
From Genetic Mutations to Molecular Basis of Heart Failure Treatment: An Overview of the Mechanism and Implication of the Novel Modulators for Cardiac Myosin

 , ,
, ,
Abstract
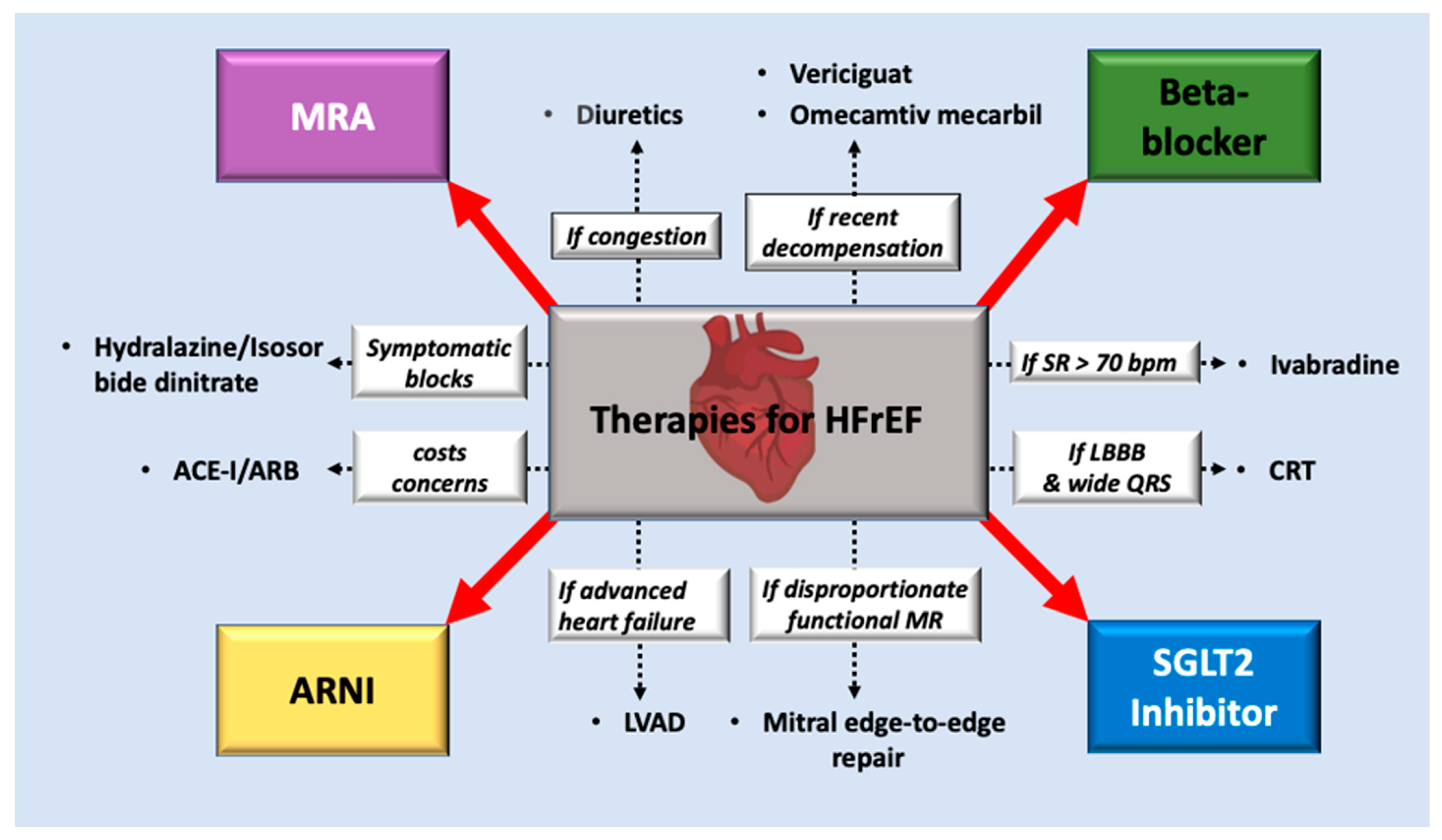
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Medications | Initial Dose | Target Dose | Trial | Year |
|---|---|---|---|---|
| ACE-I | ||||
| Enalapril [15] | 2.5 mg BID | 20 mg BID | CONSENSUS | 1987 |
| Fosinopril [16] | 5–10 mg daily | 40 mg daily | FEST | 1995 |
| Captopril [17] | 6.25–25 mg TID | 50 mg TID | SAVE | 1992 |
| Enalapril [18] | 2.5 mg BID | 20 mg BID | SOLVD | 1991 |
| Perindopril [19] | 2 mg daily | 16 mg daily | PEP-CHF | 2006 |
| Ramipril [20] | 1.25–2.5 mg daily | 10 mg daily | AIRE | 1993 |
| Trandolapril [21] | 1 mg daily | 4 mg daily | TRACE | 1995 |
| ARB | ||||
| Candesartan [22] | 4–8 mg daily | 32 mg daily | CHARM | 2004 |
| Losartan [23] | 25–50 mg daily | 150 mg daily | ELITE II | 2000 |
| Valsartan [24] | 20–40 mg BID | 160 mg BID | Val-HeFT | 2001 |
| Beta Blockers | ||||
| Bisoprolol [25] | 1.25 mg daily | 10 mg daily | CIBIS-II | 1999 |
| Carvedilol [26] | 3.125 mg BID | 50 mg BID | Carvedilol Heart Failure Study | 1996 |
| Metoprolol [27] | 12.5–25 mg daily | 200 mg daily | MERIT-HF | 1999 |
| Nebivolol [28] | 1.25 mg daily | 10 mg daily | SENIORS | 2005 |
| MRA | ||||
| Spironolactone [29] | 12.5 mg daily | 25 mg daily | RALES | 1999 |
| Eplerenone [30] | 25 mg daily | 50 mg daily | EMPHASIS-HF | 2010 |
| Vasodilators | ||||
| Hydralazine +Isosorbide dinitrate [31] | 25 mg TID20 mg TID | 300 mg daily160 mg daily | V-HeFT | 1986 |
| If Inhibitor | ||||
| Ivabradine [11] | 5 mg BID | 7.5 mg BID | SHIFT | 2010 |
| ARNI | ||||
| Sacubitril/valsartan [10] | 49 mg/51 mg BID | 97 mg/103 mg BID | PARADIGM-HF | 2014 |
| SGLT-2 inhibitors | ||||
| Dapagliflozin [12] | 5 mg daily | 10 mg daily | DAPA-HF | 2019 |
| Empagliflozin [32] | 10 mg daily | 10 mg daily | EMPEROR-Reduced | 2020 |
| Guanylate cyclase stimulator | ||||
| Vericiguat [13] | 2.5 mg daily | 10 mg daily | VICTORIA | 2020 |
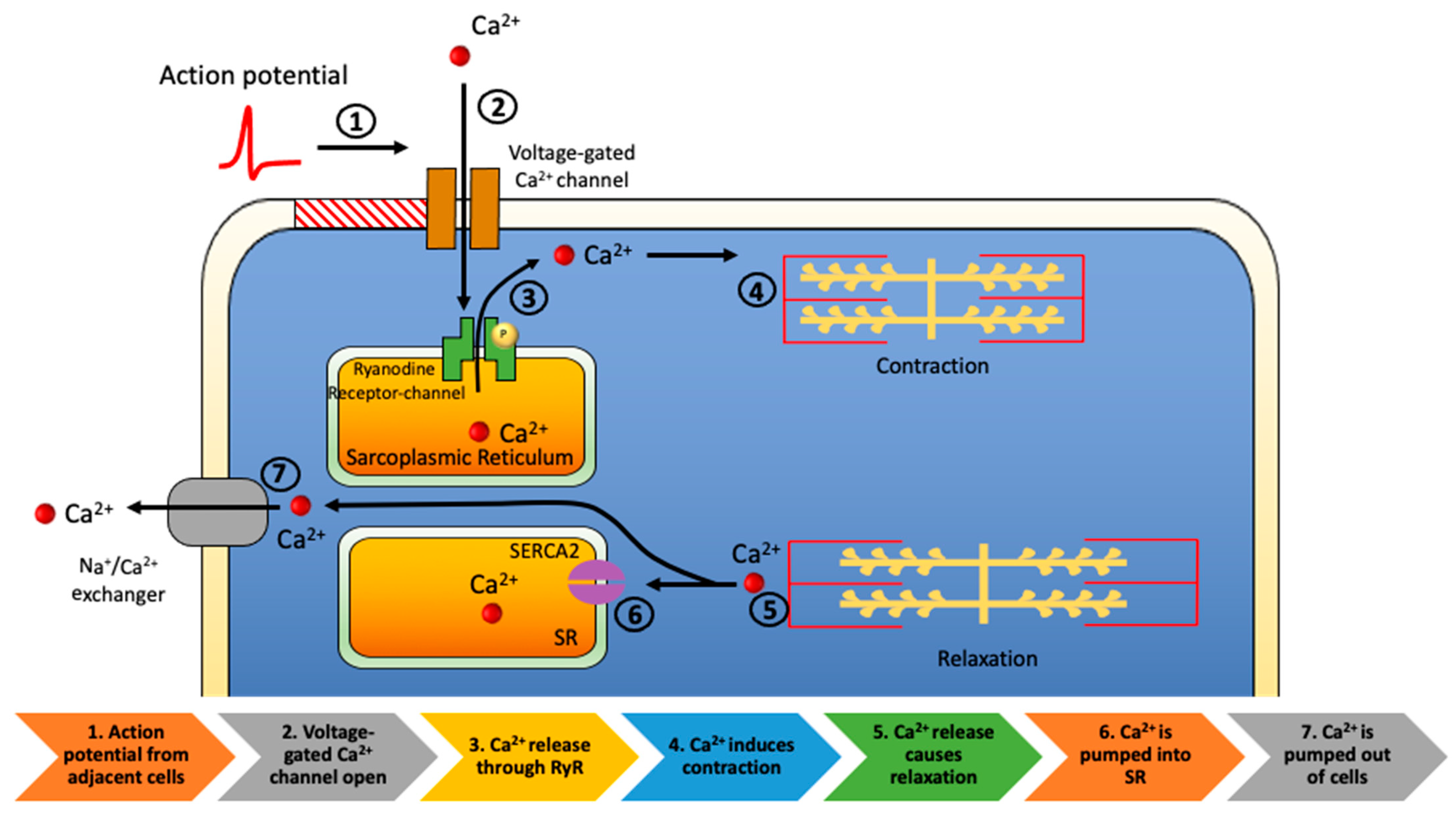
2. Cardiomyocyte Abnormalities in HF
3. Molecular Basis of Hereditary Cardiomyopathy
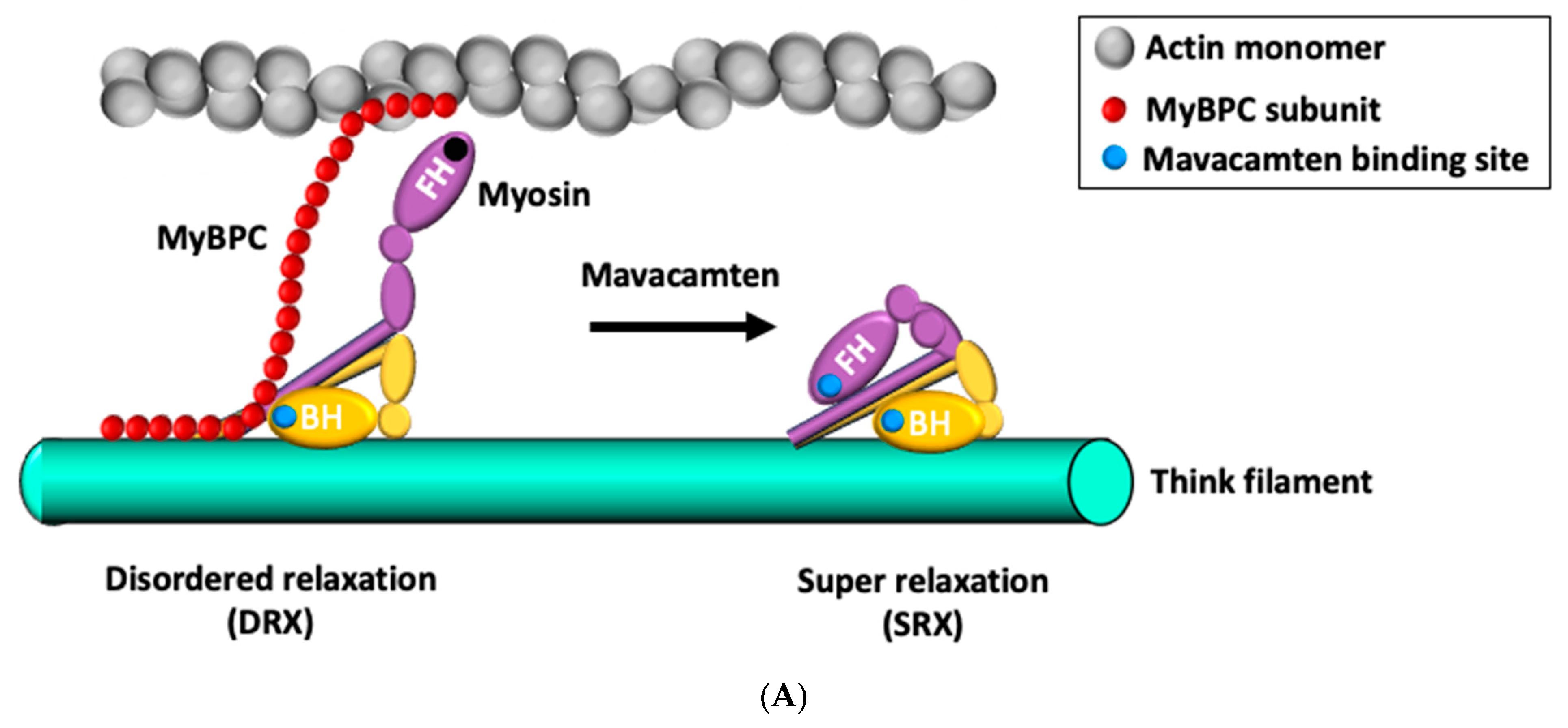
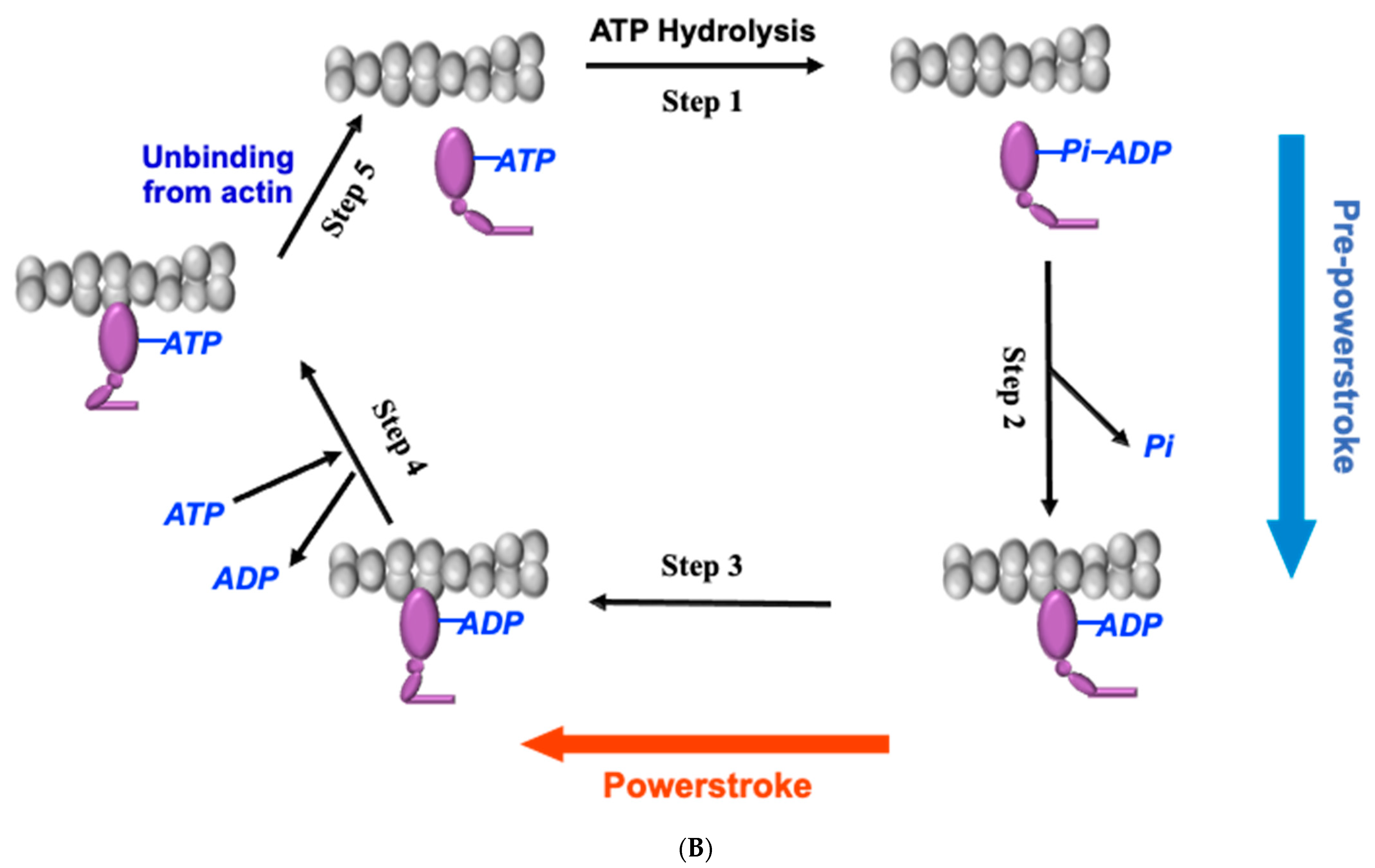
4. Small-Molecule Modulator of Cardiac Myosin
5. Novel Selective Cardiac Myosin Activator
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Groenewegen, A.; Rutten, F.H.; Mosterd, A.; Hoes, A.W. Epidemiology of heart failure. Eur. J. Heart Fail. 2020, 22, 1342–1356. [Google Scholar] [CrossRef]
- Ponikowski, P.; Anker, S.D.; AlHabib, K.F.; Cowie, M.R.; Force, T.L.; Hu, S.; Jaarsma, T.; Krum, H.; Rastogi, V.; Rohde, L.E.; et al. Heart failure: Preventing disease and death worldwide. ESC Heart Fail. 2014, 1, 4–25. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.S.P.; Voors, A.A.; Piotr, P.; McMurray, J.J.V.; Solomon, S.D. Time to rename the middle child of heart failure: Heart failure with mildly reduced ejection fraction. Eur. Heart J. 2020, 41, 2353–2355. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.D.; McMurray, J.J.V.; Anand, I.S.; Ge, J.; Lam, C.S.P.; Maggioni, A.P.; Martinez, F.; Packer, M.; Pfeffer, M.A.; Pieske, B.; et al. Angiotensin–Neprilysin Inhibition in Heart Failure with Preserved Ejection Fraction. N. Engl. J. Med. 2019, 381, 1609–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, G.; Gibbs, C.R.; Davies, M.K.; Lip, G.Y. ABC of heart failure. Pathophysiology. BMJ 2000, 320, 167–170. [Google Scholar] [CrossRef]
- Hartupee, J.; Mann, D.L. Neurohormonal activation in heart failure with reduced ejection fraction. Nat. Rev. Cardiol. 2017, 14, 30–38. [Google Scholar] [CrossRef] [Green Version]
