
PAI-1 in Diabetes: Pathophysiology and Role as a Therapeutic Target


Abstract
:1. Introduction
1.1. Fibrinolysis in Diabetes
1.2. PAI-1 Structure and Function
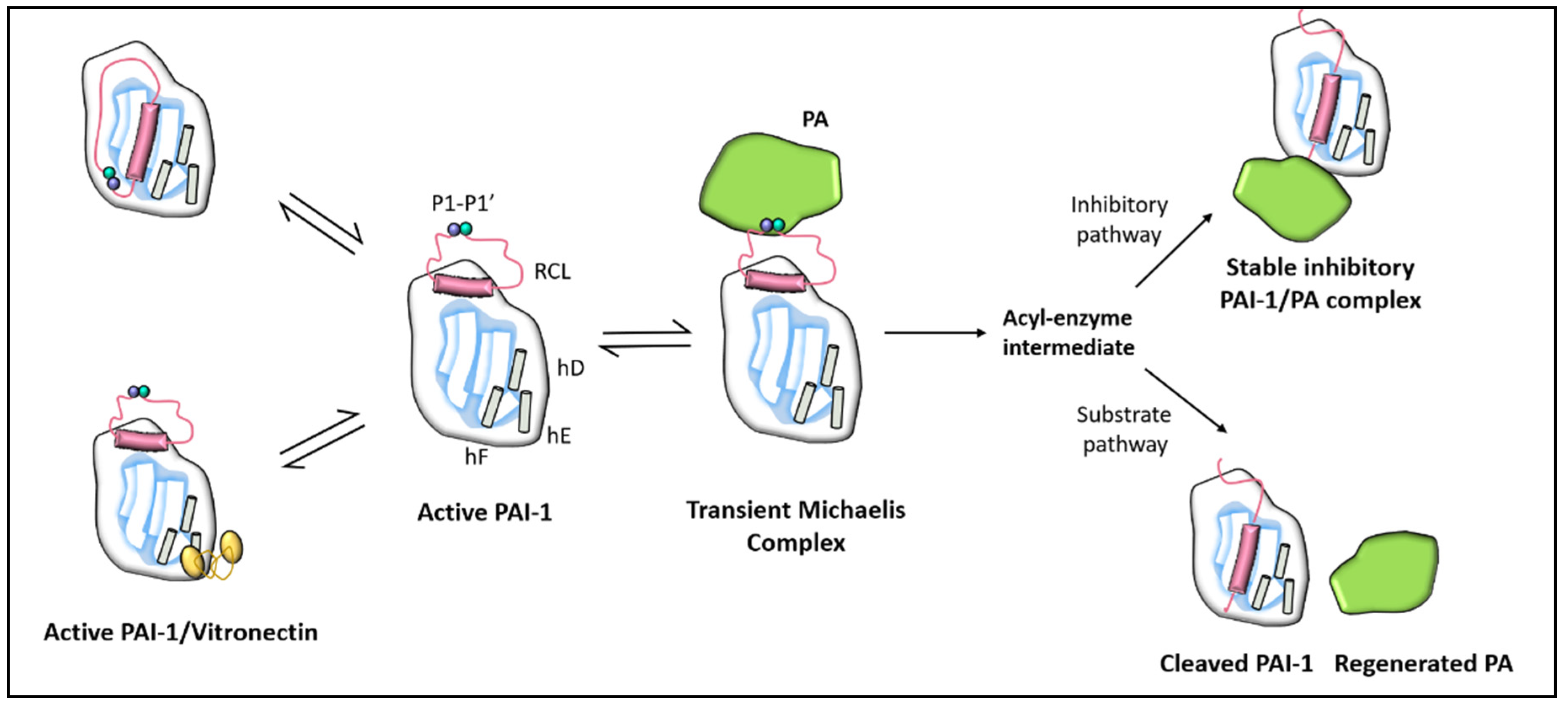
1.2.1. PAI-1 Structure
1.2.2. PAI-1 Function
1.3. PAI-1 Production and Regulation
1.3.1. Source of PAI-1
1.3.2. Regulation of PAI-1
1.4. Role of PAI-1 in Diabetes
1.5. Role of PAI-1 in CVD
1.6. Role of PAI-1 in Diabetic Retinopathy and Chronic Kidney Disease
2. Targeting PAI-1 for Potential Therapies
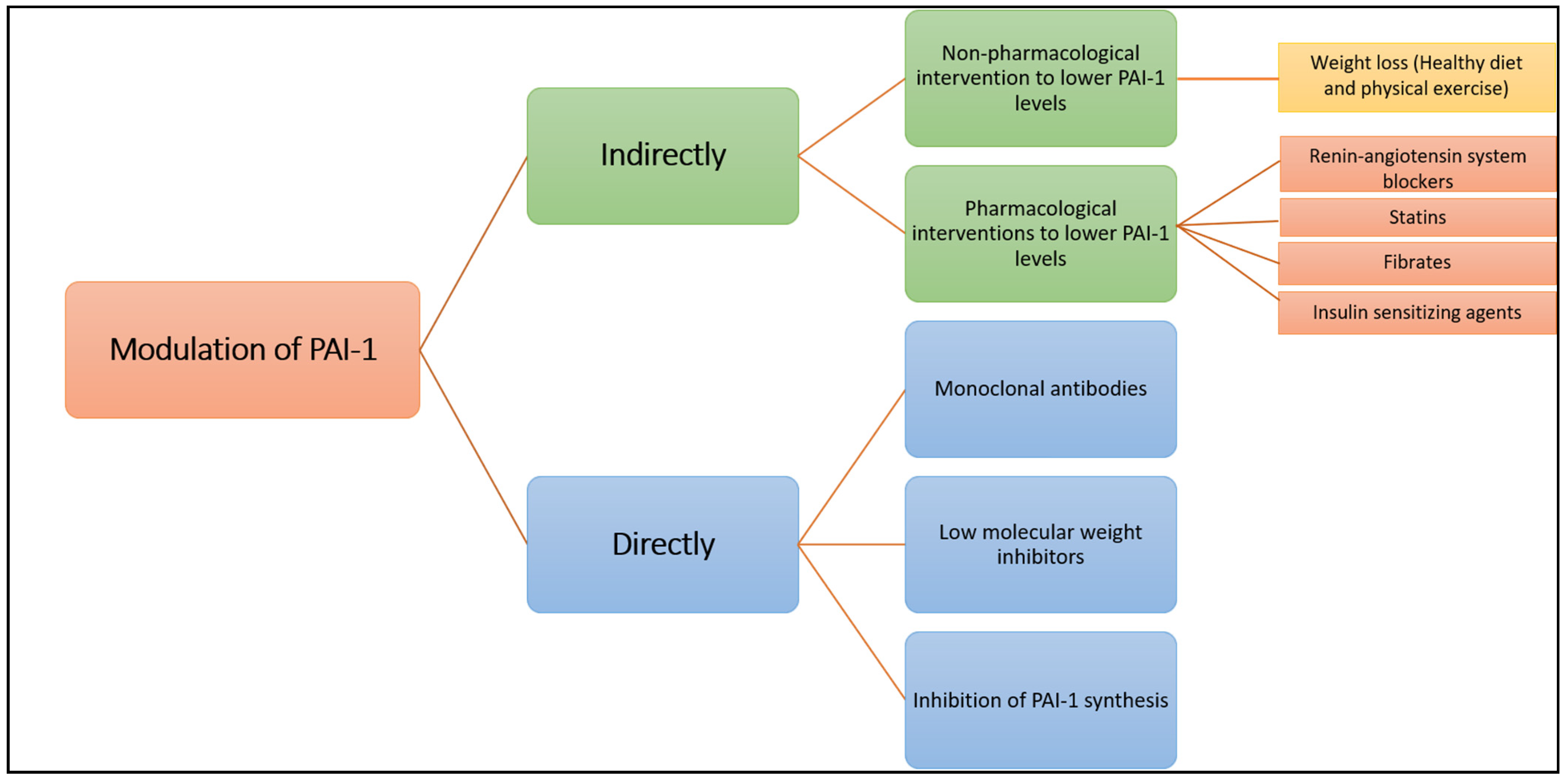
2.1. Indirect Modulation of PAI-1 Production or Function
2.1.1. A. Non-Pharmacological Intervention to Lower PAI-1 Levels
Weight Loss (Healthy Diet and Physical Exercise)
2.1.2. Pharmacological Interventions to Lower PAI-1 Levels
Renin-Angiotensin System Blockers
Statins
Fibrates
Insulin-Sensitising Agents
2.2. Direct Modulation of PAI-1 Production or Activity
2.2.1. Inhibition of PAI-1 Synthesis
2.2.2. Interfering with Serpin-Proteinase Interaction
Monoclonal Antibodies
Low Molecular Weight Inhibitors
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Narayan, K.M.V.; Boyle, J.P.; Thompson, T.J.; Sorensen, S.W.; Williamson, D.F. Lifetime Risk for Diabetes Mellitus in the United States. JAMA 2003, 290, 1884–1890. [Google Scholar] [CrossRef] [Green Version]
- Kannel, W.B.; McGee, D.L. Diabetes and Glucose Tolerance as Risk Factors for Cardiovascular Disease: The Framingham Study. Diabetes Care 1979, 2, 120–126. [Google Scholar] [CrossRef]
- Alzahrani, S.H.; Ajjan, R. Review article: Coagulation and fibrinolysis in diabetes. Diabetes Vasc. Dis. Res. 2010, 7, 260–273. [Google Scholar] [CrossRef]
- Kearney, K.; Tomlinson, D.; Smith, K.; Ajjan, R. Hypofibrinolysis in diabetes: A therapeutic target for the reduction of cardiovascular risk. Cardiovasc. Diabetol. 2017, 16, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Sumaya, W.; Wallentin, L.; James, S.K.; Siegbahn, A.; Gabrysch, K.; Himmelmann, A.; Ajjan, R.A.; Storey, R.F. Impaired Fibrinolysis Predicts Adverse Outcome in Acute Coronary Syndrome Patients with Diabetes: A PLATO Sub-Study. Thromb. Haemost. 2020, 120, 412–422. [Google Scholar] [CrossRef] [Green Version]
- Hess, K.; Alzahrani, S.H.; Mathai, M.; Schroeder, V.; Carter, A.M.; Howell, G.J.; Koko, T.; Strachan, M.W.J.; Price, J.; Smith, K.A.; et al. A novel mechanism for hypofibrinolysis in diabetes: The role of complement C3. Diabetologia 2012, 55, 1103–1113. [Google Scholar] [CrossRef]
- Sprengers, E.; Kluft, C. Plasminogen activator inhibitors. Blood 1987, 69, 381–387. [Google Scholar] [CrossRef] [Green Version]
- Schneider, D.J. Abnormalities of coagulation, platelet function, and fibrinolysis associated with syndromes of insulin resistance. Coron. Artery Dis. 2005, 16, 473–476. [Google Scholar] [CrossRef]
- Skurk, T.; Hauner, H. Obesity and impaired fibrinolysis: Role of adipose production of plasminogen activator inhibitor-1. Int. J. Obes. 2004, 28, 1357–1364. [Google Scholar] [CrossRef] [Green Version]
- Aso, Y. Plasminogen activator inhibitor (PAI)-1 in vascular inflammation and thrombosis. Front. Biosci. 2007, 12, 2957–2966. [Google Scholar] [CrossRef] [Green Version]
- Gils, A.; Declerck, P.J. The structural basis for the pathophysiological relevance of PAI-1 in cardiovascular diseases and the development of potential PAI-1 inhibitors. Thromb. Haemost. 2004, 91, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Durand, M.K.; Bødker, J.S.; Christensen, A.; Dupont, D.M.; Hansen, M.; Jensen, J.K.; Kjelgaard, S.; Mathiasen, L.; Pedersen, K.E.; Skeldal, S.; et al. Plasminogen activator inhibitor-1 and tumour growth, invasion, and metastasis. Thromb. Haemost. 2004, 91, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Binder, B.R.; Christ, G.; Gruber, F.; Grubic, N.; Hufnagl, P.; Krebs, M.; Mihaly, J.; Prager, G.W. Plasminogen activator inhibitor 1: Physiological and pathophysiological roles. Physiology 2002, 17, 56–61. [Google Scholar] [CrossRef]
- Placencio, V.R.; Declerck, Y.A. Plasminogen Activator Inhibitor-1 in Cancer: Rationale and Insight for Future Therapeutic Testing. Cancer Res. 2015, 75, 2969–2974. [Google Scholar] [CrossRef] [Green Version]
