
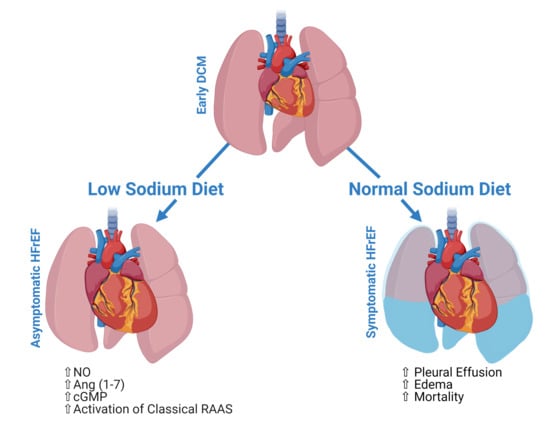
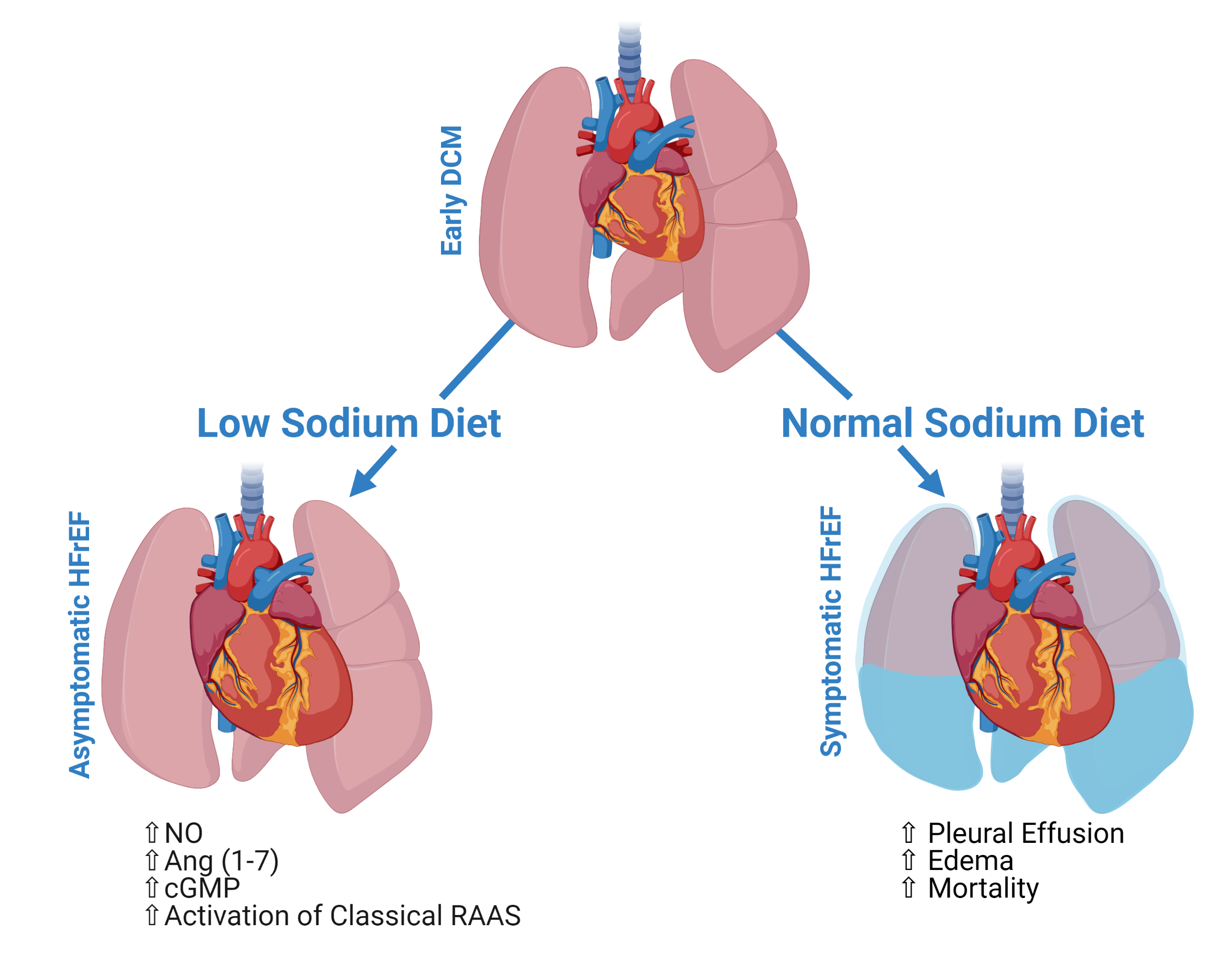
A Low-Sodium Diet Boosts Ang (1–7) Production and NO-cGMP Bioavailability to Reduce Edema and Enhance Survival in Experimental Heart Failure

 ,
,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
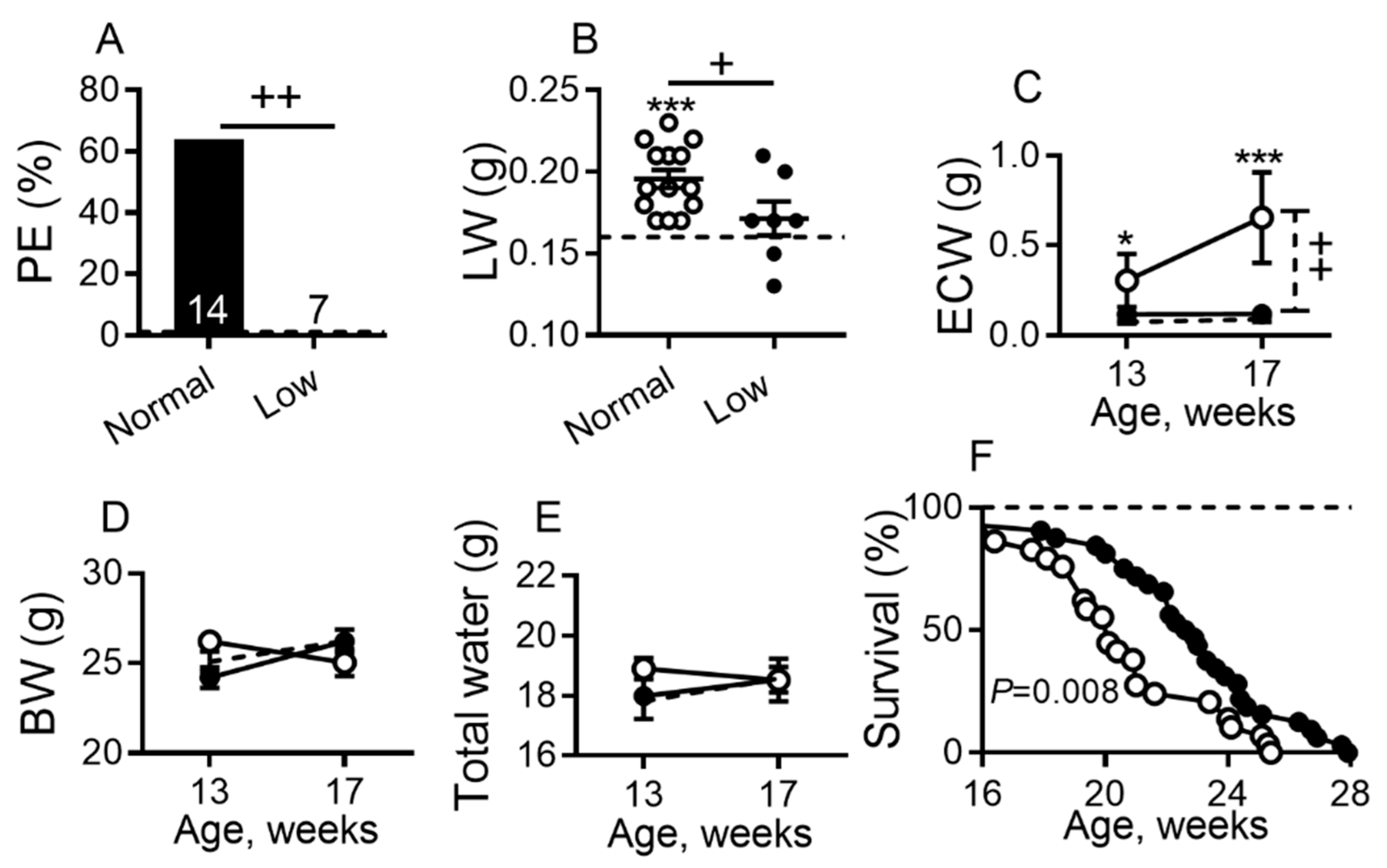
2.1. Dietary Sodium Restriction Suppresses Development of Pleural Effusion, Systemic Edema and Prolongs Survival
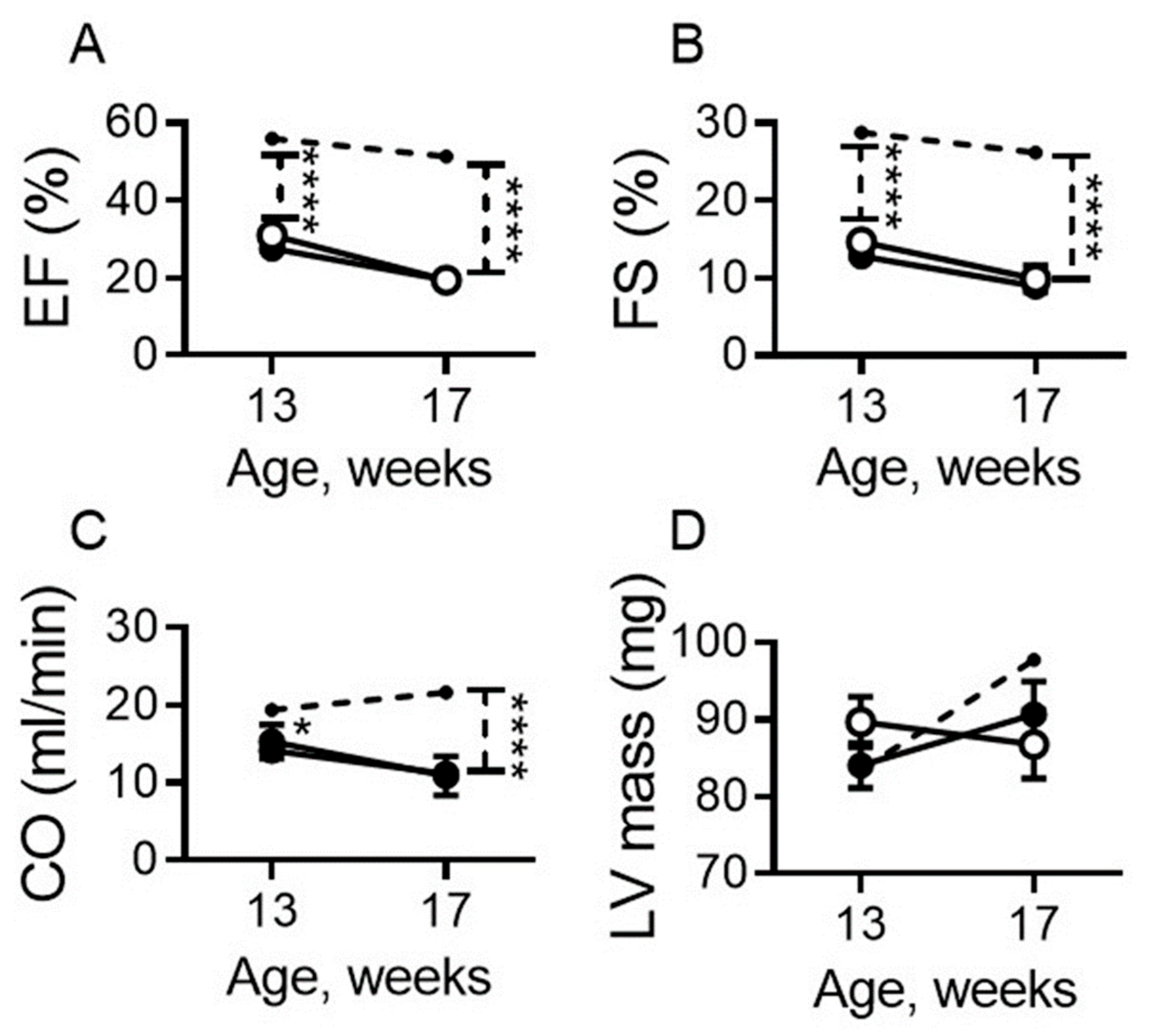
2.2. Dietary Sodium Restriction Does Not Modulate Systolic Function
2.3. Dietary Sodium Restriction Stimulates Systemic Classical Renin-Angiotensin-Aldosterone System (RAAS) and Counter-Regulatory RAAS without Affecting Blood Pressure and Renal Function
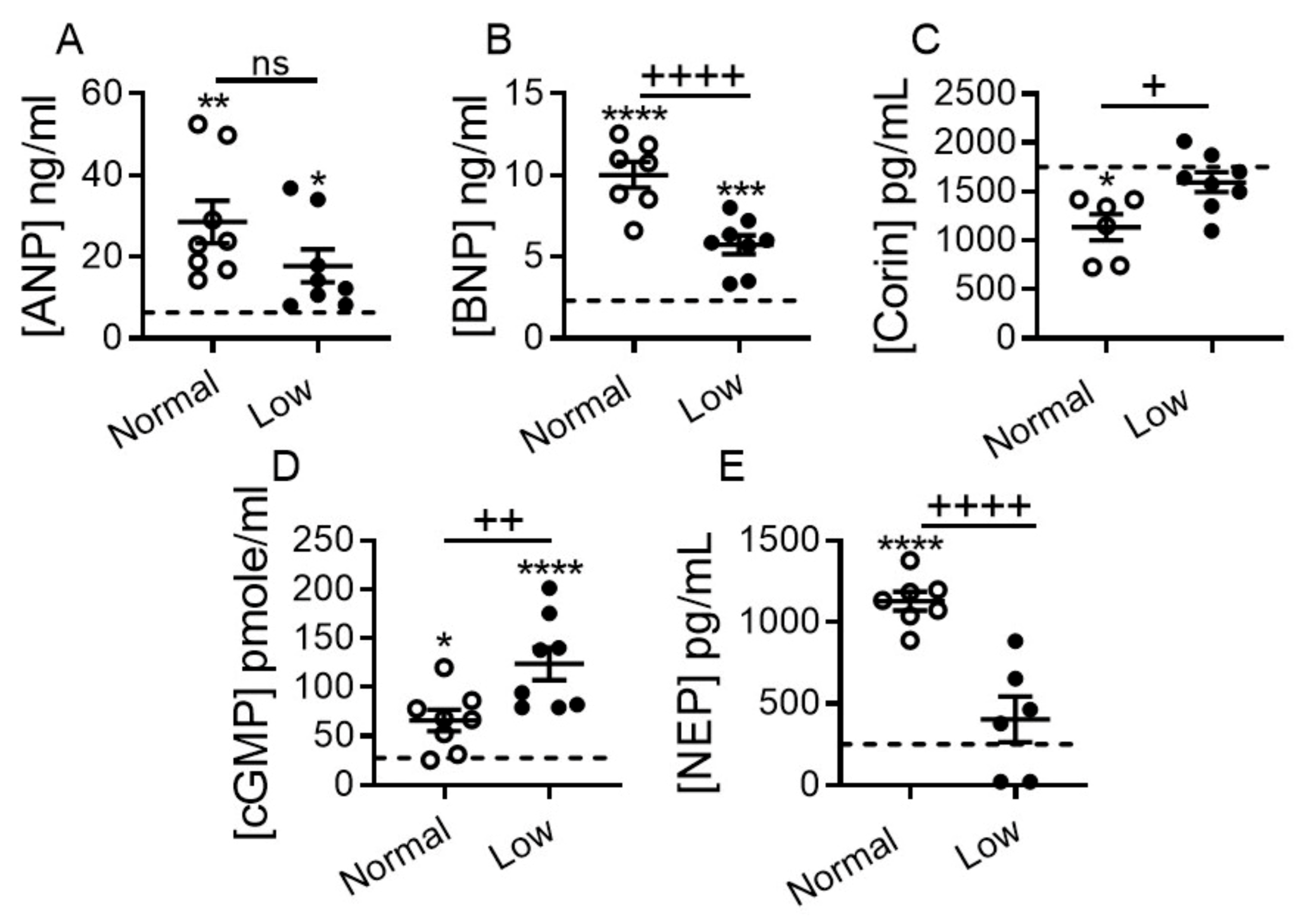
2.4. Impact of Dietary Sodium Restriction on Systemic Levels of Natriuretic Peptide System Components and 3′-5′-Cyclic Guanosine Monophosphate (cGMP) Generation
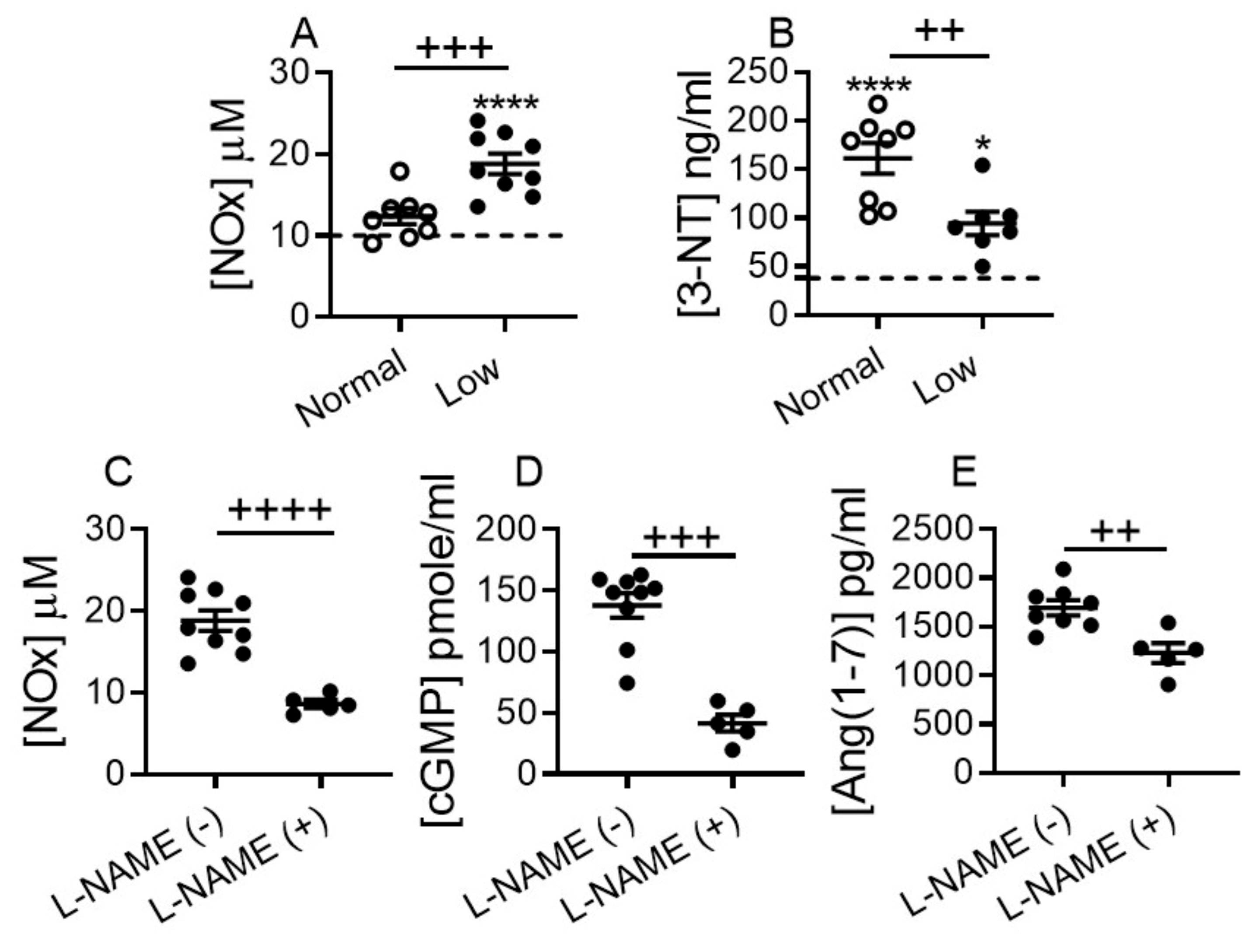
2.5. Dietary Sodium Restriction Stimulates Systemic Ang (1–7), NO and cGMP Generation
2.6. Impact of Dietary Sodium Restriction on Cardiac Expression of Nitric Oxide Synthases (NOS) and Phosphodiesterases
3. Discussion
4. Materials and Methods
4.1. Experimental Mice
4.2. Mouse Diets with Different Sodium Concentration
4.3. Pleural Effusion and Lung Assessment
4.4. Extracellular Water Analysis by Quantitative Magnetic Resonance
4.5. Cardiac Echocardiography
4.6. Blood Pressure Measurements
4.7. Renal Evaluation
4.8. Plasma Biomarker Measurements
4.9. Nitric Oxide (NO) Detection
4.10. Suppression of NO Production by N-Nitro-L-Arginine Methyl Ester (L-NAME)
4.11. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.12. Statistical Analysis
5. Conclusions and Translational Perspectives
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T. 2013 ACCF/AHA guideline for the management of heart failure: A report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2013, 128, e240–e327. [Google Scholar] [CrossRef]
- Weintraub, R.G.; Semsarian, C.; Macdonald, P. Dilated cardiomyopathy. Lancet 2017, 390, 400–414. [Google Scholar] [CrossRef]
- Schrier, R.W.; Abraham, W.T. Hormones and hemodynamics in heart failure. N. Engl. J. Med. 1999, 341, 577–585. [Google Scholar] [CrossRef]