- George, J.; Struthers, A.D.; Lang, C.C. Modulation of the renin-angiotensin-aldosterone system in heart failure. Curr. Atheroscler. Rep. 2014, 16, 403. [Google Scholar] [CrossRef]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.-P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E.; Colvin, M.M.; Drazner, M.H.; Filippatos, G.S.; Fonarow, G.C.; Givertz, M.M.; et al. 2017 ACC/AHA/HFSA Focused Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure. J. Am. Coll. Cardiol. 2017, 70, 776–803. [Google Scholar] [CrossRef]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef] [Green Version]
- Swedberg, K.; Komajda, M.; Böhm, M.; Borer, J.S.; Ford, I.; Dubost-Brama, A.; Lerebours, G.; Tavazzi, L. Ivabradine and outcomes in chronic heart failure (SHIFT): A randomised placebo-controlled study. Lancet 2010, 376, 875–885. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, P.W.; Pieske, B.; Anstrom, K.J.; Ezekowitz, J.; Hernandez, A.F.; Butler, J.; Lam, C.S.P.; Ponikowski, P.; Voors, A.A.; Jia, G.; et al. Vericiguat in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2020, 382, 1883–1893. [Google Scholar] [CrossRef]
- Weintraub, R.G.; Semsarian, C.; Macdonald, P. Dilated cardiomyopathy. Lancet 2017, 390, 400–414. [Google Scholar] [CrossRef]
- CONSENSUS Trial Study Group. Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). N. Engl. J. Med. 1987, 316, 1429–1435. [Google Scholar] [CrossRef]
- Erhardt, L.; MacLean, A.; Ilgenfritz, J.; Gelperin, K.; Blumenthal, M. Fosinopril attenuates clinical deterioration and improves exercise tolerance in patients with heart failure. Fosinopril Efficacy/Safety Trial (FEST) Study Group. Eur. Heart J. 1995, 16, 1892–1899. [Google Scholar] [CrossRef]
- Pfeffer, M.A.; Braunwald, E.; Moyé, L.A.; Basta, L.; Brown, E.J.; Cuddy, T.E.; Davis, B.R.; Geltman, E.M.; Goldman, S.; Flaker, G.C.; et al. Effect of Captopril on Mortality and Morbidity in Patients with Left Ventricular Dysfunction after Myocardial Infarction. N. Engl. J. Med. 1992, 327, 669–677. [Google Scholar] [CrossRef] [Green Version]
- The SOLVD Investigators. Effect of Enalapril on Survival in Patients with Reduced Left Ventricular Ejection Fractions and Congestive Heart Failure. N. Engl. J. Med. 1991, 325, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Cleland, J.G.; Tendera, M.; Adamus, J.; Freemantle, N.; Polonski, L.; Taylor, J. The perindopril in elderly people with chronic heart failure (PEP-CHF) study. Eur. Heart J. 2006, 27, 2338–2345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Acute Infarction Ramipril Efficacy (AIRE) Study Investigators. Effect of ramipril on mortality and morbidity of survivors of acute myocardial infarction with clinical evidence of heart failure. Lancet 1993, 342, 821–828. [Google Scholar]
- Køber, L.; Torp-Pedersen, C.; Carlsen, J.E.; Bagger, H.; Eliasen, P.; Lyngborg, K.; Videbæk, J.; Cole, D.S.; Auclert, L.; Pauly, N.C.; et al. A Clinical Trial of the Angiotensin-Converting-Enzyme Inhibitor Trandolapril in Patients with Left Ventricular Dysfunction after Myocardial Infarction. N. Engl. J. Med. 1995, 333, 1670–1676. [Google Scholar] [CrossRef] [PubMed]
- Young, J.B.; Dunlap, M.E.; Pfeffer, M.A.; Probstfield, J.L.; Cohen-Solal, A.; Dietz, R.; Granger, C.B.; Hradec, J.; Kuch, J.; McKelvie, R.S.; et al. Mortality and morbidity reduction with Candesartan in patients with chronic heart failure and left ventricular systolic dysfunction: Results of the CHARM low-left ventricular ejection fraction trials. Circulation 2004, 110, 2618–2626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitt, B.; Poole-Wilson, P.A.; Segal, R.; Martinez, F.A.; Dickstein, K.; Camm, A.J.; Konstam, M.A.; Riegger, G.; Klinger, G.H.; Neaton, J.; et al. Effect of losartan compared with captopril on mortality in patients with symptomatic heart failure: Randomised trial—The Losartan Heart Failure Survival Study ELITE II. Lancet 2000, 355, 1582–1587. [Google Scholar] [CrossRef]
