
Glucose Variability: How Does It Work?




Abstract
:1. Introduction
2. Biochemical and Pathophysiological Abnormalities Induced by Excessive Glucose Fluctuations
2.1. Oxidative Stress and Non-Enzymatic Glycation
2.2. Chronic Low-Grade Inflammation
2.3. Endothelial Dysfunction and Vascular Remodeling
2.4. Platelet Activation and Hypercoagulability
2.5. Impaired Angiogenesis
2.6. Renal Fibrosis
2.7. Beta Cell Dysfunction
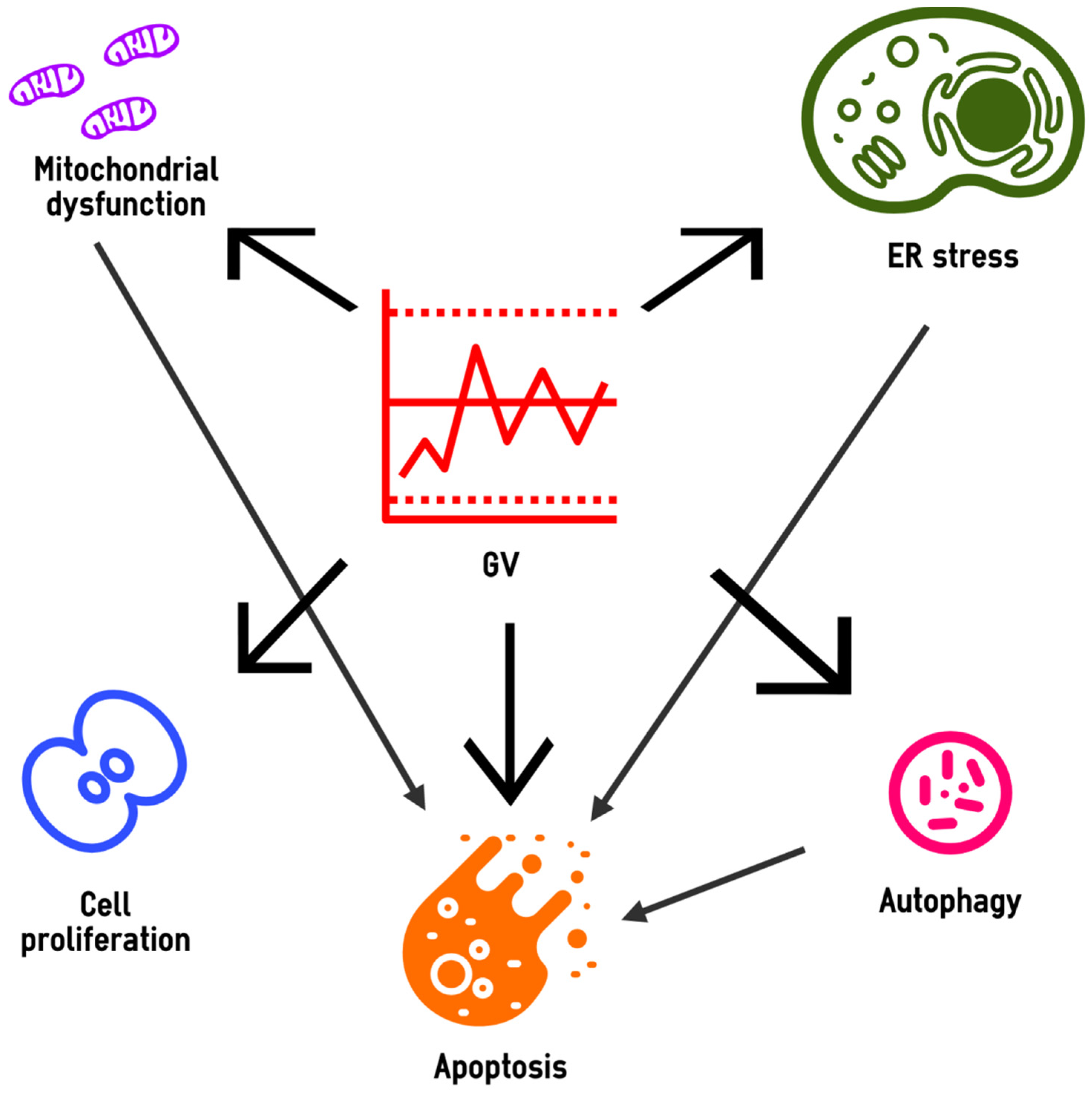
3. Cell Biology under High-GV Conditions
3.1. Altered Mitochondrial Homeostasis
3.2. Endoplasmic Reticulum Stress
3.3. Autophagy
3.4. Apoptosis
3.5. Cell Proliferation
4. Molecular Mechanisms of the High GV Effects in the Target Cells
4.1. Gene Expression
4.2. Epigenetic Modifications
4.3. Signaling Pathways
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AGEs | Advanced glycation end-products |
| AMPK | AMP-activated protein kinase |
| ATF4 | Activating transcription factor 4 |
| BMP | Bone morphogenetic protein |
| CARM1 | Coactivator-associated arginine methyltransferase 1 |
| CGM | Continuous glucose monitoring |
| CHOP | C/EBP homologous protein |
| CHG | Constantly high glucose |
| DNMT3b | DNA (cytosine-5-)-methyltransferase 3 beta |
| eNOS | Endothelial nitric oxide synthase |
| EPCs | Endothelial progenitor cells |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinase |
| GLUT4 | Glucose transporter type 4 |
| GSK3β | Glycogen synthase kinase 3 beta |
| GV | Glucose variability |
| HbA1c | Glycated hemoglobin A1c |
| hsCRP | High-sensitivity C-reactive protein |
| HMGB1 | High-mobility group box 1 |
| HUVECs | Human umbilical vein endothelial cells |
| ICAM-1 | Intercellular adhesion molecules 1 |
| IHG | Intermittently high glucose |
| INS-1 | Insulinoma cells |
| IRF5 | Interferon regulatory factor 5 |
| JNK | c-Jun N-terminal kinase |
| LAMP | Lysosomal-associated membrane protein |
| LC3 | Microtubule-associated proteins 1A/1B light chain 3B |
| MAGE | Mean amplitude of glucose excursions |
| MAPK | Mitogen-activated protein kinase |
| MCP-1 | Monocyte chemoattractant protein 1 |
| miRNAs | small single-stranded non-coding RNAs |
| mTOR | Mechanistic target of rapamycin |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NF-κB | Nuclear factor kB |
| PAI-1 | Plasminogen activator inhibitor-1 |
| PI3K | Phosphoinositide-3-kinase |
| PKC | Protein kinase C |
| ROS | Reactive oxygen species |
| RUNX2 | Runt-related transcription factor 2 |
| SHC1 | SHC-transforming protein 1 |
| SKP2 | S-phase kinase-associated protein 2 |
| SD | Standard deviation |
| sICAM-1 | Soluble intercellular adhesion molecules 1 |
| SIRT1 | Sirtuin 1 |
| SGLT1 | Sodium-glucose cotransporter 1 |
| SOD2 | Superoxide dismutase 2 |
| T1D | Type 1 diabetes |
| T2D | Type 2 diabetes |
| TGF-β1 | Transforming growth factor beta 1 |
| TLR4 | Toll-like receptor 4 |
| TNF-α | Tumor necrosis factor α |
| TSP-1 | Thrombospondin-1 |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labeling |
| VCAM-1 | Vascular cell adhesion molecules 1 |
| VEGF | Vascular endothelial growth factor |
| VSMCs | Vascular smooth muscle cells |
References
- Rodbard, D. Glucose variability: A review of clinical applications and research developments. Diabetes Technol. Ther. 2018, 20, S25–S215. [Google Scholar] [CrossRef] [PubMed]
- Umpierrez, G.E.; Kovatchev, B.P. Glycemic variability: How to measure and its clinical implication for type 2 diabetes. Am. J. Med. Sci. 2018, 356, 518–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danne, T.; Nimri, R.; Battelino, T.; Bergenstal, R.M.; Close, K.L.; De Vries, J.H.; Garg, S.; Heinemann, L.; Hirsch, I.; Amiel, S.A.; et al. International consensus on use of continuous glucose monitoring. Diabetes Care 2017, 40, 1631–1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceriello, A. Glucose variability and diabetic complications: Is it time to treat? Diabetes Care 2020, 43, 1169–1171. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Sun, B.; Huang, S.; Zhu, C.; Bian, M. Glycemic variability: Adverse clinical outcomes and how to improve it? Cardiovasc. Diabetol. 2020, 19, 102. [Google Scholar] [CrossRef] [PubMed]
- Wadén, J.; Forsblom, C.; Thorn, L.M.; Gordin, D.; Saraheimo, M.; Groop, P.-H.; Finnish Diabetic Nephropathy Study Group. A1C variability predicts incident cardiovascular events, microalbuminuria, and overt diabetic nephropathy in patients with type 1 diabetes. Diabetes 2009, 58, 2649–2655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luk, A.O.Y.; Ma, R.C.W.; Lau, E.S.H.; Yang, X.; Lau, W.W.Y.; Yu, L.W.L.; Chow, F.C.C.; Chan, J.C.N.; So, W.-Y. Risk Association of HbA1c variability with chronic kidney disease and cardiovascular disease in type 2 diabetes: Prospective analysis of the hong kong diabetes registry. Diabetes Metab. Res. Rev. 2013, 29, 384–390. [Google Scholar] [CrossRef]
- Li, S.; Tang, X.; Luo, Y.; Wu, B.; Huang, Z.; Li, Z.; Peng, L.; Ling, Y.; Zhu, J.; Zhong, J.; et al. Impact of long-term glucose variability on coronary atherosclerosis progression in patients with type 2 diabetes: A 2.3 year follow-up study. Cardiovasc. Diabetol. 2020, 19, 146. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, Y.; Arima, H.; Zoungas, S.; Ninomiya, T.; Cooper, M.; Hamet, P.; Mancia, G.; Poulter, N.; Harrap, S.; Woodward, M.; et al. Impact of visit-to-visit glycemic variability on the risks of macrovascular and microvascular events and all-cause mortality in type 2 diabetes: The ADVANCE trial. Diabetes Care 2014, 37, 2359–2365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinman, B.; Marso, S.P.; Poulter, N.R.; Emerson, S.S.; Pieber, T.R.; Pratley, R.E.; Lange, M.; Brown-Frandsen, K.; Moses, A.; Ocampo Francisco, A.M.; et al. Day-to-day fasting glycaemic variability in DEVOTE: Associations with severe hypoglycaemia and cardiovascular outcomes (DEVOTE 2). Diabetologia 2018, 61, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.J.; Schwenke, D.C.; Bahn, G.; Reaven, P.; VADT Investigators. Glycemic variation and cardiovascular risk in the veterans affairs diabetes trial. Diabetes Care 2018, 41, 2187–2194. [Google Scholar] [CrossRef] [Green Version]
- Kaze, A.D.; Santhanam, P.; Erqou, S.; Ahima, R.S.; Echouffo-Tcheugui, J.B. Long-term variability of glycemic markers and risk of all-cause mortality in type 2 diabetes: The look AHEAD study. BMJ Open Diabetes Res. Care 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Ge, Q.M.; Dong, Y.; Zhang, H.M.; Su, Q. Effects of intermittent high glucose on oxidative stress in endothelial cells. Acta Diabetol. 2010, 47 (Suppl. S1), 97–103. [Google Scholar] [CrossRef]
- Sun, L.-Q.; Xue, B.; Li, X.-J.; Wang, X.; Qu, L.; Zhang, T.-T.; Zhao, J.; Wang, B.-A.; Zou, X.-M.; Mu, Y.-M.; et al. Inhibitory effects of salvianolic acid b on apoptosis of schwann cells and its mechanism induced by intermittent high glucose. Life Sci. 2012, 90, 99–108. [Google Scholar] [CrossRef]