- Weber, K.T. Aldosterone in congestive heart failure. N. Engl. J. Med. 2001, 345, 1689–1697. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.; Georgiopoulou, V.V.; Kalogeropoulos, A.P.; Dunbar, S.B.; Reilly, C.M.; Sands, J.M.; Fonarow, G.C.; Jessup, M.; Gheorghiade, M.; Yancy, C.; et al. Dietary sodium intake in heart failure. Circulation 2012, 126, 479–485. [Google Scholar] [CrossRef]
- Jun, M.; Neal, B. Low dietary sodium in heart failure: A need for scientific rigour. Heart 2014, 100, e2. [Google Scholar] [CrossRef]
- Cogswell, M.E.; Mugavero, K.; Bowman, B.A.; Frieden, T.R. Dietary Sodium and Cardiovascular Disease Risk—Measurement Matters. N. Engl. J. Med. 2016, 6, 580–586. [Google Scholar] [CrossRef] [Green Version]
- Babcock, M.C.; Robinson, A.T.; Migdal, K.U.; Watso, J.C.; Wenner, M.M.; Stocker, S.D.; Farquhar, W.B. Reducing Dietary Sodium to 1000 mg per Day Reduces Neurovascular Transduction Without Stimulating Sympathetic Outflow. Hypertension 2019, 73, 587–593. [Google Scholar] [CrossRef]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 140, e596–e646. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.; Adamopoulos, S.; Anker, S.D.; Auricchio, A.; Bohm, M.; Dickstein, K.; Falk, V.; Filippatos, G.; Fonseca, C.; Gomez-Sanchez, M.A.; et al. ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur. J. Heart Fail. 2012, 14, 803–869. [Google Scholar] [CrossRef]
- Doukky, R.; Avery, E.; Mangla, A.; Collado, F.M.; Ibrahim, Z.; Poulin, M.F.; Richardson, D.; Powell, L.H. Impact of Dietary Sodium Restriction on Heart Failure Outcomes. JACC Heart Fail. 2016, 4, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D.; Fahimi, S.; Singh, G.M.; Micha, R.; Khatibzadeh, S.; Engell, R.E.; Lim, S.; Danaei, G.; Ezzati, M.; Powles, J.; et al. Global sodium consumption and death from cardiovascular causes. N. Engl. J. Med. 2014, 371, 624–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahtani, K.R.; Heneghan, C.; Onakpoya, I.; Tierney, S.; Aronson, J.K.; Roberts, N.; Hobbs, F.D.R.; Nunan, D. Reduced Salt Intake for Heart Failure: A Systematic Review. JAMA Intern. Med. 2018, 178, 1693–1700. [Google Scholar] [CrossRef]