- Fjellström, O.; Deinum, J.; Sjögren, T.; Johansson, C.; Geschwindner, S.; Nerme, V.; Legnehed, A.; McPheat, J.; Olsson, K.; Bodin, C.; et al. Characterization of a Small Molecule Inhibitor of Plasminogen Activator Inhibitor Type 1 That Accelerates the Transition into the Latent Conformation. J. Biol. Chem. 2013, 288, 873–885. [Google Scholar] [CrossRef] [Green Version]
- Simone, T.M.; Higgins, P.J. Low Molecular Weight Antagonists of Plasminogen Activator Inhibitor-1: Therapeutic Potential in Cardiovascular Disease. Mol. Med. Ther. 2012, 1, 101. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.; A Huntington, J.; Pannu, N.S.; Carrell, R.W.; Read, R.J. How vitronectin binds PAI-1 to modulate fibrinolysis and cell migration. Nat. Struct. Mol. Biol. 2003, 10, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Blouse, G.E.; Dupont, D.M.; Schar, C.R.; Jensen, J.K.; Minor, K.H.; Anagli, J.Y.; Gårdsvoll, H.; Ploug, M.; Peterson, C.B.; Andreasen, P.A. Interactions of Plasminogen Activator Inhibitor-1 with Vitronectin Involve an Extensive Binding Surface and Induce Mutual Conformational Rearrangements. Biochemistry 2009, 48, 1723–1735. [Google Scholar] [CrossRef]
- Schar, C.R.; Blouse, G.E.; Minor, K.H.; Peterson, C.B. A Deletion Mutant of Vitronectin Lacking the Somatomedin B Domain Exhibits Residual Plasminogen Activator Inhibitor-1-binding Activity. J. Biol. Chem. 2008, 283, 10297–10309. [Google Scholar] [CrossRef] [Green Version]
- Mertens, I.; van Gaal, L.F. Obesity, haemostasis and the fibrinolytic system. Obes. Rev. 2002, 3, 85–101. [Google Scholar] [CrossRef]
- van de Craen, B.; Declerck, P.J.; Gils, A. The Biochemistry, Physiology and Pathological roles of PAI-1 and the requirements for PAI-1 inhibition in vivo. Thromb. Res. 2012, 130, 576–585. [Google Scholar] [CrossRef]
- Cesari, M.; Pahor, M.; Incalzi, R.A. REVIEW: Plasminogen Activator Inhibitor-1 (PAI-1): A Key Factor Linking Fibrinolysis and Age-Related Subclinical and Clinical Conditions. Cardiovasc. Ther. 2010, 28, e72–e91. [Google Scholar] [CrossRef] [Green Version]
- Charlton, P. The status of plasminogen activator inhibitor-1 as a therapeutic target. Expert Opin. Investig. Drugs 1997, 6, 539–554. [Google Scholar] [CrossRef]
- Tjärnlund-Wolf, A.; Brogren, H.; Lo, E.H.; Wang, X. Plasminogen Activator Inhibitor-1 and Thrombotic Cerebrovascular Diseases. Stroke 2012, 43, 2833–2839. [Google Scholar] [CrossRef] [PubMed]
- Aso, Y. Fibrinolysis and diabetic vascular disease: Roles of plasminogen activator inhibitor-1 and thrombin-activatable fibrinolysis inhibitor. Future Lipidol. 2006, 1, 429–440. [Google Scholar] [CrossRef]
- Mansfield, M.W.; Stickland, M.H.; Grant, P.J. Plasminogen Activator Inhibitor-1 (PAI-1) Promoter Polymorphism and Coronary Artery Disease in Non-Insulin-Dependent Diabetes. Thromb. Haemost. 1995, 74, 1032–1034. [Google Scholar] [CrossRef]
- Raji, M.A.; Snih, S.A.; Ray, L.A.; Patel, K.V.; Markides, K.S. Cognitive status and incident disability in older Mexican Americans: Findings from the Hispanic established population for the epidemiological study of the elderly. Ethn. Dis. 2004, 14, 26–31. [Google Scholar] [PubMed]
- Lutsey, P.L.; Cushman, M.; Steffen, L.M.; Green, D.; Barr, R.G.; Herrington, D.; Ouyang, P.; Folsom, A.R. Plasma hemostatic factors and endothelial markers in four racial/ethnic groups: The MESA study. J. Thromb. Haemost. 2006, 4, 2629–2635. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurti, C.; Tang, D.B.; Barr, C.F.; Alving, B.M. Plasminogen Activator and Plasminogen Activator Inhibitor Activities in a Reference Population. Am. J. Clin. Pathol. 1988, 89, 747–752. [Google Scholar] [CrossRef]
- Trost, S.; Pratley, R.E.; Sobel, B.E. Impaired fibrinolysis and risk for cardiovascular disease in the metabolic syndrome and type 2 diabetes. Curr. Diabetes Rep. 2006, 6, 47–54. [Google Scholar] [CrossRef]