- Sun, L.-Q.; Chen, Y.-Y.; Wang, X.; Li, X.-J.; Xue, B.; Qu, L.; Zhang, T.-T.; Mu, Y.-M.; Lu, J.-M. The protective effect of alpha lipoic acid on schwann cells exposed to constant or intermittent high glucose. Biochem. Pharmacol. 2012, 84, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Xu, Y.; Deng, H.; Sun, S.; Dai, Z.; Sun, Y. Intermittent high glucose exacerbates the aberrant production of adiponectin and resistin through mitochondrial superoxide overproduction in adipocytes. J. Mol. Endocrinol. 2010, 44, 179–185. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, J.; Yang, L.; Chen, R.; Yang, R.; Zhang, H.; Cai, D.; Chen, H. The cytotoxic role of intermittent high glucose on apoptosis and cell viability in pancreatic beta cells. J. Diabetes Res. 2014, 2014, 712781. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-G.; Zhang, Y.-Q.; Zhao, D.-K.; Wu, J.-X.; Zhao, J.; Jiao, X.-M.; Chen, B.; Lv, X.-F. Relationship between blood glucose fluctuation and macrovascular endothelial dysfunction in type 2 diabetic patients with coronary heart disease. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 3593–3600. [Google Scholar]
- Wang, J.; Yin, H.; Huang, Y.; Guo, C.; Xia, C.; Liu, Q.; Zhang, L. Panax quinquefolius saponin of stem and leaf attenuates intermittent high glucose-induced oxidative stress injury in cultured human umbilical vein endothelial cells via PI3K/Akt/GSK-3 β Pathway. Evid. Based Complement. Altern. Med. 2013, 2013, 196283. [Google Scholar] [CrossRef]
- He, Y.-T.; Xing, S.-S.; Gao, L.; Wang, J.; Xing, Q.-C.; Zhang, W. Ginkgo biloba attenuates oxidative DNA damage of human umbilical vein endothelial cells induced by intermittent high glucose. Pharmazie 2014, 69, 203–207. [Google Scholar]
- Sun, J.; Xu, Y.; Sun, S.; Sun, Y.; Wang, X. Intermittent high glucose enhances cell proliferation and VEGF expression in retinal endothelial cells: The role of mitochondrial reactive oxygen species. Mol. Cell. Biochem. 2010, 343, 27–35. [Google Scholar] [CrossRef]
- Hu, Z.; Fang, W.; Liu, Y.; Liang, H.; Chen, W.; Wang, H. Acute glucose fluctuation promotes RAGE expression via reactive oxygen species-mediated NF-κB activation in rat podocytes. Mol. Med. Rep. 2021, 23, 1–9. [Google Scholar] [CrossRef]
- Sun, J.; Xu, Y.; Dai, Z.; Sun, Y. Intermittent high glucose stimulate MCP-l, IL-18, and PAI-1, but inhibit adiponectin expression and secretion in adipocytes dependent of ROS. Cell Biochem. Biophys. 2009, 55, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.-Q.; Li, H.-L.; Gao, L.; Pan, L.; Zhao, J.-J.; Li, G.-W. Involvement of chronic stresses in rat islet and INS-1 cell glucotoxicity induced by intermittent high glucose. Mol. Cell. Endocrinol. 2008, 291, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Shen, H.; Liu, H.; Wang, Y.; Bai, Y.; Han, P. Acute blood glucose fluctuation enhances rat aorta endothelial cell apoptosis, oxidative stress and pro-inflammatory cytokine expression in vivo. Cardiovasc. Diabetol. 2016, 15, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.-M.; Hsieh, C.-J.; Huang, J.-C.; Huang, I.-C. Acute and chronic fluctuations in blood glucose levels can increase oxidative stress in type 2 diabetes mellitus. Acta Diabetol. 2012, 49 (Suppl. S1), S171–S177. [Google Scholar] [CrossRef]
- Wentholt, I.M.E.; Kulik, W.; Michels, R.P.J.; Hoekstra, J.B.L.; De Vries, J.H. Glucose fluctuations and activation of oxidative stress in patients with type 1 diabetes. Diabetologia 2008, 51, 183–190. [Google Scholar] [CrossRef] [Green Version]
- Siegelaar, S.E.; Barwari, T.; Kulik, W.; Hoekstra, J.B.; De Vries, J.H. No relevant relationship between glucose variability and oxidative stress in well-regulated type 2 diabetes patients. J. Diabetes Sci. Technol. 2011, 5, 86–92. [Google Scholar] [CrossRef] [Green Version]
- Kohata, Y.; Ohara, M.; Nagaike, H.; Fujikawa, T.; Osaka, N.; Goto, S.; Fukase, A.; Kushima, H.; Hiromura, M.; Terasaki, M.; et al. Association of hemoglobin A1c, 1,5-Anhydro-D-Glucitol and glycated albumin with oxidative stress in type 2 diabetes mellitus patients: A cross-sectional study. Diabetes Ther. 2020, 11, 655–665. [Google Scholar] [CrossRef] [Green Version]
- Klimontov, V.V.; Myakina, N.E. Glucose variability indices predict the episodes of nocturnal hypoglycemia in elderly type 2 diabetic patients treated with insulin. Diabetes Metab. Syndr. 2017, 11, 119–124. [Google Scholar] [CrossRef]
- Gómez, A.M.; Henao, D.C.; Imitola-Madero, A.; Taboada, L.B.; Cruz, V.; Robledo Gómez, M.A.; Rondón, M.; Muñoz-Velandia, O.; García-Jaramillo, M.; León Vargas, F.M. Defining high glycemic variability in type 1 diabetes: Comparison of multiple indexes to identify patients at risk of hypoglycemia. Diabetes Technol. Ther. 2019, 21, 430–439. [Google Scholar] [CrossRef]
- Ceriello, A.; Novials, A.; Ortega, E.; La Sala, L.; Pujadas, G.; Testa, R.; Bonfigli, A.R.; Esposito, K.; Giugliano, D. Evidence that hyperglycemia after recovery from hypoglycemia worsens endothelial function and increases oxidative stress and inflammation in healthy control subjects and subjects with type 1 diabetes. Diabetes 2012, 61, 2993–2997. [Google Scholar] [CrossRef] [Green Version]
- Ceriello, A.; Novials, A.; Ortega, E.; Pujadas, G.; La Sala, L.; Testa, R.; Bonfigli, A.R.; Genovese, S. Hyperglycemia following recovery from hypoglycemia worsens endothelial damage and thrombosis activation in type 1 diabetes and in healthy controls. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 116–123. [Google Scholar] [CrossRef]
- Fishman, S.L.; Sonmez, H.; Basman, C.; Singh, V.; Poretsky, L. The role of advanced glycation end-products in the development of coronary artery disease in patients with and without diabetes mellitus: A review. Mol. Med. 2018, 24, 59. [Google Scholar] [CrossRef] [PubMed]
- Testa, R.; Bonfigli, A.R.; Prattichizzo, F.; La Sala, L.; De Nigris, V.; Ceriello, A. The “Metabolic Memory” theory and the early treatment of hyperglycemia in prevention of diabetic complications. Nutrients 2017, 9, 437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamagishi, S.-I.; Nakamura, N.; Matsui, T. Glycation and cardiovascular disease in diabetes: A perspective on the concept of metabolic memory. J. Diabetes 2017, 9, 141–148. [Google Scholar] [CrossRef]
- Schisano, B.; Tripathi, G.; McGee, K.; McTernan, P.G.; Ceriello, A. Glucose oscillations, more than constant high glucose, induce P53 activation and a metabolic memory in human endothelial cells. Diabetologia 2011, 54, 1219–1226. [Google Scholar] [CrossRef] [Green Version]
- Corgnali, M.; Piconi, L.; Ihnat, M.; Ceriello, A. Evaluation of gliclazide ability to attenuate the hyperglycaemic “memory” induced by high glucose in isolated human endothelial cells. Diabetes Metab. Res. Rev. 2008, 24, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Domingueti, C.P.; Dusse, L.M.S.; das Carvalho, M.G.; de Sousa, L.P.; Gomes, K.B.; Fernandes, A.P. Diabetes mellitus: The linkage between oxidative stress, inflammation, hypercoagulability and vascular complications. J. Diabetes Complicat. 2016, 30, 738–745. [Google Scholar] [CrossRef]
- Nguyen, D.V.; Shaw, L.C.; Grant, M.B. Inflammation in the pathogenesis of microvascular complications in diabetes. Front. Endocrinol. 2012, 3, 170. [Google Scholar] [CrossRef] [Green Version]
