al. RELAX Trial. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: A randomized clinical trial. JAMA 2013, 309, 1268–1277. [Google Scholar] [CrossRef] [PubMed]
- Pieske, B.; Butler, J.; Filippatos, G.; Lam, C.; Maggioni, A.P.; Ponikowski, P.; Shah, S.; Solomon, S.; Kraigher-Krainer, E.; Samano, E.T.; et al. Rationale and design of the SOluble guanylate Cyclase stimulatoR in heArT failurE Studies (SOCRATES) Investigators and Coordinators. Eur. J. Heart Fail. 2014, 16, 1026–1038. [Google Scholar] [CrossRef]
- Green, C.P.; Porter, C.B.; Bresnahan, D.R.; Spertus, J.A. Development and evaluation of the Kansas city cardiomyopathy questionnaire: A new health status measure for heart failure. J. Am. Coll. Cardiol. 2000, 35, 1245–1255. [Google Scholar] [CrossRef] [Green Version]
- Wagdy, K.; Hassan, M. NEAT HFpEF: Organic nitrates fail to deliver. Glob. Cardiol. Sci. Pr. 2016, 2016, e201601. [Google Scholar] [CrossRef] [Green Version]
- Reddy, Y.N.V.; Lewis, G.D.; Shah, S.J.; LeWinter, M.; Semigran, M.; Davila-Roman, V.G.; Anstrom, K.; Hernandez, A.; Braunwald, E.; Redfield, M.M.; et al. INDIE-HFpEF (Inorganic Nitrite Delivery to Improve Exercise Capacity in Heart Failure with Preserved Ejection Fraction): Rationale and Design. Circ. Heart Fail. 2017, 10, e003862. [Google Scholar] [CrossRef] [Green Version]
- Daubert, M.A.; Yow, E.; Dunn, G.; Marchev, S.; Barnhart, H.; Douglas, P.S.; O’Connor, C.; Goldstein, S.; Udelson, J.E.; Sabbah, H.N. Novel mitochondria-targeting peptide in heart failure treatment: A randomized, placebo-controlled trial of elamipretide. Circ. Heart Fail. 2017, 10, e004389. [Google Scholar] [CrossRef]
- Van Tassell, B.W.; Raleigh, J.M.; Abbate, A. Targeting interleukin-1 in heart failure and inflammatory heart disease. Curr. Heart Fail. Rep. 2015, 12, 33–41. [Google Scholar] [CrossRef]
- Van Tassell, B.W.; Arena, R.; Biondi-Zoccai, G.; Canada, J.M.; Oddi, C.; Abouzaki, N.A.; Jahangiri, A.; Appa Falcao, R.; Kontos, M.C.; Bharat Shah, K.; et al. Effects of interleukin-1 blockade with anakinra on aerobic exercise capacity in patients with heart failure and preserved ejection fraction (from the D-HART pilot study). Am. J. Cardiol. 2014, 113, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Yamagami, K.; Oka, T.; Wang, Q.; Ishizu, T.; Lee, J.K.; Miwa, K.; Akazawa, H.; Naito, A.T.; Sakata, Y.; Komuro, I. Pirfenidone exhibits cardioprotective effects by regulating myocardial fibrosis and vascular permeability in pressure-overloaded hearts. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H512–H522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrtovec, B.; Frljak, S.; Jaklic, M.; Poglajen, G.; Zemljic, G.; Cerar, A.; Haddad, F.; Wu, J.C. A pilot trial of CD34+ cell therapy in heart failure with preserved ejection fraction. Circulation 2017, 136, A15952. [Google Scholar]
- Calcagno, S.; Infusino, F.; Salvi, N.; Taccheri, T.; Colantonio, R.; Bruno, E.; Birtolo, L.I.; Severino, P.; Lavalle, C.; Pucci, M.; et al. The Role of Ranolazine for the Treatment of Residual Angina beyond the Percutaneous Coronary Revascularization. J. Clin. Med. 2020, 9, 2110. [Google Scholar] [CrossRef] [PubMed]