- Fattal, P.; Schneider, D.; Sobel, B.; Billadello, J. Post-transcriptional regulation of expression of plasminogen activator inhibitor type 1 mRNA by insulin and insulin-like growth factor 1. J. Biol. Chem. 1992, 267, 12412–12415. [Google Scholar] [CrossRef]
- Lee, Y.H.; Nair, S.; Rousseau, E.; Allison, D.B.; Page, G.P.; Tataranni, P.A.; Bogardus, C.; Permana, P.A. Microarray profiling of isolated abdominal subcutaneous adipocytes from obese vs. non-obese Pima Indians: Increased expression of inflammation-related genes. Diabetologia 2005, 48, 1776–1783. [Google Scholar] [CrossRef] [Green Version]
- Skurk, T.; Lee, Y.-M.; Hauner, H. Angiotensin II and Its Metabolites Stimulate PAI-1 Protein Release From Human Adipocytes in Primary Culture. Hypertension 2001, 37, 1336–1340. [Google Scholar] [CrossRef] [Green Version]
- Aso, Y.; Matsumoto, S.; Fujiwara, Y.; Tayama, K.; Inukai, T.; Takemura, Y. Impaired fibrinolytic compensation for hypercoagulability in obese patients with type 2 diabetes: Association with increased plasminogen activator inhibitor-1. Metab. Clin. Exp. 2002, 51, 471–476. [Google Scholar] [CrossRef]
- Ågren, A.; Jörneskog, G.; Elgue, G.; Henriksson, P.; Wallén, H.; Wiman, B. Increased Incorporation of Antiplasmin into the Fibrin Network in Patients with Type 1 Diabetes. Diabetes Care 2014, 37, 2007–2014. [Google Scholar] [CrossRef] [Green Version]
- Alessi, M.C.; Juhan-Vague, I.; Kooistra, T.; Declerck, P.J.; Collen, D. Insulin Stimulates the Synthesis of Plasminogen Activator Inhibitor 1 by the Human Hepatocellular Cell Line Hep G2. Thromb. Haemost. 1988, 60, 491–494. [Google Scholar] [CrossRef]
- Schneider, D.J.; Sobel, B.E. Augmentation of synthesis of plasminogen activator inhibitor type 1 by insulin and insulin-like growth factor type I: Implications for vascular disease in hyperinsulinemic states. Proc. Natl. Acad. Sci. USA 1991, 88, 9959–9963. [Google Scholar] [CrossRef] [Green Version]
- Nordt, T.K.; Schneider, D.J.; E Sobel, B. Augmentation of the synthesis of plasminogen activator inhibitor type-1 by precursors of insulin. A potential risk factor for vascular disease. Circulation 1994, 89, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Cusi, K.; Maezono, K.; Osman, A.; Pendergrass, M.; Patti, M.E.; Pratipanawatr, T.; DeFronzo, R.A.; Kahn, C.R.; Mandarino, L.J. Insulin resistance differentially affects the PI 3-kinase—And MAP kinase—Mediated signaling in human muscle. J. Clin. Investig. 2000, 105, 311–320. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.S.; Goldstein, J.L. Selective versus Total Insulin Resistance: A Pathogenic Paradox. Cell Metab. 2008, 7, 95–96. [Google Scholar] [CrossRef] [Green Version]
- Muniyappa, R.; Montagnani, M.; Koh, K.K.; Quon, M.J. Cardiovascular Actions of Insulin. Endocr. Rev. 2007, 28, 463–491. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, W.A.; Quiñones, M.J. Role of endothelial dysfunction in insulin resistance. Am. J. Cardiol. 2003, 92, 10–17. [Google Scholar] [CrossRef]
- Montagnani, M.; Golovchenko, I.; Kim, I.; Koh, G.Y.; Goalstone, M.L.; Mundhekar, A.N.; Johansen, M.; Kucik, D.F.; Quon, M.J.; Draznin, B. Inhibition of Phosphatidylinositol 3-Kinase Enhances Mitogenic Actions of Insulin in Endothelial Cells. J. Biol. Chem. 2002, 277, 1794–1799. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-Q.; Su, M.; Walia, R.R.; Hao, Q.; Covington, J.W.; Vaughan, D.E. Sp1 Sites Mediate Activation of the Plasminogen Activator Inhibitor-1 Promoter by Glucose in Vascular Smooth Muscle Cells. J. Biol. Chem. 1998, 273, 8225–8231. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Billadello, J.J.; Schneider, D.J. Identification and localization of a fatty acid response region in the human plasminogen activator inhibitor-1 gene. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2696–2701. [Google Scholar] [CrossRef] [Green Version]
- Schneider, D.J.; Sobel, B.E. Synergistic augmentation of expression of plasminogen activator inhibitor type-1 induced by insulin, very-low-density lipoproteins, and fatty acids. Coron. Artery Dis. 1996, 7, 813–818. [Google Scholar] [CrossRef]