- Piconi, L.; Quagliaro, L.; Assaloni, R.; Da Ros, R.; Maier, A.; Zuodar, G.; Ceriello, A. Constant and intermittent high glucose enhances endothelial cell apoptosis through mitochondrial superoxide overproduction. Diabetes Metab. Res. Rev. 2006, 22, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Quagliaro, L.; Piconi, L.; Assaloni, R.; Da Ros, R.; Maier, A.; Zuodar, G.; Ceriello, A. Intermittent high glucose enhances ICAM-1, VCAM-1 and e-selectin expression in human umbilical vein endothelial cells in culture: The distinct role of protein kinase C and mitochondrial superoxide production. Atherosclerosis 2005, 183, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Li-Bo, Y.; Wen-Bo, Q.; Xiao-Hong, L.; You-Lun, F.; Tie, Z. Intermittent high glucose promotes expression of proinflammatory cytokines in monocytes. Inflamm. Res. 2011, 60, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Bi, Y.; Jin, G.; Gan, H.; Yu, L. High and fluctuating glucose levels increase the expression and secretion of interleukin-18 in mouse peritoneal macrophages. Mol. Med. Rep. 2015, 12, 2715–2720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Rashed, F.; Sindhu, S.; Arefanian, H.; Al Madhoun, A.; Kochumon, S.; Thomas, R.; Al-Kandari, S.; Alghaith, A.; Jacob, T.; Al-Mulla, F.; et al. Repetitive intermittent hyperglycemia drives the m1 polarization and inflammatory responses in THP-1 macrophages through the mechanism involving the TLR4-IRF5 pathway. Cells 2020, 9, 1892. [Google Scholar] [CrossRef] [PubMed]
- Ratter, J.M.; Rooijackers, H.M.M.; Tack, C.J.; Hijmans, A.G.M.; Netea, M.G.; de Galan, B.E.; Stienstra, R. Proinflammatory effects of hypoglycemia in humans with or without diabetes. Diabetes 2017, 66, 1052–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratter, J.M.; Rooijackers, H.M.M.; Jacobs, C.W.M.; de Galan, B.E.; Tack, C.J.; Stienstra, R. Hypoglycaemia induces recruitment of non-classical monocytes and cytotoxic lymphocyte subsets in type 1 diabetes. Diabetologia 2018, 61, 2069–2071. [Google Scholar] [CrossRef] [Green Version]
- Ceriello, A.; Novials, A.; Ortega, E.; Canivell, S.; La Sala, L.; Pujadas, G.; Esposito, K.; Giugliano, D.; Genovese, S. Glucagon-like peptide 1 reduces endothelial dysfunction, inflammation, and oxidative stress induced by both hyperglycemia and hypoglycemia in type 1 diabetes. Diabetes Care 2013, 36, 2346–2350. [Google Scholar] [CrossRef] [Green Version]
- Ceriello, A.; Novials, A.; Ortega, E.; Canivell, S.; Pujadas, G.; La Sala, L.; Bucciarelli, L.; Rondinelli, M.; Genovese, S. Vitamin C further improves the protective effect of GLP-1 on the ischemia-reperfusion-like effect induced by hyperglycemia post-hypoglycemia in type 1 diabetes. Cardiovasc. Diabetol. 2013, 12, 97. [Google Scholar] [CrossRef] [Green Version]
- Kiec-Wilk, B.; Matejko, B.; Razny, U.; Stankiewicz, M.; Skupien, J.; Klupa, T.; Malecki, M.T. Hypoglycemic episodes are associated with inflammatory status in patients with type 1 diabetes mellitus. Atherosclerosis 2016, 251, 334–338. [Google Scholar] [CrossRef]
- Cherney, D.Z.I.; Scholey, J.W.; Sochett, E.; Bradley, T.J.; Reich, H.N. The acute effect of clamped hyperglycemia on the urinary excretion of inflammatory cytokines/chemokines in uncomplicated type 1 diabetes: A pilot study. Diabetes Care 2011, 34, 177–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eik, W.; Marcon, S.S.; Krupek, T.; Previdelli, I.T.S.; Pereira, O.C.N.; Silva, M.a.R.C.P.; Bazotte, R.B. Blood levels of pro-inflammatory and anti-inflammatory cytokines during an oral glucose tolerance test in patients with symptoms suggesting reactive hypoglycemia. Braz. J. Med. Biol. Res. 2016, 49. [Google Scholar] [CrossRef]
- Drummond, J.B.; Barbosa, I.G.; Dantzer, R.; Teixeira, A.L. The effect of insulin-induced hypoglycemia on inflammatory markers: A systematic review. Brain Behav. Immun. 2018, 73, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Kaya, A.; Koçyiğit, C.; Çatlı, G.; Özkan, E.B.; Dündar, B.N. The relationship between glycemic variability and inflammatory markers in obese children with insulin resistance and metabolic syndrome. J. Clin. Res. Pediatr. Endocrinol. 2017, 9, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Klimontov, V.V.; Tyan, N.V.; Fazullina, O.N.; Myakina, N.E.; Orlov, N.B.; Konenkov, V.I. Acute-phase serum proteins and adipocytokines in women with type 2 diabetes mellitus: Relationships with body composition and blood glucose fluctuations. Ter. Arkh. 2016, 88, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Buscemi, S.; Verga, S.; Cottone, S.; Azzolina, V.; Buscemi, B.; Gioia, D.; Cerasola, G. Glycaemic variability and inflammation in subjects with metabolic syndrome. Acta Diabetol. 2009, 46, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Ali, A.; Katare, R. Molecular complexities underlying the vascular complications of diabetes mellitus—A comprehensive review. J. Diabetes Complicat. 2020, 34, 107613. [Google Scholar] [CrossRef]
- Liao, J.; Lei, M.; Chen, X.; Liu, F. Effect of intermittent high glucose on synthesis of nitric oxide in human umbilical vein endothelial cells and its mechanism. Zhong Nan Da Xue Xue Bao. Yi Xue Ban 2010, 35, 295–300. [Google Scholar] [CrossRef]
- Kuricová, K.; Pácal, L.; Šoupal, J.; Prázný, M.; Kaňková, K. Effect of glucose variability on pathways associated with glucotoxicity in diabetes: Evaluation of a novel in vitro experimental approach. Diabetes Res. Clin. Pract. 2016, 114, 1–8. [Google Scholar] [CrossRef]
- Maeda, M.; Hayashi, T.; Mizuno, N.; Hattori, Y.; Kuzuya, M. Intermittent high glucose implements stress-induced senescence in human vascular endothelial cells: Role of superoxide production by NADPH oxidase. PLoS ONE 2015, 10, e0123169. [Google Scholar] [CrossRef]
- Horváth, E.M.; Benko, R.; Kiss, L.; Murányi, M.; Pék, T.; Fekete, K.; Bárány, T.; Somlai, A.; Csordás, A.; Szabo, C. Rapid “glycaemic Swings” induce nitrosative stress, activate Poly(ADP-Ribose) polymerase and impair endothelial function in a rat model of diabetes mellitus. Diabetologia 2009, 52, 952–961. [Google Scholar] [CrossRef] [Green Version]
- Di Flaviani, A.; Picconi, F.; Di Stefano, P.; Giordani, I.; Malandrucco, I.; Maggio, P.; Palazzo, P.; Sgreccia, F.; Peraldo, C.; Farina, F.; et al. Impact of glycemic and blood pressure variability on surrogate measures of cardiovascular outcomes in type 2 diabetic patients. Diabetes Care 2011, 34, 1605–1609. [Google Scholar] [CrossRef] [Green Version]
- Peña, A.S.; Couper, J.J.; Harrington, J.; Gent, R.; Fairchild, J.; Tham, E.; Baghurst, P. Hypoglycemia, but not glucose variability, relates to vascular function in children with type 1 diabetes. Diabetes Technol. Ther. 2012, 14, 457–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Qaissi, A.; Papageorgiou, M.; Deshmukh, H.; Madden, L.A.; Rigby, A.; Kilpatrick, E.S.; Atkin, S.L.; Sathyapalan, T. Effects of acute insulin-induced hypoglycaemia on endothelial microparticles in adults with and without type 2 diabetes. Diabetes Obes. Metab. 2019, 21, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Tiftikcioglu, B.I.; Bilgin, S.; Duksal, T.; Kose, S.; Zorlu, Y. Autonomic neuropathy and endothelial dysfunction in patients with impaired glucose tolerance or type 2 diabetes mellitus. Medicine 2016, 95, e3340. [Google Scholar] [CrossRef]