- Ni, Z.; Vaziri, N.D. Effect of salt loading on nitric oxide synthase expression in normotensive rats. Am. J. Hypertens. 2001, 14, 155–163. [Google Scholar] [CrossRef] [Green Version]
- Schweda, F. Salt feedback on the renin-angiotensin-aldosterone system. Pflügers Arch. Eur. J. Physiol. 2015, 467, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.D.; Mehta, R.M.; Tripathi, R.; Reed, G.L.; Gladysheva, I.P. Renin Activity in Heart Failure with Reduced Systolic Function-New Insights. Int. J. Mol. Sci. 2019, 20, 3182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, Y.; Nishikimi, T.; Kuwahara, K. Atrial and brain natriuretic peptides: Hormones secreted from the heart. Peptides 2019, 111, 18–25. [Google Scholar] [CrossRef]
- Sampaio, W.O.; Souza dos Santos, R.A.; Faria-Silva, R.; da Mata Machado, L.T.; Schiffrin, E.L.; Touyz, R.M. Angiotensin-(1-7) through receptor Mas mediates endothelial nitric oxide synthase activation via Akt-dependent pathways. Hypertension 2007, 49, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Patel, K.P.; Schultz, H.D. Angiotensin peptides and nitric oxide in cardiovascular disease. Antioxid Redox Signal. 2013, 19, 1121–1132. [Google Scholar] [CrossRef]
- Patel, V.B.; Zhong, J.C.; Grant, M.B.; Oudit, G.Y. Role of the ACE2/Angiotensin 1-7 Axis of the Renin-Angiotensin System in Heart Failure. Circ. Res. 2016, 118, 1313–1326. [Google Scholar] [CrossRef] [Green Version]
- Santos, R.A.S.; Sampaio, W.O.; Alzamora, A.C.; Motta-Santos, D.; Alenina, N.; Bader, M.; Campagnole-Santos, M.J. The ACE2/Angiotensin-(1-7)/MAS Axis of the Renin-Angiotensin System: Focus on Angiotensin-(1-7). Physiol. Rev. 2018, 98, 505–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ocaranza, M.P.; Riquelme, J.A.; Garcia, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.S.; Lavandero, S. Counter-regulatory renin-angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 116–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartupee, J.; Mann, D.L. Neurohormonal Activation in Heart Failure with Reduced Ejection Fraction. Nat. Rev. Cardiol. 