- Cohn, J.N.; Tognoni, G. A Randomized Trial of the Angiotensin-Receptor Blocker Valsartan in Chronic Heart Failure. N. Engl. J. Med. 2001, 345, 1667–1675. [Google Scholar] [CrossRef]
- Lechat, P.F.; Brunhuber, K.W.; Hofmann, R.; Kuhn, P.; Nesser, H.J.; Slany, J.; Weihs, W.; Wiedermann, C.; Wimmer, H.; Van Mieghem, W.; et al. The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): A randomised trial. Lancet 1999, 353, 9–13. [Google Scholar]
- Packer, M.; Bristow, M.R.; Cohn, J.N.; Colucci, W.S.; Fowler, M.B.; Gilbert, E.M.; Shusterman, N.H. The Effect of Carvedilol on Morbidity and Mortality in Patients with Chronic Heart Failure. N. Engl. J. Med. 1996, 334, 1349–1355. [Google Scholar] [CrossRef]
- MERIT-HF Study Group. Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet 1999, 353, 2001–2007. [Google Scholar]
- Flather, M.D.; Shibata, M.C.; Coats, A.J.; Van Veldhuisen, D.J.; Parkhomenko, A.; Borbola, J.; Cohen-Solal, A.; Dumitrascu, D.; Ferrari, R.; Lechat, P.; et al. Randomized trial to determine the effect of nebivolol on mortality and cardiovascular hospital admission in elderly patients with heart failure (SENIORS). Eur. Heart J. 2005, 26, 215–225. [Google Scholar] [CrossRef]
- Pitt, B.; Zannad, F.; Remme, W.J.; Cody, R.; Castaigne, A.; Perez, A.; Palensky, J.; Wittes, J. The Effect of Spironolactone on Morbidity and Mortality in Patients with Severe Heart Failure. N. Engl. J. Med. 1999, 341, 709–717. [Google Scholar] [CrossRef] [Green Version]
- Zannad, F.; McMurray, J.J.V.; Krum, H.; van Veldhuisen, D.J.; Swedberg, K.; Shi, H.; Vincent, J.; Pocock, S.J.; Pitt, B. Eplerenone in Patients with Systolic Heart Failure and Mild Symptoms. N. Engl. J. Med. 2010, 364, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Cohn, J.N.; Archibald, D.G.; Ziesche, S.; Franciosa, J.A.; Harston, W.E.; Tristani, F.E.; Dunkman, W.B.; Jacobs, W.; Francis, G.S.; Flohr, K.H.; et al. Effect of Vasodilator Therapy on Mortality in Chronic Congestive Heart Failure. N. Engl. J. Med. 1986, 314, 1547–1552. [Google Scholar] [CrossRef]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef]
- Eisner, D.A.; Caldwell, J.L.; Kistamás, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef]
- Moore, J.R.; Campbell, S.G.; Lehman, W. Structural determinants of muscle thin filament cooperativity. Arch. Biochem. Biophys. 2016, 594, 8–17. [Google Scholar] [CrossRef] [Green Version]
- Park, W.J.; Oh, J.G. SERCA2a: A prime target for modulation of cardiac contractility during heart failure. BMB Rep. 2013, 46, 237–243. [Google Scholar] [CrossRef] [Green Version]
- Burke, M.A.; Cook, S.A.; Seidman, J.G.; Seidman, C.E. Clinical and Mechanistic Insights into the Genetics of Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 68, 2871–2886. [Google Scholar] [CrossRef]
- Ingles, J.; Sarina, T.; Yeates, L.; Hunt, L.; Macciocca, I.; McCormack, L.; Winship, I.; McGaughran, J.; Atherton, J.; Semsarian, C. Clinical predictors of genetic testing outcomes in hypertrophic cardiomyopathy. Genet. Med. Off. J. Am. Coll. Med. Genet. 2013, 15, 972–977. [Google Scholar] [CrossRef] [Green Version]
- McNally, E.M.; Mestroni, L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ. Res. 2017, 121, 731–748. [Google Scholar] [CrossRef]