- Foreman, Y.D.; Brouwers, M.C.G.J.; Berendschot, T.T.J.M.; van Dongen, M.C.J.M.; Eussen, S.J.P.M.; van Greevenbroek, M.M.J.; Henry, R.M.A.; Houben, A.J.H.M.; van der Kallen, C.J.H.; Kroon, A.A.; et al. The oral glucose tolerance test-derived incremental glucose peak is associated with greater arterial stiffness and maladaptive arterial remodeling: The Maastricht study. Cardiovasc. Diabetol. 2019, 18, 152. [Google Scholar] [CrossRef]
- Zhang, L.; Sun, H.; Liu, S.; Gao, J.; Xia, J. Glycemic variability is associated with vascular calcification by the markers of endoplasmic reticulum stress-related apoptosis, Wnt1, Galectin-3 and BMP-2. Diabetol. Metab. Syndr. 2019, 11, 67. [Google Scholar] [CrossRef]
- Suslova, T.E.; Sitozhevskii, A.V.; Ogurkova, O.N.; Kravchenko, E.S.; Kologrivova, I.V.; Anfinogenova, Y.; Karpov, R.S. Platelet hemostasis in patients with metabolic syndrome and type 2 diabetes mellitus: CGMP- and NO-dependent mechanisms in the insulin-mediated platelet aggregation. Front. Physiol. 2014, 5, 501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santilli, F.; Formoso, G.; Sbraccia, P.; Averna, M.; Miccoli, R.; Di Fulvio, P.; Ganci, A.; Pulizzi, N.; Lattanzio, S.; Ciabattoni, G.; et al. Postprandial hyperglycemia is a determinant of platelet activation in early type 2 diabetes mellitus. J. Thromb. Haemost. 2010, 8, 828–837. [Google Scholar] [CrossRef]
- Nusca, A.; Tuccinardi, D.; Proscia, C.; Melfi, R.; Manfrini, S.; Nicolucci, A.; Ceriello, A.; Pozzilli, P.; Ussia, G.P.; Grigioni, F.; et al. Incremental role of glycaemic variability over HbA1c in identifying type 2 diabetic patients with high platelet reactivity undergoing percutaneous coronary intervention. Cardiovasc. Diabetol. 2019, 18, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, G.; Chen, L.; Wang, M.; Cui, W. Acute fluctuation in blood glucose has no effect on platelet aggregation rate. Clin. Lab. 2014, 60, 1071–1073. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, A.; Prince, L.R.; Novodvorsky, P.; Bernjak, A.; Thomas, M.R.; Birch, L.; Lambert, D.; Kay, L.J.; Wright, F.J.; Macdonald, I.A.; et al. Effect of hypoglycemia on inflammatory responses and the response to low-dose endotoxemia in humans. J. Clin. Endocrinol. Metab. 2019, 104, 1187–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, P.Z.; Soares, R. Neovascularization in diabetes and its complications. Unraveling the angiogenic paradox. Life Sci. 2013, 92, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Sang, Y.; Yin, T.; Wang, B.; Yang, W.; Li, X.; Li, H.; Kang, Y. MiR-1273g-3p participates in acute glucose fluctuation-induced autophagy, dysfunction, and proliferation attenuation in human umbilical vein endothelial cells. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E734–E743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biscetti, F.; Pitocco, D.; Straface, G.; Zaccardi, F.; de Cristofaro, R.; Rizzo, P.; Lancellotti, S.; Arena, V.; Stigliano, E.; Musella, T.; et al. Glycaemic variability affects ischaemia-induced angiogenesis in diabetic mice. Clin. Sci. 2011, 121, 555–564. [Google Scholar] [CrossRef]
- Hu, L.; Dai, S.-C.; Luan, X.; Chen, J.; Cannavicci, A. Dysfunction and therapeutic potential of endothelial progenitor cells in diabetes mellitus. J. Clin. Med. Res. 2018, 10, 752–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inaba, Y.; Tsutsumi, C.; Haseda, F.; Fujisawa, R.; Mitsui, S.; Sano, H.; Terasaki, J.; Hanafusa, T.; Imagawa, A. Impact of glycemic variability on the levels of endothelial progenitor cells in patients with type 1 diabetes. Diabetol. Int. 2018, 9, 113–120. [Google Scholar] [CrossRef]
- Maiorino, M.I.; Casciano, O.; Della Volpe, E.; Bellastella, G.; Giugliano, D.; Esposito, K. Reducing glucose variability with continuous subcutaneous insulin infusion increases endothelial progenitor cells in type 1 diabetes: An observational study. Endocrine 2016, 52, 244–252. [Google Scholar] [CrossRef]
- Boscari, F.; D’Anna, M.; Bonora, B.M.; Tresso, S.; Cappellari, R.; Avogaro, A.; Bruttomesso, D.; Fadini, G.P. Effects of glucose variability on hematopoietic stem/progenitor cells in patients with type 1 diabetes. J. Endocrinol. Invest. 2021, 44, 119–126. [Google Scholar] [CrossRef]
- Goth, A.; Lengyel, L.; Nadasdi, N.; Savely, C. Renal lesions due to fluctuations in blood sugar levels. Acta Med. Scand. 1957, 158, 475–480. [Google Scholar] [CrossRef]
- Takeuchi, A.; Throckmorton, D.C.; Brogden, A.P.; Yoshizawa, N.; Rasmussen, H.; Kashgarian, M. Periodic high extracellular glucose enhances production of collagens iii and iv by mesangial cells. Am. J. Physiol. 1995, 268, F13–F19. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.C.; Saunders, H.J.; Qi, W.; Pollock, C.A. Intermittent high glucose enhances cell growth and collagen synthesis in cultured human tubulointerstitial cells. Diabetologia 1999, 42, 1113–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polhill, T.S.; Saad, S.; Poronnik, P.; Fulcher, G.R.; Pollock, C.A. Short-term peaks in glucose promote renal fibrogenesis independently of total glucose exposure. Am. J. Physiol. Renal. Physiol. 2004, 287, F268–F273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, C.; Zhou, X.; Chang, Z.; Ling, H.; Cheng, X.; Li, W. Blood glucose fluctuation accelerates renal injury involved to inhibit the akt signaling pathway in diabetic rats. Endocrine 2016, 53, 81–96. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-F.; Li, T.-C.; Li, C.-I.; Liu, C.-S.; Lin, W.-Y.; Yang, S.-Y.; Chiang, J.-H.; Huang, C.-C.; Sung, F.-C.; Lin, C.-C. Visit-to-visit glucose variability predicts the development of end-stage renal disease in type 2 diabetes: 10-year follow-up of Taiwan diabetes study. Medicine 2015, 94, e1804. [Google Scholar] [CrossRef]
- Takenouchi, A.; Tsuboi, A.; Terazawa-Watanabe, M.; Kurata, M.; Fukuo, K.; Kazumi, T. Direct association of visit-to-visit HbA1c variation with annual decline in estimated glomerular filtration rate in patients with type 2 diabetes. J. Diabetes Metab. Disord. 2015, 14, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibb, F.W.; McKnight, J.A.; Clarke, C.; Strachan, M.W.J. Preserved C-peptide secretion is associated with fewer low-glucose events and lower glucose variability on flash glucose monitoring in adults with type 1 diabetes. Diabetologia 2020, 63, 906–914. [Google Scholar] [CrossRef] [Green Version]
- Christensen, M.B.; Gæde, P.; Hommel, E.; Gotfredsen, A.; Nørgaard, K. Glycaemic variability and hypoglycaemia are associated with c-peptide levels in insulin-treated type 2 diabetes. Diabetes Metab. 2020, 46, 61–65. [Google Scholar] [CrossRef]