2017, 14, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Ibebuogu, U.N.; Gladysheva, I.P.; Houng, A.K.; Reed, G.L. Decompensated heart failure is associated with reduced corin levels and decreased cleavage of pro-atrial natriuretic peptide. Circ. Heart Fail. 2011, 4, 114–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dries, D.L. Process matters: Emerging concepts underlying impaired natriuretic peptide system function in heart failure. Circ. Heart Fail. 2011, 4, 107–110. [Google Scholar] [CrossRef] [Green Version]
- Zaidi, S.S.; Ward, R.D.; Ramanathan, K.; Yu, X.; Gladysheva, I.P.; Reed, G.L. Possible Enzymatic Downregulation of the Natriuretic Peptide System in Patients with Reduced Systolic Function and Heart Failure: A Pilot Study. Biomed. Res. Int. 2018, 2018, 7279036. [Google Scholar] [CrossRef] [Green Version]
- Cotton, J.M.; Kearney, M.T.; Shah, A.M. Nitric oxide and myocardial function in heart failure: Friend or foe? Heart 2002, 88, 564–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannitsi, S.; Bougiakli, M.; Bechlioulis, A.; Naka, K. Endothelial dysfunction and heart failure: A review of the existing bibliography with emphasis on flow mediated dilation. JRSM Cardiovasc. Dis. 2019, 8, 2048004019843047. [Google Scholar] [CrossRef] [PubMed]
- Fentzke, R.C.; Korcarz, C.E.; Lang, R.M.; Lin, H.; Leiden, J.M. Dilated cardiomyopathy in transgenic mice expressing a dominant-negative CREB transcription factor in the heart. J. Clin. Investig. 1998, 101, 2415–2426. [Google Scholar] [CrossRef] [PubMed]
- Gladysheva, I.P.; Wang, D.; McNamee, R.A.; Houng, A.K.; Mohamad, A.A.; Fan, T.M.; Reed, G.L. Corin overexpression improves cardiac function, heart failure, and survival in mice with dilated cardiomyopathy. Hypertension 2013, 61, 327–332. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Gladysheva, I.P.; Fan, T.H.; Sullivan, R.; Houng, A.K.; Reed, G.L. Atrial natriuretic peptide affects cardiac remodeling, function, heart failure, and survival in a mouse model of dilated cardiomyopathy. Hypertension 2014, 63, 514–519. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, R.; Wang, D.; Sullivan, R.; Fan, T.H.; Gladysheva, I.P.; Reed, G.L. Depressed Corin Levels Indicate Early Systolic Dysfunction Before Increases of Atrial Natriuretic Peptide/B-Type Natriuretic Peptide and Heart Failure Development. Hypertension 2016, 67, 362–367. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, R.; Sullivan, R.; Fan, T.M.; Wang, D.; Sun, Y.; Reed, G.L.; Gladysheva, I.P. Enhanced heart failure, mortality and renin activation in female mice with experimental dilated cardiomyopathy. PLoS ONE 2017, 12, e0189315. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, R.D.; Mehta, R.M.; Tripathi, R.; Gladysheva, I.P.; Reed, G.L. Normalizing Plasma Renin Activity in Experimental Dilated Cardiomyopathy: Effects on Edema, Cachexia, and Survival. Int. J. Mol. Sci. 2019, 20, 3886. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, R.; Sullivan, R.D.; Fan, T.M.; Houng, A.K.; Mehta, R.M.; Reed, G.L.; Gladysheva, I.P. Cardiac-Specific Overexpression of Catalytically Inactive Corin Reduces Edema, Contractile Dysfunction, and Death in Mice with Dilated Cardiomyopathy. Int. J. Mol. Sci. 2020, 21, 203. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, R.; Sullivan, R.D.; Fan, T.M.; Mehta, R.M.; Gladysheva, I.P.; Reed, G.L. In Experimental Dilated Cardiomyopathy Heart Failure and Survival Are Adversely Affected by a Lack of Sexual Interactions. Int. J. Mol. Sci. 2020, 21, 5450. [Google Scholar] [CrossRef]
- Houser, S.R.; Margulies, K.B.; Murphy, A.M.; Spinale, F.G.; Francis, G.S.; Prabhu, S.D.; Rockman, H.A.; Kass, D.A.; Molkentin, J.D.; Sussman, M.A.; et al. Animal models of heart failure: A scientific statement from the American Heart Association. Circ. Res. 2012, 111, 131–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, R.D.; Houng, A.K.; Gladysheva, I.P.; Fan, T.M.; Tripathi, R.; Reed, G.L.; Wang, D. Corin Overexpression Reduces Myocardial Infarct Size and Modulates Cardiomyocyte Apoptotic Cell Death. Int. J. Mol. Sci. 2020, 21, 3456. [Google Scholar] [CrossRef] [PubMed]
- Spellberg, B.; Ibrahim, A.S.; Edwards, J.E., Jr.; Filler, S.G. Mice with disseminated candidiasis die of progressive sepsis. J. Infect. Dis. 2005, 192, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Eleuteri, E.; Magno, F.; Gnemmi, I.; Carbone, M.; Colombo, M.; La Rocca, G.; Anzalone, R.; Tarro Genta, F.; Zummo, G.; Di Stefano, A.; et al. Role of oxidative and nitrosative stress biomarkers in chronic heart failure. Front. Biosci. 2009, 14, 2230–2237. [Google Scholar] [CrossRef] [Green Version]