- Hughes, S.E.; McKenna, W.J. New insights into the pathology of inherited cardiomyopathy. Heart Br. Card. Soc. 2005, 91, 257–264. [Google Scholar] [CrossRef] [Green Version]
- Vikhorev, P.G.; Vikhoreva, N.N. Cardiomyopathies and Related Changes in Contractility of Human Heart Muscle. Int. J. Mol. Sci. 2018, 19, 2234. [Google Scholar] [CrossRef] [Green Version]
- Ameri, P.; Schiattarella, G.G.; Crotti, L.; Torchio, M.; Bertero, E.; Rodolico, D.; Forte, M.; Di Mauro, V.; Paolillo, R.; Chimenti, C.; et al. Novel Basic Science Insights to Improve the Management of Heart Failure: Review of the Working Group on Cellular and Molecular Biology of the Heart of the Italian Society of Cardiology. Int. J. Mol. Sci. 2020, 21, 1192. [Google Scholar] [CrossRef] [Green Version]
- Predmore, J.M.; Wang, P.; Davis, F.; Bartolone, S.; Westfall, M.V.; Dyke, D.B.; Pagani, F.; Powell, S.R.; Day, S.M. Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies. Circulation 2010, 121, 997–1004. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Ren, J.; Guo, W. Sarcomeric protein isoform transitions in cardiac muscle: A journey to heart failure. Biochim. Biophys. Acta 2015, 1852, 47–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dijk, S.J.; Holewijn, R.A.; Tebeest, A.; Dos Remedios, C.; Stienen, G.J.; van der Velden, J. A piece of the human heart: Variance of protein phosphorylation in left ventricular samples from end-stage primary cardiomyopathy patients. J. Muscle Res. Cell Motil. 2009, 30, 299–302. [Google Scholar] [CrossRef] [Green Version]
- De Tombe, P.P.; Mateja, R.D.; Tachampa, K.; Ait Mou, Y.; Farman, G.P.; Irving, T.C. Myofilament length dependent activation. J. Mol. Cell. Cardiol. 2010, 48, 851–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alamo, L.; Ware, J.S. Effects of myosin variants on interacting-heads motif explain distinct hypertrophic and dilated cardiomyopathy phenotypes. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Trivedi, D.V. The myosin mesa and the basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Nat. Struct. Mol. Biol. 2017, 24, 525–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brito, R.; Alamo, L.; Lundberg, U.; Guerrero, J.R.; Pinto, A.; Sulbarán, G.; Gawinowicz, M.A.; Craig, R.; Padrón, R. A molecular model of phosphorylation-based activation and potentiation of tarantula muscle thick filaments. J. Mol. Biol. 2011, 414, 44–61. [Google Scholar] [CrossRef] [Green Version]
- Stewart, M.A.; Franks-Skiba, K.; Chen, S.; Cooke, R. Myosin ATP turnover rate is a mechanism involved in thermogenesis in resting skeletal muscle fibers. Proc. Natl. Acad. Sci. USA 2010, 107, 430–435. [Google Scholar] [CrossRef] [Green Version]
- McNamara, J.W.; Li, A.; Smith, N.J.; Lal, S.; Graham, R.M.; Kooiker, K.B.; van Dijk, S.J.; Remedios, C.G.D.; Harris, S.P.; Cooke, R. Ablation of cardiac myosin binding protein-C disrupts the super-relaxed state of myosin in murine cardiomyocytes. J. Mol. Cell. Cardiol. 2016, 94, 65–71. [Google Scholar] [CrossRef] [Green Version]
- Walsh, R.; Buchan, R.; Wilk, A.; John, S.; Felkin, L.E.; Thomson, K.L.; Chiaw, T.H.; Loong, C.C.W.; Pua, C.J.; Raphael, C.; et al. Defining the genetic architecture of hypertrophic cardiomyopathy: Re-evaluating the role of non-sarcomeric genes. Eur. Heart J. 2017, 38, 3461–3468. [Google Scholar] [CrossRef]
- Kimura, A. Molecular basis of hereditary cardiomyopathy: Abnormalities in calcium sensitivity, stretch response, stress response and beyond. J. Hum. Genet. 2010, 55, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Redwood, C.; Robinson, P. Alpha-tropomyosin mutations in inherited cardiomyopathies. J. Muscle Res. Cell Motil. 2013, 34, 285–294. [Google Scholar] [CrossRef]