- Maier, L.S.; Layug, B.; Karwatowska-Prokopczuk, E.; Belardinelli, L.; Lee, S.; Sander, J.; Lang, C.; Wachter, R.; Edelmann, F.; Hasenfuss, G.; et al. RAnoLazIne for the Treatment of Diastolic Heart Failure in Patients with Preserved Ejection Fraction: The RALI-DHF Proof-of-Concept Study. JACC Heart Fail. 2013, 1, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Maack, C.; Lehrke, M.; Backs, J.; Heinzel, F.R.; Hulot, J.S.; Marx, N.; Paulus, W.J.; Rossignol, P.; Taegtmeyer, H.; Bauersachs, J.; et al. Heart failure and diabetes: Metabolic alterations and therapeutic interventions: A state-of-the-art review from the Translational Research Committee of the Heart Failure Association-European Society of Cardiology. Eur. Heart J. 2018, 39, 4243–4254. [Google Scholar] [CrossRef] [Green Version]
- Nieminen, M.S.; Buerke, M.; Parissis, J.; Ben-Gal, T.; Pollesello, P.; Kivikko, M.; Karavidas, A.; Severino, P.; Comín-Colet, J.; Wikström, G.; et al. Pharmaco-economics of levosimendan in cardiology: A European perspective. Int. J. Cardiol. 2015, 199, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Parissis, J.T.; Paraskevaidis, I.; Bistola, V.; Farmakis, D.; Panou, F.; Kourea, K.; Nikolaou, M.; Filippatos, G.; Kremastinos, D. Effects of levosimendan on right ventricular function in patients with advanced heart failure. Am. J. Cardiol. 2006, 98, 1489–1492. [Google Scholar] [CrossRef]
- Böhm, M.; Perez, A.C.; Jhund, P.S.; Reil, J.C.; Komajda, M.; Zile, M.R.; McKelvie, R.S.; Anand, I.S.; Massie, B.M.; Carson, P.E.; et al. Relationship between heart rate and mortality and morbidity in the irbesartan patients with heart failure and preserved systolic function trial (I-Preserve). Eur. J. Heart Fail. 2014, 16, 778–787. [Google Scholar] [CrossRef]
- Simpson, J.; Castagno, D.; Doughty, R.N.; Poppe, K.K.; Earle, N.; Squire, I.; Richards, M.; Andersson, B.; Ezekowitz, J.A.; Komajda, M.; et al. Is heart rate a risk marker in patients with chronic heart failure and concomitant atrial fibrillation? Results from the MAGGIC meta-analysis. Eur. J. Heart Fail. 2015, 17, 1182–1191. [Google Scholar] [CrossRef] [Green Version]
- Wachter, R.; Schmidt-Schweda, S.; Westermann, D.; Post, H.; Edelmann, F.; Kasner, M.; Lüers, C.; Steendijk, P.; Hasenfuss, G.; Tschöpe, C.; et al. Blunted frequency-dependent upregulation of cardiac output is related to impaired relaxation in diastolic heart failure. Eur. Heart J. 2009, 30, 3027–3036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reil, J.-C.; Hohl, M.; Reil, G.-H.; Granzier, H.L.; Kratz, M.T.; Kazakov, A.; Fries, P.; Muller, A.; Lenski, M.; Custodis, F.; et al. Heart rate reduction by If-inhibition improves vascular stiffness and left ventricular systolic and diastolic function in a mouse model of heart failure with preserved ejection fraction. Eur. Heart J. 2013, 34, 2839–2849. [Google Scholar] [CrossRef] [PubMed]
- Komajda, M.; Isnard, R.; Cohen-Solal, A.; Metra, M.; Pieske, B.; Ponikowski, P.; Voors, A.A.; Dominjon, F.; Henon-Goburdhun, C.; Pannaux, M.; et al. Effect of ivabradine in patients with heart failure with preserved ejection fraction: The EDIFY randomized placebo-controlled trial. Eur. J. Heart Fail. 2017, 19, 1495–1503. [Google Scholar] [CrossRef] [Green Version]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. EMPA-REG OUTCOME Investigators. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Gallo, L.A.; Wright, E.M.; Vallon, V. Probing SGLT2 as a therapeutic target for diabetes: Basic physiology and consequences. Diab Vasc. Dis. Res. 2015, 12, 78–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, J.; Hamo, C.E.; Filippatos, G.; Pocock, S.J.; Bernstein, R.A.; Brueckmann, M.; Cheung, A.K.; George, J.T.; Green, J.B.; Januzzi, J.L.; et al. EMPEROR Trials Program. The potential role and rationale for treatment of heart failure with sodium-glucose co-transporter 2 inhibitors. Eur. J. Heart Fail. 2017, 19, 1390–1400. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.; McMurray, J.J.V.; Cherney, D.Z.I. The metabolo-diuretic promise of sodium-dependent glucose cotransporter 2 inhibition: The search for the sweet spot in heart failure. JAMA Cardiol. 2017, 2, 939–994. [Google Scholar] [CrossRef]
- Pabel, S.; Wagner, S.; Bollenberg, H.; Bengel, P.; Kovacs, A.; Schach, C.; Tirilomis, P.; Mustroph, J.; Renner, A.; Gummert, J.; et al. Empagliflozin directly improves diastolic function in human heart failure. Eur. J. Heart Fail. 2018, 20, 1690–1700. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Trial Name (Years) | Drug (Posology) | Sample Size | ClinicalTrials.gov Identifier | Follow up Duration | Results |
|---|---|---|---|---|---|
| RAAS and Neprylisin Pathway | |||||
| I-PRESERVE (2002–2008) | Irbesartan (Oral, from 75 to 300 mg daily vs. placebo) | 4128 | NCT00095238 | 49.5 months | Irbesartan did not improve outcomes (death from any cause or hospitalization for CV cause) |
| CHARM-PRESERVED (1999–2003) | Candesartan(32 mg once daily vs. placebo) | 3023 | NCT00634712 | 36.6 months | Candesartan did not improve outcomes (cardiovascular mortality or hospitalization due to congestive HF) |
| TOPCAT(2006–2013) | Spironolactone(Oral, 15 mg to 45 mg daily vs. placebo) | 3445 | NCT00094302 | 39 months | Spironolactone did not significantly reduce the incidence of the primary composite outcome of death from CV causes, aborted cardiac arrest, or hospitalization for the HF management |
| ALDO-DHF (2007–2012) | Spironolactone(Oral, 25 mg daily vs. placebo) | 422 | ISRCTN94726526 | 12 months | Long-term aldosterone receptor blockade improved left ventricular diastolic function but did not affect maximal exercise capacity, symptoms or quality of life |
| PARAGON-HF (2019–2019) | Sacubitril/Valsartan(Oral. Two periods:(1) a single-blind treatment from 3 to 8 weeks with valsartan 80 mg bid, followed by sacubitril/valsartan 100 mg bid(2) a double-blind randomized treatment with sacubitril/valsartan 200 mg bid or valsartan 160 mg bid | 4822 | NCT01920711 | 35 months | Sacubitril–valsartan did not result in a significantly lower rate of total hospitalizations for HF and death from CV causes |
| Oxidative stress and Nitric oxide pathway | |||||
| KNO3CK OUT-HFpEF (2016–2022) | Potassium Nitrate (KNO3)(Oral, 6 millimoles of inorganic nitrate per capsule, three times daily for 6 weeks vs. placebo) | 76 | NCT02840799 | N/A | Outcome: VO2 (ongoing study) |
| INDIE-HFpEF (2016–2018) | Inorganic nitrite or nitrate preparations(Nebulized sodium nitrite at 46 mg then 80 mg three times per day vs. placebo) | 105 | NCT02742129 | 17 months | Administration of inhaled inorganic nitrite for 4 weeks, compared with placebo, did not result in significant improvement in exercise capacity and VO2 |
| SOCRATES-PRESERVED (2013–2015) | Vericiguat(Oral, 2.5 mg once daily for 2 weeks, up-titration to 5 mg orally once daily for 2 weeks, up-titration to 10 mg orally once daily for 8 weeks vs. placebo) | 477 | NCT01951638 | 16 weeks | Vericiguat, did not change NT-proBNP levels at 12 weeks compared with placebo but it was associated with improvements in quality of life |