- Gomes, E.R.; Lara, A.A.; Almeida, P.W.; Guimaraes, D.; Resende, R.R.; Campagnole-Santos, M.J.; Bader, M.; Santos, R.A.; Guatimosim, S. Angiotensin-(1-7) prevents cardiomyocyte pathological remodeling through a nitric oxide/guanosine 3’,5’-cyclic monophosphate-dependent pathway. Hypertension 2010, 55, 153–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gohlke, P.; Pees, C.; Unger, T. AT2 receptor stimulation increases aortic cyclic GMP in SHRSP by a kinin-dependent mechanism. Hypertension 1998, 31, 349–355. [Google Scholar] [CrossRef] [Green Version]
- Kraehling, J.R.; Sessa, W.C. Contemporary Approaches to Modulating the Nitric Oxide-cGMP Pathway in Cardiovascular Disease. Circ. Res. 2017, 120, 1174–1182. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.L.; Borgeson, D.D.; Grantham, J.A.; Luchner, A.; Redfield, M.M.; Burnett, J.C., Jr. Dietary sodium modulation of aldosterone activation and renal function during the progression of experimental heart failure. Eur. J. Heart Fail. 2015, 17, 144–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, W.; Seth, D.M.; Prieto, M.C.; Kobori, H.; Navar, L.G. Activation of the renin-angiotensin system by a low-salt diet does not augment intratubular angiotensinogen and angiotensin II in rats. Am. J. Physiol. Renal Physiol. 2013, 304, F505–F514. [Google Scholar] [CrossRef] [Green Version]
- Nishikimi, T.; Nakagawa, Y.; Minamino, N.; Ikeda, M.; Tabei, K.; Fujishima, A.; Takayama, K.; Akimoto, K.; Yamada, C.; Nakao, K.; et al. Pro-B-type natriuretic peptide is cleaved intracellularly: Impact of distance between O-glycosylation and cleavage sites. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 309, R639–R649. [Google Scholar] [CrossRef] [Green Version]
- Semenov, A.G.; Tamm, N.N.; Seferian, K.R.; Postnikov, A.B.; Karpova, N.S.; Serebryanaya, D.V.; Koshkina, E.V.; Krasnoselsky, M.I.; Katrukha, A.G. Processing of pro-B-type natriuretic peptide: Furin and corin as candidate convertases. Clin. Chem. 2010, 56, 1166–1176. [Google Scholar] [CrossRef] [Green Version]
- Nougue, H.; Pezel, T.; Picard, F.; Sadoune, M.; Arrigo, M.; Beauvais, F.; Launay, J.M.; Cohen-Solal, A.; Vodovar, N.; Logeart, D. Effects of sacubitril/valsartan on neprilysin targets and the metabolism of natriuretic peptides in chronic heart failure: A mechanistic clinical study. Eur. J. Heart Fail. 2019, 21, 598–605. [Google Scholar] [CrossRef]