- Miyashita, Y.; Shirai, K.; Itoh, Y.; Sasaki, H.; Totsuka, M.; Murano, T.; Watanabe, H. Low lipoprotein lipase mass in preheparin serum of type 2 diabetes mellitus patients and its recovery with insulin therapy. Diabetes Res. Clin. Pract. 2002, 56, 181–187. [Google Scholar] [CrossRef]
- Banfi, C.; Mussoni, L.; Risé, P.; Cattaneo, M.G.; Vicentini, L.; Battaini, F.; Galli, C.; Tremoli, E. Very Low Density Lipoprotein—Mediated Signal Transduction and Plasminogen Activator Inhibitor Type 1 in Cultured HepG2 Cells. Circ. Res. 1999, 85, 208–217. [Google Scholar] [CrossRef] [Green Version]
- Kitagawa, N.; Yano, Y.; Gabazza, E.C.; Bruno, N.E.; Araki, R.; Matsumoto, K.; Katsuki, A.; Hori, Y.; Nakatani, K.; Taguchi, O.; et al. Different metabolic correlations of thrombin-activatable fibrinolysis inhibitor and plasminogen activator inhibitor-1 in non-obese type 2 diabetic patients. Diabetes Res. Clin. Pract. 2006, 73, 150–157. [Google Scholar] [CrossRef]
- Soares, A.L.; Rosário, P.W.; Borges, M.A.R.; Sousa, M.O.; Fernandes, A.P.S.M.; Carvalho, M.D.G.M. PAI-1 and D-Dimer in Type 2 Diabetic Women With Asymptomatic Macrovascular Disease Assessed by Carotid Doppler. Clin. Appl. Thromb. Hemost. 2009, 16, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Le, D.S.N.; Miles, R.; Savage, P.J.; Cornell, E.; Tracy, R.P.; Knowler, W.C.; Krakoff, J. The association of plasma fibrinogen concentration with diabetic microvascular complications in young adults with early-onset of type 2 diabetes. Diabetes Res. Clin. Pract. 2008, 82, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Romuk, E.; Jagosz, J.; Skrzep-Poloczek, B.; Wojciechowska, C.; Strojek, K.; Sędek, Ł.; Birkner, E. Evaluation of VCAM-1 and PAI-1 concentration in diabetes mellitus patients. Exp. Clin. Diabetol. 2008, 8, 85–88. [Google Scholar]
- Sahli, D.; Eriksson, J.W.; Boman, K.; Svensson, M.K. Tissue plasminogen activator (tPA) activity is a novel and early marker of asymptomatic LEAD in type 2 diabetes. Thromb. Res. 2009, 123, 701–706. [Google Scholar] [CrossRef]
- Erem, C.; Hacıhasanoğlu, A.; Çelik, Ş.; Ovalı, E.; Ersöz, H.Ö.; Ukinç, K.; Deger, O.; Telatar, M. Coagulation and Fibrinolysis Parameters in Type 2 Diabetic Patients with and without Diabetic Vascular Complications. Med. Princ. Pract. 2004, 14, 22–30. [Google Scholar] [CrossRef]
- Verkleij, C.J.N.; de Bruijn, R.E.; Meesters, E.W.; Gerdes, V.E.; Meijers, J.C.M.; Marx, P.F. The Hemostatic System in Patients with Type 2 Diabetes with and without Cardiovascular Disease. Clin. Appl. Thromb. 2010, 17, E57–E63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krekora, K.; Vitacolonna, E.; Di Castelnuovo, A.; D’Orazio, A.; de Lucia, D.; Dooijewaard, G.; Capani, F.; Donati, M.; Iacoviello, L. Decrease in urokinase-type plasminogen activator (u-PA) levels in patients with non-insulin dependent diabetes mellitus. Fibrinolysis Proteolysis 1997, 11, 215–219. [Google Scholar] [CrossRef]
- Hernández, C.; Chacón, P.; García-Pascual, L.; Mesa, J.; Simó, R. Relationship Between Lipoprotein(a) Phenotypes and Plaminogen Activator Inhibitor Type 1 in Diabetic Patients. Thromb. Res. 2000, 99, 119–127. [Google Scholar] [CrossRef]
- Johansson, L.; Jansson, J.-H.; Boman, K.; Nilsson, T.K.; Stegmayr, B.; Hallmans, G. Tissue Plasminogen Activator, Plasminogen Activator Inhibitor-1, and Tissue Plasminogen Activator/Plasminogen Activator Inhibitor-1 Complex as Risk Factors for the Development of a First Stroke. Stroke 2000, 31, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Spiess, B.D. Ischemia-A Coagulation Problem? J. Cardiovasc. Pharmacol. 1996, 27, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Hamsten, A.; Eriksson, P. Fibrinolysis and atherosclerosis: An update. Fibrinolysis 1994, 8, 253–262. [Google Scholar] [CrossRef]
- Brazionis, L.; Rowley, K.; Jenkins, A.; Itsiopoulos, C.; O’Dea, K. Plasminogen activator inhibitor-1 activity in type 2 diabetes: A different relationship with coronary heart disease and diabetic retinopathy. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 786–791. [Google Scholar] [CrossRef]