| NEAT-HFpEF (2014–2016) | Isosorbide mononitrate(6-week dose-escalation regimen of isosorbide mononitrate, from 30 mg to 60 mg to 120 mg once daily vs. placebo) | 110 | NCT02053493 | 6 weeks | Patients who received isosorbide mononitrate were less active and did not have better quality of life or submaximal exercise capacity than patients who received placebo |
| Inflammation pathway and calcium handling | |||||
| D-HART (2014–2017) | Anakinra(Subcutaneous, Interleukin-1 blockade, 100 mg subcutaneously once daily for 12 weeks vs. placebo) | 60 | NCT02173548 | 12 weeks | Anakinra significantly reduced the systemic inflammatory response and improved the aerobic exercise capacity of patients with HFpEF and elevated plasma CRP levels. |
| HELP (2018–2020) | Levosimendan(Injectable Solution 0.075–0.1 µg/kg/min for 24 h weekly vs. placebo) | 38 | NCT03541603 | 6 weeks | Levosimendan infusion did not affect exercise-PCWP but did reduce PCWP incorporating data from rest and exercise, in tandem with increased 6 min-walking-test |
| Fibrosis pathway | |||||
| PIROUETTE (2017–2020) | Pirfenidone(Oral, 801 mg three times daily vs. placebo | 129 | NCT02932566 | 12 months | Change in myocardial ECV from baseline to 52 weeks |
| SGLT-2 inhibition | |||||
| EMPEROR-Preserved (2017–2021) | Empagliflozin(Oral, 10 mg daily vs. placebo) | 5988 | NCT03057951 | 20 months | Time to first event of adjudicated CV death or HHF (ongoing) |
| DELIVER (2018–2022) | Dapagliflozin (Oral, 10 mg daily vs. placebo) | 6263 | NCT03619213 | 27 months | Composite of CV death, HHF and urgent HF visit (ongoing) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Severino, P.; D’Amato, A.; Prosperi, S.; Fanisio, F.; Birtolo, L.I.; Costi, B.; Netti, L.; Chimenti, C.; Lavalle, C.; Maestrini, V.; et al. Myocardial Tissue Characterization in Heart Failure with Preserved Ejection Fraction: From Histopathology and Cardiac Magnetic Resonance Findings to Therapeutic Targets. Int. J. Mol. Sci. 2021, 22, 7650. https://doi.org/10.3390/ijms22147650
Severino P, D’Amato A, Prosperi S, Fanisio F, Birtolo LI, Costi B, Netti L, Chimenti C, Lavalle C, Maestrini V, et al. Myocardial Tissue Characterization in Heart Failure with Preserved Ejection Fraction: From Histopathology and Cardiac Magnetic Resonance Findings to Therapeutic Targets. International Journal of Molecular Sciences. 2021; 22(14):7650. https://doi.org/10.3390/ijms22147650
Chicago/Turabian StyleSeverino, Paolo, Andrea D’Amato, Silvia Prosperi, Francesca Fanisio, Lucia Ilaria Birtolo, Bettina Costi, Lucrezia Netti, Cristina Chimenti, Carlo Lavalle, Viviana Maestrini, and et al. 2021. "Myocardial Tissue Characterization in Heart Failure with Preserved Ejection Fraction: From Histopathology and Cardiac Magnetic Resonance Findings to Therapeutic Targets" International Journal of Molecular Sciences 22, no. 14: 7650. https://doi.org/10.3390/ijms22147650
APA StyleSeverino, P., D’Amato, A., Prosperi, S., Fanisio, F., Birtolo, L. I., Costi, B., Netti, L., Chimenti, C., Lavalle, C., Maestrini, V., Mancone, M., & Fedele, F. (2021). Myocardial Tissue Characterization in Heart Failure with Preserved Ejection Fraction: From Histopathology and Cardiac Magnetic Resonance Findings to Therapeutic Targets. International Journal of Molecular Sciences, 22(14), 7650. https://doi.org/10.3390/ijms22147650