
NMRK2 Gene Is Upregulated in Dilated Cardiomyopathy and Required for Cardiac Function and NAD Levels during Aging

 , , and
, , and 
Abstract
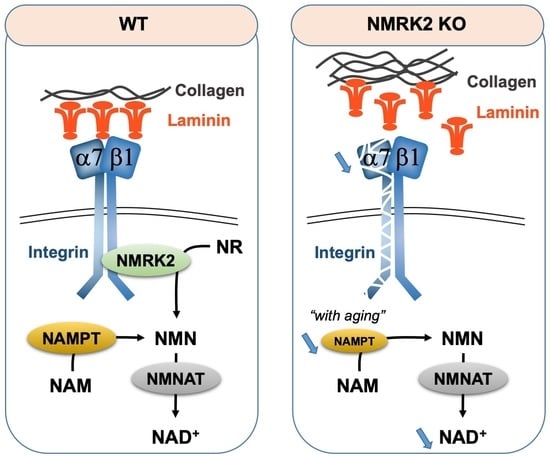
:
1. Introduction
2. Results
2.1. Nmrk2 Upregulation Is a Common Signature in Mouse Models of DCM and in Humans
2.2. Nmrk2 Is a Striated Muscle Specific Protein Associated to the Sarcolemna in Control Hearts and Delocalized to the Cytosol When Overexpressed in the Failing Heart of SRFHKO Mice
2.3. Eccentric Cardiac Remodeling in Young Adult Mice Lacking Nmrk2 upon Pressure Overload
2.4. Nmrk2 Mutants Develop Eccentric Cardiac Remodeling and Subclinical LV Dysfunction with Aging
2.5. Nmrk2 Is Required to Preserve NAD Levels in the Heart at Late Age
2.6. Laminin Disorganization and Cardiac Fibrosis in Nmrk2-KO Mice
3. Discussion
3.1. Role of Nmrk2 in NAD Biosynthesis
3.2. Role of NMRK2 in Laminin Deposition and Integrin Stabilization
4. Materials and Methods
4.1. Study Approval
4.2. Transverse Aortic Constriction (TAC)
4.3. Statistics
4.4. Supplemental Material and Method
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pinto, Y.M.; Elliott, P.M.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Böhm, M.; Duboc, D.; Gimeno, J.; de Groote, P.; Imazio, M.; et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: A position statement of the ESC working group on myocardial and pericardial diseases. Eur. Heart J. 2016, 37, 1850–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merlo, M.; Cannatá, A.; Vitagliano, A.; Zambon, E.; Lardieri, G.; Sinagra, G. Clinical management of dilated cardiomyopathy: Current knowledge and future perspectives. Expert Rev. Cardiovasc. Ther. 2016, 14, 137–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, J.; Frese, K.S.; Peil, B.; Kloos, W.; Keller, A.; Nietsch, R.; Feng, Z.; Müller, S.; Kayvanpour, E.; Vogel, B.; et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur. Heart J. 2015, 36, 1123–1135a. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, S. The failing heart—An engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef] [Green Version]
- Raghow, R. An “Omics” Perspective on Cardiomyopathies and Heart Failure. Trends Mol. Med. 2016, 22, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Louzao-Martinez, L.; Vink, A.; Harakalova, M.; Asselbergs, F.W.; Verhaar, M.C.; Cheng, C. Characteristic adaptations of the extracellular matrix in dilated cardiomyopathy. Int. J. Cardiol. 2016, 220, 634–646. [Google Scholar] [CrossRef]
- Burke, M.A.; Chang, S.; Wakimoto, H.; Gorham, J.M.; Conner, D.A.; Christodoulou, D.C.; Parfenov, M.G.; DePalma, S.R.; Eminaga, S.; Konno, T.; et al. Molecular profiling of dilated cardiomyopathy that progresses to heart failure. JCI Insight 2016, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parlakian, A.; Charvet, C.; Escoubet, B.; Mericskay, M.; Molkentin, J.D.; Gary-Bobo, G.; De Windt, L.J.; Ludosky, M.