- Azad, N.; Agrawal, L.; Emanuele, N.V.; Klein, R.; Bahn, G.D.; McCarren, M.; Reaven, P.; Hayward, R.; Duckworth, W. Association of PAI-1 and Fibrinogen with Diabetic Retinopathy in the Veterans Affairs Diabetes Trial (VADT). Diabetes Care 2013, 37, 501–506. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.-L.; Chen, S. Plasma Plasminogen Activator Inhibitor-1 Is Associated with End-Stage Proliferative Diabetic Retinopathy in the Northern Chinese Han Population. Exp. Diabetes Res. 2012, 2012, 350852. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Menicucci, G.; Giebel, S.; Colombo, E.; McGuire, P. Plasminogen Activator Inhibitor-1 (PAI-1) in Early Diabetic Retinopathy and Retinal Neovascularization. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2367. [Google Scholar]
- Eddy, A.A.; Fogo, A.B. Plasminogen Activator Inhibitor-1 in Chronic Kidney Disease: Evidence and Mechanisms of Action. J. Am. Soc. Nephrol. 2006, 17, 2999–3012. [Google Scholar] [CrossRef] [Green Version]
- Eddy, A.A. Plasminogen activator inhibitor-1 and the kidney. Am. J. Physiol. Ren. Physiol. 2002, 283, F209–F220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, B.Y.; Uddin, J.; Park, J.H.; Lee, J.H.; Lee, H.B.; Miyata, T.; Ha, H. Novel Plasminogen Activator Inhibitor-1 Inhibitors Prevent Diabetic Kidney Injury in a Mouse Model. PLoS ONE 2016, 11, e0157012. [Google Scholar] [CrossRef]
- Nicholas, S.B.; Aguiniga, E.; Ren, Y.; Kim, J.; Wong, J.; Govindarajan, N.; Noda, M.; Wang, W.; Kawano, Y.; Collins, A.; et al. Plasminogen activator inhibitor-1 deficiency retards diabetic nephropathy. Kidney Int. 2005, 67, 1297–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGill, H.C., Jr.; McMahan, C.A.; Herderick, E.E.; Zieske, A.W.; Malcom, G.T.; Tracy, R.E.; Strong, J.P. Obesity accelerates the progression of coronary atherosclerosis in young men. Circulation 2002, 105, 2712–2718. [Google Scholar] [CrossRef]
- Killewich, L.A.; Macko, R.F.; Montgomery, P.S.; Wiley, L.A.; Gardner, A.W. Exercise training enhances endogenous fibrinolysis in peripheral arterial disease. J. Vasc. Surg. 2004, 40, 741–745. [Google Scholar] [CrossRef] [Green Version]
- Kokkinos, P.; Myers, J. Exercise and physical activity: Clinical outcomes and applications. Circulation 2010, 122, 1637–1648. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.J.; Agirbasli, M.A.; Williams, G.H.; Litchfield, W.R.; Vaughan, D.E. Effect of Activation and Inhibition of the Renin-Angiotensin System on Plasma PAI-1. Hypertension 1998, 32, 965–971. [Google Scholar] [CrossRef] [Green Version]
- Vaughan, D.E.; Rouleau, J.-L.; Ridker, P.M.; Arnold, J.M.O.; Menapace, F.J.; Pfeffer, M.A. Effects of Ramipril on Plasma Fibrinolytic Balance in Patients With Acute Anterior Myocardial Infarction. Circulation 1997, 96, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Cesari, M.; Kritchevsky, S.B.; Atkinson, H.H.; Penninx, B.W.; Di Bari, M.; Tracy, R.P.; Pahor, M. Angiotensin-converting enzyme inhibition and novel cardiovascular risk biomarkers: Results from the Trial of Angiotensin Converting Enzyme Inhibition and Novel Cardiovascular Risk Factors (TRAIN) study. Am. Heart J. 2009, 157, 334.e1–334.e8. [Google Scholar] [CrossRef] [Green Version]
- Skurk, T.; Lee, Y.-M.; Nicuta-Rolfs, T.-O.; Haastert, B.; Wirth, A.; Hauner, H. Effect of the angiotensin II receptor blocker candesartan on fibrinolysis in patients with mild hypertension. Diabetes Obes. Metab. 2004, 6, 56–62. [Google Scholar] [CrossRef]
- Zirlik, A.; Ernst, S.; Leugers, A.; Willecke, F.; Sobel, B.E.; Bode, C.; Nordt, T.K. Inhibition by fibrates of plasminogen activator inhibitor type-1 expression in human adipocytes and preadipocytes. Thromb. Haemost. 2009, 101, 1060–1069. [Google Scholar] [PubMed] [Green Version]
- Lindahl, T.L.; Sigurdardottir, O.; Wiman, B. Stability of plasminogen activator inhibitor 1 (PAI-1). Thromb. Haemost. 1989, 61, 748–751. [Google Scholar] [CrossRef]
- Gils, A.; Declerck, P.J. Proteinase Specificity and Functional Diversity in Point Mutants of Plasminogen Activator Inhibitor 1. J. Biol. Chem. 1997, 272, 12662–12666. [Google Scholar] [CrossRef] [Green Version]