- Zhang, Y.; Dai, J.; Han, X.; Zhao, Y.; Zhang, H.; Liu, X.; Li, W.; Ling, H.; Zhou, X.; Ying, C. Glycemic variability indices determined by self-monitoring of blood glucose are associated with β-cell function in chinese patients with type 2 diabetes. Diabetes Res. Clin. Pract. 2020, 164, 108152. [Google Scholar] [CrossRef]
- Cheong, Y.-H.; Kim, M.-K.; Son, M.-H.; Kaang, B.-K. Glucose Exposure pattern determines glucagon-like peptide 1 receptor expression and signaling through endoplasmic reticulum stress in rat insulinoma cells. Biochem. Biophys. Res. Commun. 2011, 414, 220–225. [Google Scholar] [CrossRef]
- Shao, C.; Gu, J.; Meng, X.; Zheng, H.; Wang, D. Systematic Investigation into the role of intermittent high glucose in pancreatic beta-cells. Int. J. Clin. Exp. Med. 2015, 8, 5462–5469. [Google Scholar]
- Wada, J.; Nakatsuka, A. Mitochondrial dynamics and mitochondrial dysfunction in diabetes. Acta Med. Okayama 2016, 70, 151–158. [Google Scholar] [CrossRef]
- Galvan, D.L.; Green, N.H.; Danesh, F.R. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int. 2017, 92, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-Y.; Yiang, G.-T.; Lai, T.-T.; Li, C.-J. The oxidative stress and mitochondrial dysfunction during the pathogenesis of diabetic retinopathy. Oxid. Med. Cell. Longev. 2018, 2018, 3420187. [Google Scholar] [CrossRef] [PubMed]
- Cieluch, A.; Uruska, A.; Zozulinska-Ziolkiewicz, D. Can we prevent mitochondrial dysfunction and diabetic cardiomyopathy in type 1 diabetes mellitus? Pathophysiology and treatment options. Int. J. Mol. Sci. 2020, 21, 2852. [Google Scholar] [CrossRef] [PubMed]
- Rocha, M.; Diaz-Morales, N.; Rovira-Llopis, S.; Escribano-Lopez, I.; Bañuls, C.; Hernandez-Mijares, A.; Diamanti-Kandarakis, E.; Victor, V.M. Mitochondrial dysfunction and endoplasmic reticulum stress in diabetes. Curr. Pharm. Des. 2016, 22, 2640–2649. [Google Scholar] [CrossRef]
- La Sala, L.; Mrakic-Sposta, S.; Micheloni, S.; Prattichizzo, F.; Ceriello, A. Glucose-sensing MicroRNA-21 disrupts ROS homeostasis and impairs antioxidant responses in cellular glucose variability. Cardiovasc. Diabetol. 2018, 17, 105. [Google Scholar] [CrossRef] [Green Version]
- Tao, S.; Ren, Y.; Zheng, H.; Zhao, M.; Zhang, X.; Zhu, Y.; Yang, J.; Zheng, S. Salvianolic acid B inhibits intermittent high glucose-induced INS-1 cell apoptosis through regulation of Bcl-2 proteins and mitochondrial membrane potential. Eur. J. Pharmacol. 2017, 814, 56–62. [Google Scholar] [CrossRef]
- Quincozes-Santos, A.; Bobermin, L.D.; de Assis, A.M.; Gonçalves, C.-A.; Souza, D.O. Fluctuations in glucose levels induce glial toxicity with glutamatergic, oxidative and inflammatory implications. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1–14. [Google Scholar] [CrossRef]
- Yin, X.; Zheng, F.; Pan, Q.; Zhang, S.; Yu, D.; Xu, Z.; Li, H. Glucose fluctuation increased hepatocyte apoptosis under lipotoxicity and the involvement of mitochondrial permeability transition opening. J. Mol. Endocrinol. 2015, 55, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Saito, S.; Thuc, L.C.; Teshima, Y.; Nakada, C.; Nishio, S.; Kondo, H.; Fukui, A.; Abe, I.; Ebata, Y.; Saikawa, T.; et al. Glucose fluctuations aggravate cardiac susceptibility to ischemia/reperfusion injury by modulating MicroRNAs expression. Circ. J. 2016, 80, 186–195. [Google Scholar] [CrossRef] [Green Version]
- Chareyron, I.; Christen, S.; Moco, S.; Valsesia, A.; Lassueur, S.; Dayon, L.; Wollheim, C.B.; Santo Domingo, J.; Wiederkehr, A. Augmented mitochondrial energy metabolism is an early response to chronic glucose stress in human pancreatic beta cells. Diabetologia 2020, 63, 2628–2640. [Google Scholar] [CrossRef]
- Xu, W.-L.; Liu, S.; Li, N.; Ye, L.-F.; Zha, M.; Li, C.-Y.; Zhao, Y.; Pu, Q.; Bao, J.-J.; Chen, X.-J.; et al. Quercetin antagonizes glucose fluctuation induced renal injury by inhibiting aerobic glycolysis via HIF-1α/MiR-210/ISCU/FeS pathway. Front. Med. 2021, 8, 656086. [Google Scholar] [CrossRef]
- Silva-Rodrigues, T.; de-Souza-Ferreira, E.; Machado, C.M.; Cabral-Braga, B.; Rodrigues-Ferreira, C.; Galina, A. Hyperglycemia in a type 1 diabetes mellitus model causes a shift in mitochondria coupled-glucose phosphorylation and redox metabolism in rat brain. Free Radic. Biol. Med. 2020, 160, 796–806. [Google Scholar] [CrossRef] [PubMed]
- Lemmer, I.L.; Willemsen, N.; Hilal, N.; Bartelt, A. A Guide to understanding endoplasmic reticulum stress in metabolic disorders. Mol. Metab. 2021, 47, 101169. [Google Scholar] [CrossRef] [PubMed]
- Sankrityayan, H.; Oza, M.J.; Kulkarni, Y.A.; Mulay, S.R.; Gaikwad, A.B. ER stress response mediates diabetic microvascular complications. Drug Discov. Today 2019, 24, 2247–2257. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Wang, J.J.; Zhang, S.X. Intermittent but not constant high glucose induces ER stress and inflammation in human retinal pericytes. Adv. Exp. Med. Biol. 2012, 723, 285–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikesugi, K.; Mulhern, M.L.; Madson, C.J.; Hosoya, K.-I.; Terasaki, T.; Kador, P.F.; Shinohara, T. Induction of endoplasmic reticulum stress in retinal pericytes by glucose deprivation. Curr. Eye Res. 2006, 31, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Tatsumi, Y.; Yako, H.; Sango, K.; Himeno, T.; Kondo, M.; Kato, Y.; Kamiya, H.; Nakamura, J.; Kato, K. Recurrent short-term hypoglycemia and hyperglycemia induce apoptosis and oxidative stress via the ER stress response in immortalized adult mouse schwann (IMS32) cells. Neurosci. Res. 2019, 147, 26–32. [Google Scholar] [CrossRef]
- Sage, A.T.; Holtby-Ottenhof, S.; Shi, Y.; Damjanovic, S.; Sharma, A.M.; Werstuck, G.H. Metabolic syndrome and acute hyperglycemia are associated with endoplasmic reticulum stress in human mononuclear cells. Obesity 2012, 20, 748–755. [Google Scholar] [CrossRef]
- Ichimiya, T.; Yamakawa, T.; Hirano, T.; Yokoyama, Y.; Hayashi, Y.; Hirayama, D.; Wagatsuma, K.; Itoi, T.; Nakase, H. Autophagy and autophagy-related diseases: A review. Int. J. Mol. Sci. 2020, 21, 8974. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B. Autophagy in human diseases. N. Engl. J. Med. 2020, 383, 1564–1576. [Google Scholar] [CrossRef]
- Marasco, M.R.; Linnemann, A.K. β-cell autophagy in diabetes pathogenesis. Endocrinology 2018, 159, 2127–2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muralidharan, C.; Conteh, A.M.; Marasco, M.R.; Crowder, J.J.; Kuipers, J.; de Boer, P.; Linnemann, A.K. Pancreatic beta cell autophagy is impaired in type 1 diabetes. Diabetologia 2021, 64, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Tao, T.; Xu, H. Autophagy and obesity and diabetes. Adv. Exp. Med. Biol. 2020, 1207, 445–461. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lim, Y.-M.; Lee, M.-S. The role of autophagy in systemic metabolism and human-type diabetes. Mol. Cells 2018, 41, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-A.; Wu, V.C.-C.; Wang, C.-Y. Autophagy in chronic kidney diseases. Cells 2019, 8, 61. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Livingston, M.J.; Liu, Z.; Dong, G.; Zhang, M.; Chen, J.-K.; Dong, Z. Autophagy in Diabetic kidney disease: Regulation, pathological role and therapeutic potential. Cell. Mol. Life Sci. 2018, 75, 669–688. [Google Scholar] [CrossRef]