-A.; Paulin, D.; Daegelen, D.; et al. Temporally controlled onset of dilated cardiomyopathy through disruption of the SRF gene in adult heart. Circulation 2005, 112, 2930–2939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diguet, N.; Trammell, S.A.J.; Tannous, C.; Deloux, R.; Piquereau, J.; Mougenot, N.; Gouge, A.; Gressette, M.; Manoury, B.; Blanc, J.; et al. Nicotinamide Riboside Preserves Cardiac Function in a Mouse Model of Dilated Cardiomyopathy. Circulation 2018, 137, 2256–2273. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Mayne, R.; Wu, C. A novel muscle-specific beta 1 integrin binding protein (MIBP) that modulates myogenic differentiation. J. Cell Biol. 1999, 147, 1391–1398. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Rao, H.; Burkin, D.; Kaufman, S.J.; Wu, C. The muscle integrin binding protein (MIBP) interacts with alpha7beta1 integrin and regulates cell adhesion and laminin matrix deposition. Dev. Biol. 2003, 261, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Goody, M.F.; Kelly, M.W.; Lessard, K.N.; Khalil, A.; Henry, C.A. Nrk2b-mediated NAD+ production regulates cell adhesion and is required for muscle morphogenesis in vivo: Nrk2b and NAD+ in muscle morphogenesis. Dev. Biol. 2010, 344, 809–826. [Google Scholar] [CrossRef] [Green Version]
- Goody, M.F.; Kelly, M.W.; Reynolds, C.J.; Khalil, A.; Crawford, B.D.; Henry, C.A. NAD+ biosynthesis ameliorates a zebrafish model of muscular dystrophy. PLoS Biol. 2012, 10, e1001409. [Google Scholar] [CrossRef] [Green Version]
- Bieganowski, P.; Brenner, C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell 2004, 117, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Bogan, K.L.; Brenner, C. Nicotinic acid, nicotinamide, and nicotinamide riboside: A molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu. Rev. Nutr. 2008, 28, 115–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mericskay, M. Nicotinamide adenine dinucleotide homeostasis and signalling in heart disease: Pathophysiological implications and therapeutic potential. Arch. Cardiovasc. Dis. 2016, 109, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Matasic, D.S.; Brenner, C.; London, B. Emerging potential benefits of modulating NAD+ metabolism in cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H839–H852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revollo, J.R.; Körner, A.; Mills, K.F.; Satoh, A.; Wang, T.; Garten, A.; Dasgupta, B.; Sasaki, Y.; Wolberger, C.; Townsend, R.R.; et al. Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007, 6, 363–375. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.-P.; Yamamoto, T.; Oka, S.; Sadoshima, J. The function of nicotinamide phosphoribosyltransferase in the heart. DNA Repair 2014, 23, 64–68. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, R.S.; Ratajczak, J.; Doig, C.L.; Oakey, L.A.; Callingham, R.; Da Silva Xavier, G.; Garten, A.; Elhassan, Y.S.; Redpath, P.; Migaud, M.E.; et al. Nicotinamide riboside kinases display redundancy in mediating nicotinamide mononucleotide and nicotinamide riboside metabolism in skeletal muscle cells. Mol. Metab. 2017, 6, 819–832. [Google Scholar] [CrossRef] [PubMed]