- Baluta, M.M.; Vintila, M.M. PAI-1 inhibition—Another therapeutic option for cardiovascular protection. Maedica 2015, 10, 147. [Google Scholar]
- Oishi, K.; Koyanagi, S.; Matsunaga, N.; Kadota, K.; Ikeda, E.; Hayashida, S.; Kuramoto, Y.; Shimeno, H.; Soeda, S.; Ohdo, S. Bezafibrate Induces Plasminogen Activator Inhibitor-1 Gene Expression in a CLOCK-Dependent Circadian Manner. Mol. Pharmacol. 2010, 78, 135–141. [Google Scholar] [CrossRef] [Green Version]
- Erickson, L.A.; Ginsberg, M.H.; Loskutoff, D.J. Detection and partial characterization of an inhibitor of plasminogen activator in human platelets. J. Clin. Investig. 1984, 74, 1465–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruszynska, Y.T.; Yu, J.G.; Olefsky, J.M.; Sobel, B.E. Effects of troglitazone on blood concentrations of plasminogen activator inhibitor 1 in patients with type 2 diabetes and in lean and obese normal subjects. Diabetes 2000, 49, 633–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; E Dear, A.; Knudsen, L.B.; Simpson, R.W. A long-acting glucagon-like peptide-1 analogue attenuates induction of plasminogen activator inhibitor type-1 and vascular adhesion molecules. J. Endocrinol. 2009, 201, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, P.J. The effects of metformin on the fibrinolytic system in diabetic and non-diabetic subjects. Diabete Metab. 1991, 17, 168–173. [Google Scholar] [PubMed]
- Nagi, D.; Ali, V.M.; Yudkin, J. Effect of Metformin on Intact Proinsulin and des 31,32 Proinsulin Concentrations in Subjects with Non-insulin-dependent (Type 2) Diabetes Mellitus. Diabet. Med. 1996, 13, 753–757. [Google Scholar] [CrossRef]
- Fortenberry, Y.M. Plasminogen activator inhibitor-1 inhibitors: A patent review (2006—Present). Expert Opin. Ther. Pat. 2013, 23, 801–815. [Google Scholar] [CrossRef]
- Khan, S.S.; Shah, S.J.; Klyachko, E.; Baldridge, A.S.; Eren, M.; Place, A.T.; Aviv, A.; Puterman, E.; Lloyd-Jones, D.M.; Heiman, M.; et al. A null mutation inSERPINE1protects against biological aging in humans. Sci. Adv. 2017, 3, eaao1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughan, D.E. PAI-1 antagonists: The promise and the peril. Trans. Am. Clin. Climatol. Assoc. 2011, 122, 312. [Google Scholar]
- Vinogradsky, B.; Bell, S.P.; Woodcock-Mitchell, J.; Ohtani, A.; Fujii, S. A new butadiene derivative, T-686, inhibits plasminogen activator inhibitor type-1 production in vitro by cultured human vascular endothelial cells and development of atherosclerotic lesions in vivo in rabbits. Thromb. Res. 1997, 85, 305–314. [Google Scholar] [CrossRef]
- Pawlowska, Z.; Chabielska, E.; Kobylańska, A.; Maciaszek, A.; Swiatkowska, M.; Buczko, W.; Stec, W.; Cierniewski, C. Regulation of PAI-1 Concentration in Platelets by Systemic Administration of Antisense Oligonucleotides to Rats. Thromb. Haemost. 2001, 85, 1086–1089. [Google Scholar] [CrossRef]
- Izuhara, Y.; Takahashi, S.; Nangaku, M.; Takizawa, S.; Ishida, H.; Kurokawa, K.; van Ypersele de Strihou, C.; Hirayama, N.; Miyata, T. Inhibition of plasminogen activator inhibitor-1: Its mechanism and effectiveness on coagulation and fibrosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 672–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bijnens, A.P.; Gils, A.; Stassen, J.M.; Komissarov, A.A.; Knockaert, I.; Brouwers, E.; Shore, J.D.; Declerck, P.J. The Distal Hinge of the Reactive Site Loop and Its Proximity a Target to Modulate Plasminogen Activator Inhibitor-1 Activity. J. Biol. Chem. 2001, 276, 44912–44918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levi, M.; Biemond, B.J.; van Zonneveld, A.J.; Cate, J.W.T.; Pannekoek, H. Inhibition of plasminogen activator inhibitor-1 activity results in promotion of endogenous thrombolysis and inhibition of thrombus extension in models of experimental thrombosis. Circulation 1992, 85, 305–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bijnens, A.-P.; Gils, A.; Knockaert, I.; Stassen, J.M.; Declerck, P.J. Importance of the Hinge Region between α-Helix F and the Main Part of Serpins, Based upon Identification of the Epitope of Plasminogen Activator Inhibitor Type 1 Neutralizing Antibodies. J. Biol. Chem. 2000, 275, 6375–6380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wind, T.; Jensen, M.A.; Andreasen, P.A. Epitope mapping for four monoclonal antibodies against human plasminogen activator inhibitor type-1: Implications for antibody-mediated PAI-1-neutralization and vitronectin-binding. Eur. J. Biochem. 2001, 268, 1095–1106. [Google Scholar] [CrossRef] [PubMed]