- Pasquale, F.; Syrris, P.; Kaski, J.P.; Mogensen, J.; McKenna, W.J.; Elliott, P. Long-term outcomes in hypertrophic cardiomyopathy caused by mutations in the cardiac troponin T gene. Circulation. Cardiovasc. Genet. 2012, 5, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Lossie, J.; Ushakov, D.S.; Ferenczi, M.A.; Werner, S.; Keller, S.; Haase, H.; Morano, I. Mutations of ventricular essential myosin light chain disturb myosin binding and sarcomeric sorting. Cardiovasc. Res. 2011, 93, 390–396. [Google Scholar] [CrossRef] [Green Version]
- Claes, G.R.F.; van Tienen, F.H.J.; Lindsey, P.; Krapels, I.P.C.; Helderman-van den Enden, A.T.J.M.; Hoos, M.B.; Barrois, Y.E.G.; Janssen, J.W.H.; Paulussen, A.D.C.; Sels, J.-W.; et al. Hypertrophic remodelling in cardiac regulatory myosin light chain (MYL2) founder mutation carriers. Eur. Heart J. 2015, 37, 1815–1822. [Google Scholar] [CrossRef] [Green Version]
- Kimura, A.; Harada, H.; Park, J.E.; Nishi, H.; Satoh, M.; Takahashi, M.; Hiroi, S.; Sasaoka, T.; Ohbuchi, N.; Nakamura, T.; et al. Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat. Genet. 1997, 16, 379–382. [Google Scholar] [CrossRef]
- Despond, E.A.; Dawson, J.F. Classifying Cardiac Actin Mutations Associated with Hypertrophic Cardiomyopathy. Front. Physiol. 2018, 9, 405. [Google Scholar] [CrossRef]
- Sweeney, H.L.; Straceski, A.J.; Leinwand, L.A.; Tikunov, B.A.; Faust, L. Heterologous expression of a cardiomyopathic myosin that is defective in its actin interaction. J. Biol. Chem. 1994, 269, 1603–1605. [Google Scholar] [CrossRef]
- Thierfelder, L.; Watkins, H.; MacRae, C.; Lamas, R.; McKenna, W.; Vosberg, H.P.; Seidman, J.G.; Seidman, C.E. Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: A disease of the sarcomere. Cell 1994, 77, 701–712. [Google Scholar] [CrossRef]
- Witt, C.C.; Gerull, B.; Davies, M.J.; Centner, T.; Linke, W.A.; Thierfelder, L. Hypercontractile properties of cardiac muscle fibers in a knock-in mouse model of cardiac myosin-binding protein-C. J. Biol. Chem. 2001, 276, 5353–5359. [Google Scholar] [CrossRef] [Green Version]
- Roopnarine, O. Mechanical defects of muscle fibers with myosin light chain mutants that cause cardiomyopathy. Biophys. J. 2003, 84, 2440–2449. [Google Scholar] [CrossRef] [Green Version]
- Pinto, J.R.; Parvatiyar, M.S.; Jones, M.A.; Liang, J.; Ackerman, M.J.; Potter, J.D. A functional and structural study of troponin C mutations related to hypertrophic cardiomyopathy. J. Biol. Chem. 2009, 284, 19090–19100. [Google Scholar] [CrossRef] [Green Version]
- Hooijman, P.; Stewart, M.A.; Cooke, R. A new state of cardiac myosin with very slow ATP turnover: A potential cardioprotective mechanism in the heart. Biophys. J. 2011, 100, 1969–1976. [Google Scholar] [CrossRef] [Green Version]
- Toepfer, C.N.; Garfinkel, A.C.; Venturini, G.; Wakimoto, H.; Repetti, G.; Alamo, L.; Sharma, A.; Agarwal, R.; Ewoldt, J.F.; Cloonan, P.; et al. Myosin Sequestration Regulates Sarcomere Function, Cardiomyocyte Energetics, and Metabolism, Informing the Pathogenesis of Hypertrophic Cardiomyopathy. Circulation 2020, 141, 828–842. [Google Scholar] [CrossRef]
- Kamisago, M.; Sharma, S.D.; DePalma, S.R.; Solomon, S.; Sharma, P.; McDonough, B.; Smoot, L.; Mullen, M.P.; Woolf, P.K.; Wigle, E.D.; et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N. Engl. J. Med. 2000, 343, 1688–1696. [Google Scholar] [CrossRef] [PubMed]