- Deloux, R.; Tannous, C.; Ferry, A.; Li, Z.; Mericskay, M. Aged Nicotinamide Riboside Kinase 2 Deficient Mice Present an Altered Response to Endurance Exercise Training. Front. Physiol. 2018, 9, 1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbay, E.A.; Moslehi, J.; Christensen, C.L.; Saha, S.; Tchaicha, J.H.; Ramkissoon, S.H.; Stewart, K.M.; Carretero, J.; Kikuchi, E.; Zhang, H.; et al. D-2-hydroxyglutarate produced by mutant IDH2 causes cardiomyopathy and neurodegeneration in mice. Genes Dev. 2014, 28, 479–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muchir, A.; Pavlidis, P.; Decostre, V.; Herron, A.J.; Arimura, T.; Bonne, G.; Worman, H.J. Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J. Clin. Investig. 2007, 117, 1282–1293. [Google Scholar] [CrossRef]
- Vignier, N.; Chatzifrangkeskou, M.; Morales Rodriguez, B.; Mericskay, M.; Mougenot, N.; Wahbi, K.; Bonne, G.; Muchir, A. Rescue of biosynthesis of nicotinamide adenine dinucleotide protects the heart in cardiomyopathy caused by lamin A/C gene mutation. Hum. Mol. Genet. 2018, 27, 3870–3880. [Google Scholar] [CrossRef]
- Martin, O.J.; Lai, L.; Soundarapandian, M.M.; Leone, T.C.; Zorzano, A.; Keller, M.P.; Attie, A.D.; Muoio, D.M.; Kelly, D.P. A role for peroxisome proliferator-activated receptor γ coactivator-1 in the control of mitochondrial dynamics during postnatal cardiac growth. Circ. Res. 2014, 114, 626–636. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Barrientos, T.; Mao, L.; Rockman, H.A.; Sauve, A.A.; Andrews, N.C. Lethal Cardiomyopathy in Mice Lacking Transferrin Receptor in the Heart. Cell Rep. 2015, 13, 533–545. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Singh, N.; Mullican, S.E.; Everett, L.J.; Li, L.; Yuan, L.; Liu, X.; Epstein, J.A.; Lazar, M.A. Diet-induced lethality due to deletion of the Hdac3 gene in heart and skeletal muscle. J. Biol. Chem. 2011, 286, 33301–33309. [Google Scholar] [CrossRef] [Green Version]
- Dufour, C.R.; Wilson, B.J.; Huss, J.M.; Kelly, D.P.; Alaynick, W.A.; Downes, M.; Evans, R.M.; Blanchette, M.; Giguère, V. Genome-wide orchestration of cardiac functions by the orphan nuclear receptors ERRα and gamma. Cell Metab. 2007, 5, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Ahmad, F.; Parikh, S.; Hoffman, N.E.; Rajan, S.; Verma, V.K.; Song, J.; Yuan, A.; Shanmughapriya, S.; Guo, Y.; et al. Loss of Adult Cardiac Myocyte GSK-3 Leads to Mitotic Catastrophe Resulting in Fatal Dilated Cardiomyopathy. Circ. Res. 2016, 118, 1208–1222. [Google Scholar] [CrossRef]
- Zhou, J.; Ahmad, F.; Lal, H.; Force, T. Response by Zhou et al. to Letter Regarding Article, “Loss of Adult Cardiac Myocyte GSK-3 Leads to Mitotic Catastrophe Resulting in Fatal Dilated Cardiomyopathy”. Circ. Res. 2016, 119, e29–e30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweet, M.E.; Cocciolo, A.; Slavov, D.; Jones, K.L.; Sweet, J.R.; Graw, S.L.; Reece, T.B.; Ambardekar, A.V.; Bristow, M.R.; Mestroni, L.; et al. Transcriptome analysis of human heart failure reveals dysregulated cell adhesion in dilated cardiomyopathy and activated immune pathways in ischemic heart failure. BMC Genom. 2018, 19, 812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diguet, N.; Mallat, Y.; Ladouce, R.; Clodic, G.; Prola, A.; Tritsch, E.; Blanc, J.; Larcher, J.-C.; Delcayre, C.; Samuel, J.-L.; et al. Muscle creatine kinase deficiency triggers both actin depolymerization and desmin disorganization by advanced glycation end products in dilated cardiomyopathy. J. Biol. Chem. 2011, 286, 35007–35019. [Google Scholar] [CrossRef] [Green Version]