- Korbut, A.I.; Taskaeva, I.S.; Bgatova, N.P.; Muraleva, N.A.; Orlov, N.B.; Dashkin, M.V.; Khotskina, A.S.; Zavyalov, E.L.; Konenkov, V.I.; Klein, T.; et al. SGLT2 inhibitor empagliflozin and DPP4 inhibitor linagliptin reactivate glomerular autophagy in Db/Db mice, a model of type 2 diabetes. Int. J. Mol. Sci. 2020, 21, 2987. [Google Scholar] [CrossRef] [Green Version]
- Efeyan, A.; Comb, W.C.; Sabatini, D.M. Nutrient-sensing mechanisms and pathways. Nature 2015, 517, 302–310. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- King, K.E.; Losier, T.T.; Russell, R.C. Regulation of autophagy enzymes by nutrient signaling. Trends Biochem. Sci. 2021. [Google Scholar] [CrossRef] [PubMed]
- Condon, K.J.; Sabatini, D.M. Nutrient regulation of MTORC1 at a glance. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zhao, Y. Other molecular mechanisms regulating autophagy. Adv. Exp. Med. Biol. 2019, 1206, 261–271. [Google Scholar] [PubMed]
- Elhassan, S.A.M.; Candasamy, M.; Chan, E.W.L.; Bhattamisra, S.K. Autophagy and GLUT4: The missing pieces. Diabetes Metab. Syndr. 2018, 12, 1109–1116. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Tang, M.; Liu, M.; Chen, L. Glycophagy: An emerging target in pathology. Clin. Chem. Acta 2018, 484, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Song, J.; Zhang, Y.; Ma, Y.; Yang, J.; He, G.; Chen, S. Intermittent high glucose-induced oxidative stress modulates retinal pigmented epithelial cell autophagy and promotes cell survival via increased HMGB1. BMC Ophthalmol. 2018, 18, 192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Y.; Tao, S.; Zheng, S.; Zhao, M.; Zhu, Y.; Yang, J.; Wu, Y. Salvianolic acid B improves vascular endothelial function in diabetic rats with blood glucose fluctuations via suppression of endothelial cell apoptosis. Eur. J. Pharmacol. 2016, 791, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Shen, H.; Wang, Y.; He, B.; Zhang, Y.; Bai, Y.; Du, R.; Du, Q.; Han, P. Role of the PKCβII/JNK signaling pathway in acute glucose fluctuation-induced apoptosis of rat vascular endothelial cells. Acta Diabetol. 2017, 54, 727–736. [Google Scholar] [CrossRef]
- Ying, C.; Wang, S.; Lu, Y.; Chen, L.; Mao, Y.; Ling, H.; Cheng, X.; Zhou, X. Glucose fluctuation increased mesangial cell apoptosis related to akt signal pathway. Arch. Med. Sci. 2019, 15, 730–737. [Google Scholar] [CrossRef]
- Chai, Q.; Miao, J.; Liu, M.; Zhang, Z.; Meng, Z.; Wu, W. SGLT1 Knockdown prevents glucose fluctuation-induced apoptosis of cardiomyocytes through attenuating oxidative stress and mitochondrial dysfunction. Biochem. Cell Biol. 2020. [Google Scholar] [CrossRef]
- Wang, H.; Deng, J.; Chen, L.; Ding, K.; Wang, Y. Acute glucose fluctuation induces inflammation and neurons apoptosis in hippocampal tissues of diabetic rats. J. Cell. Biochem. 2019. [Google Scholar] [CrossRef]
- Yan, T.; Zhang, Z.; Li, D. NGF Receptors and PI3K/AKT pathway involved in glucose fluctuation-induced damage to neurons and α-lipoic acid treatment. BMC Neurosci. 2020, 21, 38. [Google Scholar] [CrossRef]
- Hsieh, C.-F.; Liu, C.-K.; Lee, C.-T.; Yu, L.-E.; Wang, J.-Y. Acute glucose fluctuation impacts microglial activity, leading to inflammatory activation or self-degradation. Sci. Rep. 2019, 9, 840. [Google Scholar] [CrossRef] [Green Version]
- Xue, B.; Wang, L.; Zhang, Z.; Wang, R.; Xia, X.-X.; Han, P.-P.; Cao, L.-J.; Liu, Y.-H.; Sun, L.-Q. Puerarin may protect against schwann cell damage induced by glucose fluctuation. J. Nat. Med. 2017, 71, 472–481. [Google Scholar] [CrossRef]
- Alnahdi, A.; John, A.; Raza, H. Mitigation of glucolipotoxicity-induced apoptosis, mitochondrial dysfunction, and metabolic stress by N-acetyl cysteine in pancreatic β-cells. Biomolecules 2020, 10, 239. [Google Scholar] [CrossRef] [Green Version]
- Rani, K.; Aung, N.Y. Docosahexaenoic acid inhibits vascular smooth muscle cell proliferation induced by glucose variability. Open Biochem. J. 2017, 11, 56–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.H.; Yu, J.M.; Yoo, H.J.; Lee, S.J.; Kang, D.H.; Cho, Y.J.; Kim, D.M. Anti-proliferative effects of rutin on OLETF rat vascular smooth muscle cells stimulated by glucose variability. Yonsei Med. J. 2016, 57, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, J.; Jiang, X.; Yang, L.; Lei, L.; Cai, D.; Zhang, H.; Chen, H. GLP-1 Ameliorates the proliferation activity of INS-1 cells inhibited by intermittent high glucose concentrations through the regulation of cyclins. Mol. Med. Rep. 2014, 10, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, N.F.; Amarie, O.V.; Wu, M.; Amend, A.-L.; Rubey, M.; Gradinger, D.; Irmler, M.; Beckers, J.; Rathkolb, B.; Wolf, E.; et al. PAX6 mutation alters circadian rhythm and β cell function in mice without affecting glucose tolerance. Commun. Biol. 2020, 3, 628. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Ni, Q.; Wang, Y.; Zhang, H.; Li, W.; Nie, A.; Wang, S.; Gu, Y.; Wang, Q.; Ning, G. Raptor determines β-cell identity and plasticity independent of hyperglycemia in mice. Nat. Commun. 2020, 11, 2538. [Google Scholar] [CrossRef]
- Kiss, K.; Baghy, K.; Spisák, S.; Szanyi, S.; Tulassay, Z.; Zalatnai, A.; Löhr, J.-M.; Jesenofsky, R.; Kovalszky, I.; Firneisz, G. Chronic hyperglycemia induces trans-differentiation of human pancreatic stellate cells and enhances the malignant molecular communication with human pancreatic cancer cells. PLoS ONE 2015, 10, e0128059. [Google Scholar] [CrossRef] [Green Version]
- Regnell, S.E.; Hessner, M.J.; Jia, S.; Åkesson, L.; Stenlund, H.; Moritz, T.; La Torre, D.; Lernmark, Å. Longitudinal analysis of hepatic transcriptome and serum metabolome demonstrates altered lipid metabolism following the onset of hyperglycemia in spontaneously diabetic biobreeding rats. PLoS ONE 2017, 12, e0171372. [Google Scholar] [CrossRef]
- Felisbino, M.B.; Ziemann, M.; Khurana, I.; Okabe, J.; Al-Hasani, K.; Maxwell, S.; Harikrishnan, K.N.; de Oliveira, C.B.M.; Mello, M.L.S.; El-Osta, A. Valproic acid influences the expression of genes implicated with hyperglycaemia-induced complement and coagulation pathways. Sci. Rep. 2021, 11, 2163. [Google Scholar] [CrossRef]
- Silambarasan, M.; Tan, J.R.; Karolina, D.S.; Armugam, A.; Kaur, C.; Jeyaseelan, K. MicroRNAs in hyperglycemia induced endothelial cell dysfunction. Int. J. Mol. Sci. 2016, 17, 518. [Google Scholar] [CrossRef] [Green Version]
- Aas, V.; Hessvik, N.P.; Wettergreen, M.; Hvammen, A.W.; Hallén, S.; Thoresen, G.H.; Rustan, A.C. Chronic hyperglycemia reduces substrate oxidation and impairs metabolic switching of human myotubes. Biochim. Biophys. Acta 2011, 1812, 94–105. [Google Scholar] [CrossRef] [Green Version]
- Puri, K.; Lal, N.; Shang, R.; Ghosh, S.; Flibotte, S.; Dyer, R.; Hussein, B.; Rodrigues, B. Diabetes mellitus severity and a switch from using lipoprotein lipase to adipose-derived fatty acid results in a cardiac metabolic signature that embraces cell death. J. Am. Heart Assoc. 2019, 8, e014022. [Google Scholar] [CrossRef]