- Kärkkäinen, S.; Heliö, T.; Jääskeläinen, P.; Miettinen, R.; Tuomainen, P.; Ylitalo, K.; Kaartinen, M.; Reissell, E.; Toivonen, L.; Nieminen, M.S.; et al. Two novel mutations in the beta-myosin heavy chain gene associated with dilated cardiomyopathy. Eur. J. Heart Fail. 2004, 6, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Liang, J.; Yuan, C.C.; Kazmierczak, K.; Zhou, Z.; Morales, A.; McBride, K.L.; Fitzgerald-Butt, S.M.; Hershberger, R.E.; Szczesna-Cordary, D. Novel familial dilated cardiomyopathy mutation in MYL2 affects the structure and function of myosin regulatory light chain. FEBS J. 2015, 282, 2379–2393. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, J.P.; Debold, E.P.; Ahmad, F.; Armstrong, A.; Frederico, A.; Conner, D.A.; Mende, U.; Lohse, M.J.; Warshaw, D.; Seidman, C.E.; et al. Cardiac myosin missense mutations cause dilated cardiomyopathy in mouse models and depress molecular motor function. Proc. Natl. Acad. Sci. USA 2006, 103, 14525–14530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, C.C.; Kazmierczak, K.; Liang, J.; Zhou, Z.; Yadav, S.; Gomes, A.V.; Irving, T.C.; Szczesna-Cordary, D. Sarcomeric perturbations of myosin motors lead to dilated cardiomyopathy in genetically modified MYL2 mice. Proc. Natl. Acad. Sci. USA 2018, 115, E2338–E2347. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.N.; Potter, J.D. Sarcomeric protein mutations in dilated cardiomyopathy. Heart Fail. Rev. 2005, 10, 225–235. [Google Scholar] [CrossRef]
- Vang, S.; Corydon, T.J.; Børglum, A.D.; Scott, M.D.; Frydman, J.; Mogensen, J.; Gregersen, N.; Bross, P. Actin mutations in hypertrophic and dilated cardiomyopathy cause inefficient protein folding and perturbed filament formation. FEBS J. 2005, 272, 2037–2049. [Google Scholar] [CrossRef]
- Morimoto, S.; Lu, Q.W.; Harada, K.; Takahashi-Yanaga, F.; Minakami, R.; Ohta, M.; Sasaguri, T.; Ohtsuki, I. Ca(2+)-desensitizing effect of a deletion mutation Delta K210 in cardiac troponin T that causes familial dilated cardiomyopathy. Proc. Natl. Acad. Sci. USA 2002, 99, 913–918. [Google Scholar] [CrossRef] [Green Version]
- Kawas, R.F.; Anderson, R.L.; Ingle, S.R.B.; Song, Y.; Sran, A.S.; Rodriguez, H.M. A small-molecule modulator of cardiac myosin acts on multiple stages of the myosin chemomechanical cycle. J. Biol. Chem. 2017, 292, 16571–16577. [Google Scholar] [CrossRef] [Green Version]
- Grillo, M.P.; Erve, J.C.L.; Dick, R.; Driscoll, J.P.; Haste, N.; Markova, S.; Brun, P.; Carlson, T.J.; Evanchik, M. In vitro and in vivo pharmacokinetic characterization of mavacamten, a first-in-class small molecule allosteric modulator of beta cardiac myosin. Xenobiotica Fate Foreign Compd. Biol. Syst. 2019, 49, 718–733. [Google Scholar] [CrossRef] [PubMed]
- Green, E.M.; Wakimoto, H.; Anderson, R.L.; Evanchik, M.J.; Gorham, J.M.; Harrison, B.C.; Henze, M.; Kawas, R.; Oslob, J.D.; Rodriguez, H.M.; et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 2016, 351, 617–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivotto, I.; Oreziak, A.; Barriales-Villa, R.; Abraham, T.P.; Masri, A.; Garcia-Pavia, P.; Saberi, S.; Lakdawala, N.K.; Wheeler, M.T.; Owens, A.; et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 396, 759–769. [Google Scholar] [CrossRef]
- McNamara, J.W.; Li, A.; Lal, S.; Bos, J.M.; Harris, S.P.; van der Velden, J.; Ackerman, M.J.; Cooke, R.; Dos Remedios, C.G. MYBPC3 mutations are associated with a reduced super-relaxed state in patients with hypertrophic cardiomyopathy. PLoS ONE 2017, 12, e0180064. [Google Scholar] [CrossRef]
- Rohde, J.A.; Roopnarine, O.; Thomas, D.D.; Muretta, J.M. Mavacamten stabilizes an autoinhibited state of two-headed cardiac myosin. Proc. Natl. Acad. Sci. USA 2018, 115, E7486–E7494. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.L.; Trivedi, D.V.; Sarkar, S.S.; Henze, M.; Ma, W.; Gong, H.; Rogers, C.S.; Gorham, J.M.; Wong, F.L.; Morck, M.M.; et al. Deciphering the super relaxed state of human β-cardiac myosin and the mode of action of mavacamten from myosin molecules to muscle fibers. Proc. Natl. Acad. Sci. USA 2018, 115, E8143–E8152. [Google Scholar] [CrossRef] [Green Version]