- Brancaccio, M.; Fratta, L.; Notte, A.; Hirsch, E.; Poulet, R.; Guazzone, S.; De Acetis, M.; Vecchione, C.; Marino, G.; Altruda, F.; et al. Melusin, a muscle-specific integrin beta1-interacting protein, is required to prevent cardiac failure in response to chronic pressure overload. Nat. Med. 2003, 9, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Tomar, D.; Aryal, A.C.S.; Elmoselhi, A.B.; Thomas, M.; Elrod, J.W.; Tilley, D.G.; Force, T. Nicotinamide riboside kinase-2 alleviates ischemia-induced heart failure through P38 signaling. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165609. [Google Scholar] [CrossRef]
- Ratajczak, J.; Joffraud, M.; Trammell, S.A.J.; Ras, R.; Canela, N.; Boutant, M.; Kulkarni, S.S.; Rodrigues, M.; Redpath, P.; Migaud, M.E.; et al. NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nat. Commun. 2016, 7, 13103. [Google Scholar] [CrossRef]
- Oliviéro, P.; Chassagne, C.; Salichon, N.; Corbier, A.; Hamon, G.; Marotte, F.; Charlemagne, D.; Rappaport, L.; Samuel, J.L. Expression of laminin alpha2 chain during normal and pathological growth of myocardium in rat and human. Cardiovasc. Res. 2000, 46, 346–355. [Google Scholar] [CrossRef] [Green Version]
- Grimm, D.; Elsner, D.; Schunkert, H.; Pfeifer, M.; Griese, D.; Bruckschlegel, G.; Muders, F.; Riegger, G.A.; Kromer, E.P. Development of heart failure following isoproterenol administration in the rat: Role of the renin-angiotensin system. Cardiovasc. Res. 1998, 37, 91–100. [Google Scholar] [CrossRef]
- Guicheney, P.; Vignier, N.; Helbling-Leclerc, A.; Nissinen, M.; Zhang, X.; Cruaud, C.; Lambert, J.C.; Richelme, C.; Topaloglu, H.; Merlini, L.; et al. Genetics of laminin alpha 2 chain (or merosin) deficient congenital muscular dystrophy: From identification of mutations to prenatal diagnosis. Neuromuscul. Disord. 1997, 7, 180–186. [Google Scholar] [CrossRef]
- Nelson, I.; Stojkovic, T.; Allamand, V.; Leturcq, F.; Bécane, H.-M.; Babuty, D.; Toutain, A.; Béroud, C.; Richard, P.; Romero, N.B.; et al. Laminin α2 Deficiency-Related Muscular Dystrophy Mimicking Emery-Dreifuss and Collagen VI related Diseases. J. Neuromuscul. Dis. 2015, 2, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Carboni, N.; Marrosu, G.; Porcu, M.; Mateddu, A.; Solla, E.; Cocco, E.; Maioli, M.A.; Oppo, V.; Piras, R.; Marrosu, M.G. Dilated cardiomyopathy with conduction defects in a patient with partial merosin deficiency due to mutations in the laminin-α2-chain gene: A chance association or a novel phenotype? Muscle Nerve 2011, 44, 826–828. [Google Scholar] [CrossRef]