- Maier, K.G.; Han, X.; Sadowitz, B.; Gentile, K.L.; Middleton, F.A.; Gahtan, V. Thrombospondin-1: A proatherosclerotic protein augmented by hyperglycemia. J. Vasc. Surg. 2010, 51, 1238–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hien, T.T.; Turczyńska, K.M.; Dahan, D.; Ekman, M.; Grossi, M.; Sjögren, J.; Nilsson, J.; Braun, T.; Boettger, T.; Garcia-Vaz, E.; et al. Elevated glucose levels promote contractile and cytoskeletal gene expression in vascular smooth muscle via rho/protein kinase C and actin polymerization. J. Biol. Chem. 2016, 291, 3552–3568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rønningen, T.; Shah, A.; Reiner, A.H.; Collas, P.; Moskaug, J.Ø. Epigenetic priming of inflammatory response genes by high glucose in adipose progenitor cells. Biochem. Biophys. Res. Commun. 2015, 467, 979–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubin, A.; Salzberg, A.C.; Imamura, Y.; Grivitishvilli, A.; Tombran-Tink, J. Identification of novel targets of diabetic nephropathy and pedf peptide treatment using RNA-Seq. BMC Genom. 2016, 17, 936. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, S.; Singhal, S.; Sens, D.A.; Somji, S.; Davis, B.A.; Guyer, R.; Breen, S.; Kalonick, M.; Garrett, S.H. Elevated glucose represses lysosomal and MTOR-related genes in renal epithelial cells composed of progenitor CD133+ cells. PLoS ONE 2021, 16, e0248241. [Google Scholar] [CrossRef] [PubMed]
- Friedrichs, P.; Schlotterer, A.; Sticht, C.; Kolibabka, M.; Wohlfart, P.; Dietrich, A.; Linn, T.; Molema, G.; Hammes, H.-P. Hyperglycaemic memory affects the neurovascular unit of the retina in a diabetic mouse model. Diabetologia 2017, 60, 1354–1358. [Google Scholar] [CrossRef] [Green Version]
- Fan, S.; Yang, Z.; Liu, Y.; Zhong, J.; Zhang, S.; Xiao, Y.; Liu, X.; Yi, W.; He, C.; Hu, Y.; et al. Extensive Sub-RPE complement deposition in a nonhuman primate model of early-stage diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 2021, 62, 30. [Google Scholar] [CrossRef] [PubMed]
- Moganti, K.; Li, F.; Schmuttermaier, C.; Riemann, S.; Klüter, H.; Gratchev, A.; Harmsen, M.C.; Kzhyshkowska, J. Hyperglycemia induces mixed M1/M2 cytokine profile in primary human monocyte-derived macrophages. Immunobiology 2017, 222, 952–959. [Google Scholar] [CrossRef]
- Thiem, K.; Keating, S.T.; Netea, M.G.; Riksen, N.P.; Tack, C.J.; van Diepen, J.; Stienstra, R. Hyperglycemic memory of innate immune cells promotes in vitro proinflammatory responses of human monocytes and murine macrophages. J. Immunol. 2021, 206, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; D’Urso, A.; Toiber, D.; Sebastian, C.; Henry, R.E.; Vadysirisack, D.D.; Guimaraes, A.; Marinelli, B.; Wikstrom, J.D.; Nir, T.; et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell 2010, 140, 280–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emery, M.; Nanchen, N.; Preitner, F.; Ibberson, M.; Roduit, R. Biological characterization of gene response to insulin-induced hypoglycemia in mouse retina. PLoS ONE 2016, 11, e0150266. [Google Scholar] [CrossRef]
- Kim, J.L.; La Gamma, E.F.; Estabrook, T.; Kudrick, N.; Nankova, B.B. Whole genome expression profiling associates activation of unfolded protein response with impaired production and release of epinephrine after recurrent hypoglycemia. PLoS ONE 2017, 12, e0172789. [Google Scholar] [CrossRef] [Green Version]
- Zervou, S.; Wang, Y.-F.; Laiho, A.; Gyenesei, A.; Kytömäki, L.; Hermann, R.; Abouna, S.; Epstein, D.; Pelengaris, S.; Khan, M. Short-term hyperglycaemia causes non-reversible changes in arterial gene expression in a fully “switchable” in vivo mouse model of diabetes. Diabetologia 2010, 53, 2676–2687. [Google Scholar] [CrossRef] [Green Version]
- Saik, O.V.; Klimontov, V.V. Bioinformatic reconstruction and analysis of gene networks related to glucose variability in diabetes and its complications. IJMS 2020, 21, 8691. [Google Scholar] [CrossRef]
- El-Osta, A.; Brasacchio, D.; Yao, D.; Pocai, A.; Jones, P.L.; Roeder, R.G.; Cooper, M.E.; Brownlee, M. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med. 2008, 205, 2409–2417. [Google Scholar] [CrossRef]
- Costantino, S.; Paneni, F.; Battista, R.; Castello, L.; Capretti, G.; Chiandotto, S.; Tanese, L.; Russo, G.; Pitocco, D.; Lanza, G.A.; et al. Impact of glycemic variability on chromatin remodeling, oxidative stress, and endothelial dysfunction in patients with type 2 diabetes and with target HbA1c levels. Diabetes 2017, 66, 2472–2482. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Chung, H.; Yoon, C.; Lee, E.; Kim, T.; Kim, T.; Kwon, M.; Lee, S.; Rhee, B.; Park, J. Increase of INS-1 cell apoptosis under glucose fluctuation and the involvement of FOXO-SIRT pathway. Diabetes Res. Clin. Pract. 2012, 98, 132–139. [Google Scholar] [CrossRef]
- Aref-Eshghi, E.; Biswas, S.; Chen, C.; Sadikovic, B.; Chakrabarti, S. Glucose-induced, duration-dependent genome-wide DNA methylation changes in human endothelial cells. Am. J. Physiol. Cell Physiol. 2020, 319, C268–C276. [Google Scholar] [CrossRef]
- Abdelzaher, L.A.; Imaizumi, T.; Suzuki, T.; Tomita, K.; Takashina, M.; Hattori, Y. Astaxanthin alleviates oxidative stress insults-related derangements in human vascular endothelial cells exposed to glucose fluctuations. Life Sci. 2016, 150, 24–31. [Google Scholar] [CrossRef]
- Xiao, X.; Dong, Y.; Zhong, J.; Cao, R.; Zhao, X.; Wen, G.; Liu, J. Adiponectin protects endothelial cells from the damages induced by the intermittent high level of glucose. Endocrine 2011, 40, 386–393. [Google Scholar] [CrossRef]
- Thomas, M.C. Glycemic exposure, glycemic control, and metabolic karma in diabetic complications. Adv. Chronic Kidney Dis. 2014, 21, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liao, Z.; Lin, X.; Wu, X.; Chen, X.; Bai, X.; Zhuang, Y.; Yang, Y.; Zhang, J. Overexpression of MiR-146a Might regulate polarization transitions of BV-2 cells induced by high glucose and glucose fluctuations. Front. Endocrinol. 2019, 10, 719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Li, Y.; You, C. MiR-129-3p targeting of MCU Protects against glucose fluctuation-mediated neuronal damage via a mitochondrial-dependent intrinsic apoptotic pathway. Diabetes Metab. Syndr. Obes. 2021, 14, 153–163. [Google Scholar] [CrossRef] [PubMed]
- La Sala, L.; Cattaneo, M.; De Nigris, V.; Pujadas, G.; Testa, R.; Bonfigli, A.R.; Genovese, S.; Ceriello, A. Oscillating glucose induces MicroRNA-185 and impairs an efficient antioxidant response in human endothelial cells. Cardiovasc. Diabetol. 2016, 15, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, M.E.; El-Osta, A. Epigenetics: Mechanisms and implications for diabetic complications. Circ. Res. 2010, 107, 1403–1413. [Google Scholar] [CrossRef] [Green Version]
- Akil, A.-S.A.-S.; Jerman, L.F.; Yassin, E.; Padmajeya, S.S.; Al-Kurbi, A.; Fakhro, K.A. Reading between the (Genetic) lines: How epigenetics is unlocking novel therapies for type 1 diabetes. Cells 2020, 9, 2403. [Google Scholar] [CrossRef]
- Luo, X.; Wu, J.; Jing, S.; Yan, L.-J. Hyperglycemic stress and carbon stress in diabetic glucotoxicity. Aging Dis. 2016, 7, 90–110. [Google Scholar] [CrossRef] [Green Version]