- Metra, M.; Teerlink, J.R. Heart failure. Lancet 2017, 390, 1981–1995. [Google Scholar] [CrossRef]
- The Xamoterol in Severe Heart Failure Study Group. Xamoterol in severe heart failure. Lancet 1990, 336, 1–6. [Google Scholar] [CrossRef]
- Packer, M.; Carver, J.R.; Rodeheffer, R.J.; Ivanhoe, R.J.; DiBianco, R.; Zeldis, S.M.; Hendrix, G.H.; Bommer, W.J.; Elkayam, U.; Kukin, M.L.; et al. Effect of oral milrinone on mortality in severe chronic heart failure. The PROMISE Study Research Group. N. Engl. J. Med. 1991, 325, 1468–1475. [Google Scholar] [CrossRef]
- Packer, M.; Pitt, B.; Rouleau, J.L.; Swedberg, K.; DeMets, D.L.; Fisher, L. Long-Term Effects of Flosequinan on the Morbidity and Mortality of Patients with Severe Chronic Heart Failure: Primary Results of the PROFILE Trial After 24 Years. JACC Heart Fail. 2017, 5, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Lubsen, J.; Just, H.; Hjalmarsson, A.C.; La Framboise, D.; Remme, W.J.; Heinrich-Nols, J.; Dumont, J.M.; Seed, P. Effect of pimobendan on exercise capacity in patients with heart failure: Main results from the Pimobendan in Congestive Heart Failure (PICO) trial. Heart Br. Card. Soc. 1996, 76, 223–231. [Google Scholar] [CrossRef]
- Hampton, J.R.; van Veldhuisen, D.J.; Kleber, F.X.; Cowley, A.J.; Ardia, A.; Block, P.; Cortina, A.; Cserhalmi, L.; Follath, F.; Jensen, G.; et al. Randomised study of effect of ibopamine on survival in patients with advanced severe heart failure. Second Prospective Randomised Study of Ibopamine on Mortality and Efficacy (PRIME II) Investigators. Lancet 1997, 349, 971–977. [Google Scholar] [CrossRef] [Green Version]
- Feldman, A.M.; Oren, R.M.; Abraham, W.T.; Boehmer, J.P.; Carson, P.E.; Eichhorn, E.; Gilbert, E.M.; Kao, A.; Leier, C.V.; Lowes, B.D.; et al. Low-dose oral enoximone enhances the ability to wean patients with ultra-advanced heart failure from intravenous inotropic support: Results of the oral enoximone in intravenous inotrope-dependent subjects trial. Am. Heart J. 2007, 154, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Metra, M.; Eichhorn, E.; Abraham, W.T.; Linseman, J.; Böhm, M.; Corbalan, R.; DeMets, D.; De Marco, T.; Elkayam, U.; Gerber, M.; et al. Effects of low-dose oral enoximone administration on mortality, morbidity, and exercise capacity in patients with advanced heart failure: The randomized, double-blind, placebo-controlled, parallel group ESSENTIAL trials. Eur. Heart J. 2009, 30, 3015–3026. [Google Scholar] [CrossRef] [PubMed]
- Malik, F.I.; Hartman, J.J.; Elias, K.A.; Morgan, B.P.; Rodriguez, H.; Brejc, K.; Anderson, R.L.; Sueoka, S.H.; Lee, K.H.; Finer, J.T.; et al. Cardiac myosin activation: A potential therapeutic approach for systolic heart failure. Science 2011, 331, 1439–1443. [Google Scholar] [CrossRef] [Green Version]
- Planelles-Herrero, V.J.; Hartman, J.J. Mechanistic and structural basis for activation of cardiac myosin force production by omecamtiv mecarbil. Nat. Commun. 2017, 8, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Psotka, M.A.; Gottlieb, S.S.; Francis, G.S.; Allen, L.A.; Teerlink, J.R.; Adams, K.F., Jr.; Rosano, G.M.C.; Lancellotti, P. Cardiac Calcitropes, Myotropes, and Mitotropes: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 2345–2353. [Google Scholar] [CrossRef] [PubMed]