- Burkin, D.J.; Kaufman, S.J. The α7β1 integrin in muscle development and disease. Cell Tissue Res. 1999, 296, 183–190. [Google Scholar] [CrossRef]
- Ginsberg, M.H. Integrin activation. BMB Rep. 2014, 47, 655–659. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Gruszczynska-Biegala, J.; Zolkiewska, A. ADP-ribosylation of integrin alpha7 modulates the binding of integrin α7β1 to laminin. Biochem. J. 2005, 385, 309–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cardiac Parameters | Baseline | TAC Day 15 | ANOVA | ||
|---|---|---|---|---|---|
| Wt | Nmrk2-KO | Wt | Nmrk2-KO | ||
| N | 3 | 5 | 3 | 5 | |
| IVSThD (cm) | 0.06 ± 0.006 | 0.06 ± 0.005 | 0.1 ± 0 *** | 0.09 ± 0.008 ***, # | i, ¶¶¶ |
| LVEDD (cm) | 0.34 ± 0.006 | 0.34 ± 0.028 | 0.35 ± 0.015 | 0.39 ± 0.043 | |
| LVPWThD (cm) | 0.07 ± 0.006 | 0.08 ± 0.008 | 0.09 ± 0.007 | 0.08 ± 0.007 | ¶ |
| IVSThS (cm) | 0.11 ± 0.006 | 0.11 ± 0.007 | 0.14 ± 0 * | 0.12 ± 0.013 # | ¶, § |
| LVESD (cm) | 0.18 ± 0.006 | 0.19 ± 0.024 | 0.2 ± 0.017 | 0.27 ± 0.065 | |
| LVPWThS (cm) | 0.11 ± 0.010 | 0.12 ± 0.008 | 0.14 ± 0.006 | 0.11 ± 0.010 ## | i, § |
| EDV (mL) | 0.1 ± 0 | 0.11 ± 0.023 | 0.11 ± 0.0 | 0.16 ± 0.051 | |
| ESV (mL) | 0.02 ± 0 | 0.02 ± 0.007 | 0.02 ± 0.007 | 0.06 ± 0.041 | |
| LVEF (%) | 81.7 ± 0.87 | 81.8 ± 2.47 | 80.6 ± 2.79 | 65.8 ± 13.98 * | |
| FS (%) | 44.5 ± 1.12 | 44.54 ± 2.58 | 43.3 ± 2.73 | 32.1 ± 9.54 *, # | |
| SV (mL) | 0.08 ± 0 | 0.09 ± 0.018 | 0.09 ± 0.006 | 0.1 ± 0.019 | |
| h/r | 0.41 ± 0.021 | 0.42 ± 0.048 | 0.55 ± 0.020 * | 0.44 ± 0.049 ## | ¶, § |
| HR (bpm) | 610 ± 12.4 | 613 ± 15.1 | 566 ± 12.9 | 541 ± 55.6 | ¶ |
| LVMI (mg/g) | 2.92 ± 0.320 | 3.49 ± 0.441 | 5.01 ± 0.747 §§ | 5.56 ± 1.059 ### | ¶¶ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tannous, C.; Deloux, R.; Karoui, A.; Mougenot, N.; Burkin, D.; Blanc, J.; Coletti, D.; Lavery, G.; Li, Z.; Mericskay, M. NMRK2 Gene Is Upregulated in Dilated Cardiomyopathy and Required for Cardiac Function and NAD Levels during Aging. Int. J. Mol. Sci. 2021, 22, 3534. https://doi.org/10.3390/ijms22073534
Tannous C, Deloux R, Karoui A, Mougenot N, Burkin D, Blanc J, Coletti D, Lavery G, Li Z, Mericskay M. NMRK2 Gene Is Upregulated in Dilated Cardiomyopathy and Required for Cardiac Function and NAD Levels during Aging. International Journal of Molecular Sciences. 2021; 22(7):3534. https://doi.org/10.3390/ijms22073534
Chicago/Turabian StyleTannous, Cynthia, Robin Deloux, Ahmed Karoui, Nathalie Mougenot, Dean Burkin, Jocelyne Blanc, Dario Coletti, Gareth Lavery, Zhenlin Li, and Mathias Mericskay. 2021. "NMRK2 Gene Is Upregulated in Dilated Cardiomyopathy and Required for Cardiac Function and NAD Levels during Aging" International Journal of Molecular Sciences 22, no. 7: 3534. https://doi.org/10.3390/ijms22073534
APA StyleTannous, C., Deloux, R., Karoui, A., Mougenot, N., Burkin, D., Blanc, J., Coletti, D., Lavery, G., Li, Z., & Mericskay, M. (2021). NMRK2 Gene Is Upregulated in Dilated Cardiomyopathy and Required for Cardiac Function and NAD Levels during Aging. International Journal of Molecular Sciences, 22(7), 3534. https://doi.org/10.3390/ijms22073534