- Stoop, A.; Jespers, L.; Lasters, I.; Eldering, E.; Pannekoek, H. High-density mutagenesis by combined DNA shuffling and phage display to assign essential amino acid residues in protein-protein interactions: Application to study structure-function of plasminogen activation inhibitor 1 (PAI-I). J. Mol. Biol. 2000, 301, 1135–1147. [Google Scholar] [CrossRef] [PubMed]
- van de Craen, B.; Scroyen, I.; Abdelnabi, R.; Brouwers, E.; Lijnen, H.R.; Declerck, P.J.; Gils, A. Characterization of a panel of monoclonal antibodies toward mouse PAI-1 that exert a significant profibrinolytic effect in vivo. Thromb. Res. 2011, 128, 68–76. [Google Scholar] [CrossRef]
- Biemond, B.J.; Levi, M.; Coronel, R.; Janse, M.J.; Ten Cate, J.W.; Pannekoek, H. Thrombolysis and reocclusion in experimental jugular vein and coronary artery thrombosis: Effects of a plasminogen activator inhibitor type 1-neutralizing monoclonal antibody. Circulation 1995, 91, 1175–1181. [Google Scholar] [CrossRef] [PubMed]
- Naessens, D.; Gils, A.; Compernolle, G.; Declerck, P. Elucidation of the epitope of a latency-inducing antibody: Identification of a new molecular target for PAI-1 inhibition. Thromb. Haemost. 2003, 90, 52–58. [Google Scholar] [CrossRef]
- Gorlatova, N.V.; Elokdah, H.; Fan, K.; Crandall, D.L.; Lawrence, D.A. Mapping of a conformational epitope on plasminogen activator inhibitor-1 by random mutagenesis: Implications for serpin function. J. Biol. Chem. 2003, 278, 16329–16335. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, D.A.; Olson, S.T.; Muhammad, S.; Day, D.E.; Kvassman, J.-O.; Ginsburg, D.; Shore, J.D. Partitioning of Serpin-Proteinase Reactions between Stable Inhibition and Substrate Cleavage Is Regulated by the Rate of Serpin Reactive Center Loop Insertion into β-Sheet A. J. Biol. Chem. 2000, 275, 5839–5844. [Google Scholar] [CrossRef] [Green Version]
- Kvassman, J.-O.; Lawrence, D.A.; Shore, J.D. The Acid Stabilization of Plasminogen Activator Inhibitor-1 Depends on Protonation of a Single Group That Affects Loop Insertion into β-Sheet A. J. Biol. Chem. 1995, 270, 27942–27947. [Google Scholar] [CrossRef] [Green Version]
- Ngo, T.-H.; Zhou, Y.; Stassen, J.-M.; Declerck, P. Importance of N-Terminal Residues in Plasminogen Activator Inhibitor 1 on its Antibody Induced Latency Transition. Thromb. Haemost. 2002, 88, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Dupont, D.M.; Blouse, G.E.; Hansen, M.; Mathiasen, L.; Kjelgaard, S.; Jensen, J.K.; Christensen, A.; Gils, A.; Declerck, P.J.; Andreasen, P.A.; et al. Evidence for a Pre-latent Form of the Serpin Plasminogen Activator Inhibitor-1 with a Detached β-Strand 1C. J. Biol. Chem. 2006, 281, 36071–36081. [Google Scholar] [CrossRef] [Green Version]
- Folkes, A.; Roe, M.B.; Sohal, S.; Golec, J.; Faint, R.; Brooks, T.; Charlton, P. Synthesis and in vitro evaluation of a series of diketopiperazine inhibitors of plasminogen activator inhibitor-1. Bioorg. Med. Chem. Lett. 2001, 11, 2589–2592. [Google Scholar] [CrossRef]
- Bryans, J.; Charlton, P.; Chicarelli-Robinson, I.; Collins, M.; Faint, R.; Latham, C.; Shaw, I.; Trew, S. Inhibition of Plasminogen Activator Inhibitor-1 Activity by Two Diketopiperazines, XR330 and XR334 Produced by Streptomyces sp. J. Antibiot. 1996, 49, 1014–1021. [Google Scholar] [CrossRef]
- Charlton, P.A.; Faint, R.W.; Bent, F.; Bryans, J.; Chicarelli-Robinson, I.; Mackie, I.; Machin, S.; Bevan, P. Evaluation of a low molecular weight modulator of human plasminogen activator inhibitor-1 activity. Thromb. Haemost. 1996, 76, 808–815. [Google Scholar] [CrossRef]