- Suryavanshi, S.V.; Kulkarni, Y.A. NF-Κβ: A potential target in the management of vascular complications of diabetes. Front. Pharmacol. 2017, 8, 798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Chen, Y.; Chen, H.; Li, L.; Yao, J.; Jiang, Q.; Lin, X.; Wen, J.; Lin, L. The effect of NF-ΚB pathway on proliferation and apoptosis of human umbilical vein endothelial cells induced by intermittent high glucose. Mol. Cell Biochem. 2011, 347, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-Y.; Qian, L.-L.; Wang, N.; Miao, L.-F.; Ma, X.; Dang, S.-P.; Wu, Y.; Liu, X.-Y.; Li, X.-Y.; Chai, Q.; et al. Glucose fluctuations promote vascular BK channels dysfunction via PKCα/NF-ΚB/MuRF1 signaling. J. Mol. Cell. Cardiol. 2020, 145, 14–24. [Google Scholar] [CrossRef]
- Schultze, S.M.; Hemmings, B.A.; Niessen, M.; Tschopp, O. PI3K/AKT, MAPK and AMPK Signalling: Protein kinases in glucose homeostasis. Expert Rev. Mol. Med. 2012, 14, e1. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.-Y.; Miao, L.-F.; Qian, L.-L.; Wang, N.; Qi, M.-M.; Zhang, Y.-M.; Dang, S.-P.; Wu, Y.; Wang, R.-X. Molecular mechanisms of glucose fluctuations on diabetic complications. Front. Endocrinol. 2019, 10, 640. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Are oxidative stress-activated signaling pathways mediators of insulin resistance and -cell dysfunction? Diabetes 2003, 52, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Yarza, R.; Vela, S.; Solas, M.; Ramirez, M.J. C-Jun N-terminal kinase (JNK) Signaling as a therapeutic target for Alzheimer’s disease. Front. Pharmacol. 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bretón-Romero, R.; Feng, B.; Holbrook, M.; Farb, M.G.; Fetterman, J.L.; Linder, E.A.; Berk, B.D.; Masaki, N.; Weisbrod, R.M.; Inagaki, E.; et al. Endothelial dysfunction in human diabetes is mediated by Wnt5a-JNK signaling. Arter. Thromb. Vasc. Biol. 2016, 36, 561–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Z.; Lei, M. Involvement of JNK signal transduction pathway in endothelial cell apoptosis induced by intermittent high glucose. Zhong Nan Da Xue Xue Bao. Yi Xue Ban 2010, 35, 616–621. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-Y.; Wang, N.; Qian, L.-L.; Miao, L.-F.; Dang, S.-P.; Wu, Y.; Wang, R.-X. Glucose fluctuations promote aortic fibrosis through the ROS/P38 MAPK/Runx2 signaling pathway. J. Vasc. Res. 2020, 57, 24–33. [Google Scholar] [CrossRef]
- Lan, H.Y.; Chung, A.C.K. Transforming growth factor-β and smads. Contrib. Nephrol. 2011, 170, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.-H.; Hung, C.-C.; Hsu, H.-H.; Jing, Y.-H.; Yang, C.-W.; Chen, J.-K. Resveratrol ameliorates early diabetic nephropathy associated with suppression of augmented TGF-β/Smad and ERK1/2 signaling in streptozotocin-induced diabetic rats. Chem. Biol. Interact. 2011, 190, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Gao, W.; Dang, Y.; Liu, X.; Li, Y.; Peng, X.; Ye, X. Both ERK/MAPK and TGF-Beta/Smad signaling pathways play a role in the kidney fibrosis of diabetic mice accelerated by blood glucose fluctuation. J. Diabetes Res. 2013, 2013, 463740. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Cheng, X.; Liu, L.; Zhao, D.; Dang, Y. Blood glucose fluctuation affects skin collagen metabolism in the diabetic mouse by inhibiting the mitogen-activated protein kinase and smad pathways. Clin. Exp. Dermatol. 2013, 38, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.-M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef]
- González, A.; Hall, M.N.; Lin, S.-C.; Hardie, D.G. AMPK and TOR: The yin and yang of cellular nutrient sensing and growth control. Cell Metab. 2020, 31, 472–492. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Ren, L.; Zhi, L.; Yu, Z.; Lv, F.; Xu, F.; Peng, W.; Bai, X.; Cheng, K.; Quan, L.; et al. Negative regulation of AMPK signaling by high glucose via E3 ubiquitin ligase MG53. Mol. Cell 2021, 81, 629–637.e5. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.-Y.; Zhao, M.; Yi, T.-N.; Zhang, J. Globular adiponectin protects human umbilical vein endothelial cells against apoptosis through adiponectin receptor 1/adenosine monophosphate-activated protein kinase pathway. Chin. Med. J. 2011, 124, 2540–2547. [Google Scholar] [PubMed]
- Gleason, C.E.; Lu, D.; Witters, L.A.; Newgard, C.B.; Birnbaum, M.J. The role of AMPK and MTOR in nutrient sensing in pancreatic beta-cells. J. Biol. Chem. 2007, 282, 10341–10351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leprivier, G.; Rotblat, B. How does MTOR sense glucose starvation? AMPK is the usual suspect. Cell Death Discov. 2020, 6, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Zhang, C.-S.; Feng, J.-W.; Wei, X.; Zhang, C.; Xie, C.; Wu, Y.; Hawley, S.A.; Atrih, A.; Lamont, D.J.; et al. Aldolase is a sensor for both low and high glucose, linking to AMPK and MTORC1. Cell Res. 2021, 31, 478–481. [Google Scholar] [CrossRef]
- Ali, M.; Bukhari, S.A.; Ali, M.; Lee, H.-W. Upstream signalling of MTORC1 and its hyperactivation in type 2 diabetes (T2D). BMB Rep. 2017, 50, 601–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, Z.; Maiese, K. Mammalian target of rapamycin signaling in diabetic cardiovascular disease. Cardiovasc. Diabetol. 2012, 11, 45. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zheng, Y.; Zhou, Y.; Liu, Y.; Xie, M.; Meng, W.; An, M. The expression and significance of MTORC1 in diabetic retinopathy. BMC Ophthalmol. 2020, 20, 297. [Google Scholar] [CrossRef]
- Yasuda-Yamahara, M.; Kume, S.; Maegawa, H. Roles of MTOR in diabetic kidney disease. Antioxidants 2021, 10, 321. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Effect | Pathways | Refs. |
|---|---|---|
| Oxidative stress in endothelial and neural cells | PKC/NF-κB, PI3K/Akt, p38MAPK | [99,134,174,175] |
| Endothelial dysfunction and apoptosis | PI3K/Akt, NF-κB, PKC/JNK | [19,58,129,176,184] |
| Proliferation of VSMCs | MAPK (ERK1/2), PI3K/Akt, NF-κB | [138] |
| Vascular low-grade inflammation | NF-κB and p38 MAPK | [162] |
| Renal fibrosis | MAPK (ERK1/2) and TGF- β/Smad | [188] |
| Aortic fibrosis | TGF-β/Smad, NF-κB, p38 MAPK and Runx2 | [185] |
| Neuronal apoptosis and neurodegeneration | PI3K/Akt, NF-κB | [133,134] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klimontov, V.V.; Saik, O.V.; Korbut, A.I. Glucose Variability: How Does It Work? Int. J. Mol. Sci. 2021, 22, 7783. https://doi.org/10.3390/ijms22157783
Klimontov VV, Saik OV, Korbut AI. Glucose Variability: How Does It Work? International Journal of Molecular Sciences. 2021; 22(15):7783. https://doi.org/10.3390/ijms22157783
Chicago/Turabian StyleKlimontov, Vadim V., Olga V. Saik, and Anton I. Korbut. 2021. "Glucose Variability: How Does It Work?" International Journal of Molecular Sciences 22, no. 15: 7783. https://doi.org/10.3390/ijms22157783
APA StyleKlimontov, V. V., Saik, O. V., & Korbut, A. I. (2021). Glucose Variability: How Does It Work? International Journal of Molecular Sciences, 22(15), 7783. https://doi.org/10.3390/ijms22157783