- Bayes-Genis, A.; Barallat, J.; Richards, A.M. A Test in Context: Neprilysin: Function, Inhibition, and Biomarker. J. Am. Coll. Cardiol. 2016, 68, 639–653. [Google Scholar] [CrossRef]
- Kocks, M.J.; Lely, A.T.; Boomsma, F.; de Jong, P.E.; Navis, G. Sodium status and angiotensin-converting enzyme inhibition: Effects on plasma angiotensin-(1-7) in healthy man. J. Hypertens 2005, 23, 597–602. [Google Scholar] [CrossRef]
- Li, J.; White, J.; Guo, L.; Zhao, X.; Wang, J.; Smart, E.J.; Li, X.A. Salt inactivates endothelial nitric oxide synthase in endothelial cells. J. Nutr. 2009, 139, 447–451. [Google Scholar] [CrossRef]
- O’Neill, J.; Corbett, A.; Johns, E.J. Dietary sodium intake modulates renal excretory responses to intrarenal angiotensin (1-7) administration in anesthetized rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R260–R266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inaba, S.; Iwai, M.; Furuno, M.; Kanno, H.; Senba, I.; Okayama, H.; Mogi, M.; Higaki, J.; Horiuchi, M. Role of angiotensin-converting enzyme 2 in cardiac hypertrophy induced by nitric oxide synthase inhibition. J. Hypertens. 2011, 29, 2236–2245. [Google Scholar] [CrossRef] [PubMed]
- Eleuteri, E.; Di Stefano, A.; Ricciardolo, F.L.; Magno, F.; Gnemmi, I.; Colombo, M.; Anzalone, R.; Cappello, F.; La Rocca, G.; Tarro Genta, F.; et al. Increased nitrotyrosine plasma levels in relation to systemic markers of inflammation and myeloperoxidase in chronic heart failure. Int. J. Cardiol. 2009, 135, 386–390. [Google Scholar] [CrossRef]
- Jablonski, K.L.; Racine, M.L.; Geolfos, C.J.; Gates, P.E.; Chonchol, M.; McQueen, M.B.; Seals, D.R. Dietary sodium restriction reverses vascular endothelial dysfunction in middle-aged/older adults with moderately elevated systolic blood pressure. J. Am. Coll. Cardiol. 2013, 61, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, R.A.; Castro, C.H.; Gava, E.; Pinheiro, S.V.; Almeida, A.P.; Paula, R.D.; Cruz, J.S.; Ramos, A.S.; Rosa, K.T.; Irigoyen, M.C.; et al. Impairment of in vitro and in vivo heart function in angiotensin-(1-7) receptor MAS knockout mice. Hypertension 2006, 47, 996–1002. [Google Scholar] [CrossRef] [Green Version]