- Szentandrassy, N.; Horvath, B.; Vaczi, K.; Kistamas, K.; Masuda, L.; Magyar, J.; Banyasz, T.; Papp, Z.; Nanasi, P.P. Dose-dependent electrophysiological effects of the myosin activator omecamtiv mecarbil in canine ventricular cardiomyocytes. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2016, 67, 483–489. [Google Scholar]
- Teerlink, J.R.; Felker, G.M.; McMurray, J.J.V.; Ponikowski, P.; Metra, M.; Filippatos, G.S.; Ezekowitz, J.A.; Dickstein, K.; Cleland, J.G.F.; Kim, J.B.; et al. Acute Treatment with Omecamtiv Mecarbil to Increase Contractility in Acute Heart Failure: The ATOMIC-AHF Study. J. Am. Coll. Cardiol. 2016, 67, 1444–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teerlink, J.R.; Felker, G.M.; McMurray, J.J.; Solomon, S.D.; Adams, K.F., Jr.; Cleland, J.G.; Ezekowitz, J.A.; Goudev, A.; Macdonald, P.; Metra, M.; et al. Chronic Oral Study of Myosin Activation to Increase Contractility in Heart Failure (COSMIC-HF): A phase 2, pharmacokinetic, randomised, placebo-controlled trial. Lancet 2016, 388, 2895–2903. [Google Scholar] [CrossRef] [Green Version]
- Biering-Sørensen, T.; Querejeta Roca, G.; Hegde, S.M.; Shah, A.M.; Claggett, B.; Mosley, T.H., Jr.; Butler, K.R., Jr.; Solomon, S.D. Left ventricular ejection time is an independent predictor of incident heart failure in a community-based cohort. Eur. J. Heart Fail. 2018, 20, 1106–1114. [Google Scholar] [CrossRef] [Green Version]
- Alhakak, A.S.; Sengeløv, M.; Jørgensen, P.G.; Bruun, N.E.; Johnsen, C.; Abildgaard, U.; Iversen, A.Z.; Hansen, T.F.; Teerlink, J.R.; Malik, F.I.; et al. Left ventricular systolic ejection time is an independent predictor of all-cause mortality in heart failure with reduced ejection fraction. Eur. J. Heart Fail. 2021, 23, 240–249. [Google Scholar] [CrossRef]
- Teerlink, J.R.; Diaz, R.; Felker, G.M.; McMurray, J.J.V.; Metra, M.; Solomon, S.D.; Legg, J.C.; Büchele, G.; Varin, C.; Kurtz, C.E.; et al. Omecamtiv Mecarbil in Chronic Heart Failure with Reduced Ejection Fraction: Rationale and Design of GALACTIC-HF. JACC Heart Fail. 2020, 8, 329–340. [Google Scholar] [CrossRef]
- Teerlink, J.R.; Diaz, R.; Felker, G.M.; McMurray, J.J.V.; Metra, M.; Solomon, S.D.; Adams, K.F.; Anand, I.; Arias-Mendoza, A.; Biering-Sørensen, T.; et al. Cardiac Myosin Activation with Omecamtiv Mecarbil in Systolic Heart Failure. N. Engl. J. Med. 2020, 384, 105–116. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-J.; Chien, C.-S.; Chiang, C.-E.; Chen, C.-H.; Cheng, H.-M. From Genetic Mutations to Molecular Basis of Heart Failure Treatment: An Overview of the Mechanism and Implication of the Novel Modulators for Cardiac Myosin. Int. J. Mol. Sci. 2021, 22, 6617. https://doi.org/10.3390/ijms22126617
Chen Y-J, Chien C-S, Chiang C-E, Chen C-H, Cheng H-M. From Genetic Mutations to Molecular Basis of Heart Failure Treatment: An Overview of the Mechanism and Implication of the Novel Modulators for Cardiac Myosin. International Journal of Molecular Sciences. 2021; 22(12):6617. https://doi.org/10.3390/ijms22126617
Chicago/Turabian StyleChen, Yu-Jen, Chian-Shiu Chien, Chern-En Chiang, Chen-Huan Chen, and Hao-Min Cheng. 2021. "From Genetic Mutations to Molecular Basis of Heart Failure Treatment: An Overview of the Mechanism and Implication of the Novel Modulators for Cardiac Myosin" International Journal of Molecular Sciences 22, no. 12: 6617. https://doi.org/10.3390/ijms22126617
APA StyleChen, Y. -J., Chien, C. -S., Chiang, C. -E., Chen, C. -H., & Cheng, H. -M. (2021). From Genetic Mutations to Molecular Basis of Heart Failure Treatment: An Overview of the Mechanism and Implication of the Novel Modulators for Cardiac Myosin. International Journal of Molecular Sciences, 22(12), 6617. https://doi.org/10.3390/ijms22126617