- Friederich, P.W.; Levi, M.; Biemond, B.J.; Charlton, P.; Templeton, D.; van Zonneveld, A.J.; Bevan, P.; Pannekoek, H.; Cate, J.W.T. Novel low-molecular-weight inhibitor of PAI-1 (XR5118) promotes endogenous fibrinolysis and reduces postthrombolysis thrombus growth in rabbits. Circulation 1997, 96, 916–921. [Google Scholar]
- Björquist, P.; Ehnebom, J.; Inghardt, T.; Hansson, L.; Lindberg, M.; Linschoten, M.; Strömqvist, M.; Deinum, J. Identification of the Binding Site for a Low-Molecular-Weight Inhibitor of Plasminogen Activator Inhibitor Type 1 by Site-Directed Mutagenesis. Biochemistry 1998, 37, 1227–1234. [Google Scholar] [CrossRef]
- Gils, A.; Stassen, J.-M.; Nar, H.; Kley, J.; Wienen, W.; Ries, U.; Declerck, P. Characterization and Comparative Evaluation of a Novel PAI-1 Inhibitor. Thromb. Haemost. 2002, 88, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Elokdah, H.; Abou-Gharbia, M.; Hennan, J.K.; McFarlane, G.; Mugford, C.P.; Krishnamurthy, G.; Crandall, D.L. Tiplaxtinin, a Novel, Orally Efficacious Inhibitor of Plasminogen Activator Inhibitor-1: Design, Synthesis, and Preclinical Characterization. J. Med. Chem. 2004, 47, 3491–3494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; Eren, M.; Vaughan, U.E.; Schleimer, R.P.; Cho, S.H. A Plasminogen Activator Inhibitor-1 Inhibitor Reduces Airway Remodeling in a Murine Model of Chronic Asthma. Am. J. Respir. Cell Mol. Biol. 2012, 46, 842–846. [Google Scholar] [CrossRef] [PubMed]
- Rupin, A.; Gaertner, R.; Mennecier, P.; Richard, I.; Benoist, A.; de Nanteuil, G.; Verbeuren, T.J. S35225 is a direct inhibitor of Plasminogen Activator Inhibitor type-1 activity in the blood. Thromb. Res. 2008, 122, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Cale, J.M.; Li, S.-H.; Warnock, M.; Su, E.J.; North, P.R.; Sanders, K.L.; Puscau, M.M.; Emal, C.D.; Lawrence, D.A. Characterization of a Novel Class of Polyphenolic Inhibitors of Plasminogen Activator Inhibitor-1. J. Biol. Chem. 2010, 285, 7892–7902. [Google Scholar] [CrossRef] [Green Version]
- Vousden, K.A.; Lundqvist, T.; Popovic, B.; Naiman, B.; Carruthers, A.M.; Newton, P.; Johnson, D.J.D.; Pomowski, A.; Wilkinson, T.; Dufner, P.; et al. Discovery and characterisation of an antibody that selectively modulates the inhibitory activity of plasminogen activator inhibitor-1. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Country | No. of T2D/Controls | Mean ± SD Median (IQR) of T2D | Mean ± SD Median (IQR) of Control | p-Value |
|---|---|---|---|---|---|
| Kitagawa (2006) [49] | Japan | 47/31 | 82.7 ± 54.5 ng/mL | 52.9 ± 51.7 ng/mL | <0.05 |
| Soares (2010) [50] | Brazil | 25/12 | 108.8 ± 48.5 ng/mL | 37.6 ± 33.2 ng/mL | <0.05 |
| Le (2008) [51] | U.S.A. | 104/59 | 39(24–59) ng/mL | 31(18–53) ng/mL | NS |
| Romuk (2008) [52] | Poland | 20/21 | 10.57 ± 5.8 IU/mL | 3.90 ± 1.7 IU/mL | <0.0001 |
| Sahli (2009) [53] | Sweden | 69/80 | 39.7 ± 35.3 IU/mL | 10.5 ± 12.3 IU/mL | <0.0001 |
| Erem (2005) [54] | Turkey | 92/40 | 44.6 ± 10.6 ng/mL | 21.4 ± 5.8 ng/mL | <0.0001 |
| Verkleij (2011) [55] | Netherlands | 207/100 | 98 ± 102 ng/mL | 57 ± 38 ng/mL | 0.038 |
| Krekora (1997) [56] | Italy | 59/50 | 107 ± 28 ng/mL | 29.1 ± 17 ng/mL | <0.001 |
| Hernandez (2000) [57] | Spain | 41/40 | 51.3 ± 29 ng/mL | 23 ± 16.2ng/mL | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altalhi, R.; Pechlivani, N.; Ajjan, R.A. PAI-1 in Diabetes: Pathophysiology and Role as a Therapeutic Target. Int. J. Mol. Sci. 2021, 22, 3170. https://doi.org/10.3390/ijms22063170
Altalhi R, Pechlivani N, Ajjan RA. PAI-1 in Diabetes: Pathophysiology and Role as a Therapeutic Target. International Journal of Molecular Sciences. 2021; 22(6):3170. https://doi.org/10.3390/ijms22063170
Chicago/Turabian StyleAltalhi, Rawan, Nikoletta Pechlivani, and Ramzi A. Ajjan. 2021. "PAI-1 in Diabetes: Pathophysiology and Role as a Therapeutic Target" International Journal of Molecular Sciences 22, no. 6: 3170. https://doi.org/10.3390/ijms22063170
APA StyleAltalhi, R., Pechlivani, N., & Ajjan, R. A. (2021). PAI-1 in Diabetes: Pathophysiology and Role as a Therapeutic Target. International Journal of Molecular Sciences, 22(6), 3170. https://doi.org/10.3390/ijms22063170