- Magalhaes, G.S.; Rodrigues-Machado, M.D.G.; Motta-Santos, D.; Campagnole-Santos, M.J.; Santos, R.A.S. Activation of Ang-(1-7)/Mas Receptor Is a Possible Strategy to Treat Coronavirus (SARS-CoV-2) Infection. Front. Physiol. 2020, 11, 730. [Google Scholar] [CrossRef]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Gladysheva, I.P.; Sullivan, R.D.; Fan, T.M.; Mehta, R.M.; Tripathi, R.; Sun, Y.; Reed, G.L. Increases in plasma corin levels following experimental myocardial infarction reflect the severity of ischemic injury. PLoS ONE 2018, 13, e0202571. [Google Scholar] [CrossRef]
- Erdfelder, E.; Faul, F.; Buchner, A. GPOWER: A general power analysis program. Behav. Res. Methods Instrum. Comput. 1996, 28, 1–11. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tripathi, R.; Sullivan, R.D.; Fan, T.-H.M.; Mehta, R.M.; Gladysheva, I.P.; Reed, G.L. A Low-Sodium Diet Boosts Ang (1–7) Production and NO-cGMP Bioavailability to Reduce Edema and Enhance Survival in Experimental Heart Failure. Int. J. Mol. Sci. 2021, 22, 4035. https://doi.org/10.3390/ijms22084035
Tripathi R, Sullivan RD, Fan T-HM, Mehta RM, Gladysheva IP, Reed GL. A Low-Sodium Diet Boosts Ang (1–7) Production and NO-cGMP Bioavailability to Reduce Edema and Enhance Survival in Experimental Heart Failure. International Journal of Molecular Sciences. 2021; 22(8):4035. https://doi.org/10.3390/ijms22084035
Chicago/Turabian StyleTripathi, Ranjana, Ryan D. Sullivan, Tai-Hwang M. Fan, Radhika M. Mehta, Inna P. Gladysheva, and Guy L. Reed. 2021. "A Low-Sodium Diet Boosts Ang (1–7) Production and NO-cGMP Bioavailability to Reduce Edema and Enhance Survival in Experimental Heart Failure" International Journal of Molecular Sciences 22, no. 8: 4035. https://doi.org/10.3390/ijms22084035
APA StyleTripathi, R., Sullivan, R. D., Fan, T. -H. M., Mehta, R. M., Gladysheva, I. P., & Reed, G. L. (2021). A Low-Sodium Diet Boosts Ang (1–7) Production and NO-cGMP Bioavailability to Reduce Edema and Enhance Survival in Experimental Heart Failure. International Journal of Molecular Sciences, 22(8), 4035. https://doi.org/10.3390/ijms22084035