
Tlr2 Gene Deletion Delays Retinal Degeneration in Two Genetically Distinct Mouse Models of Retinitis Pigmentosa


Abstract
:1. Introduction
2. Results
2.1. Tlr2 and TLR-Adaptor Gene Expression during RP-Associated Retinal Degeneration
2.2. Effect of Tlr2 Deletion on Vision Loss
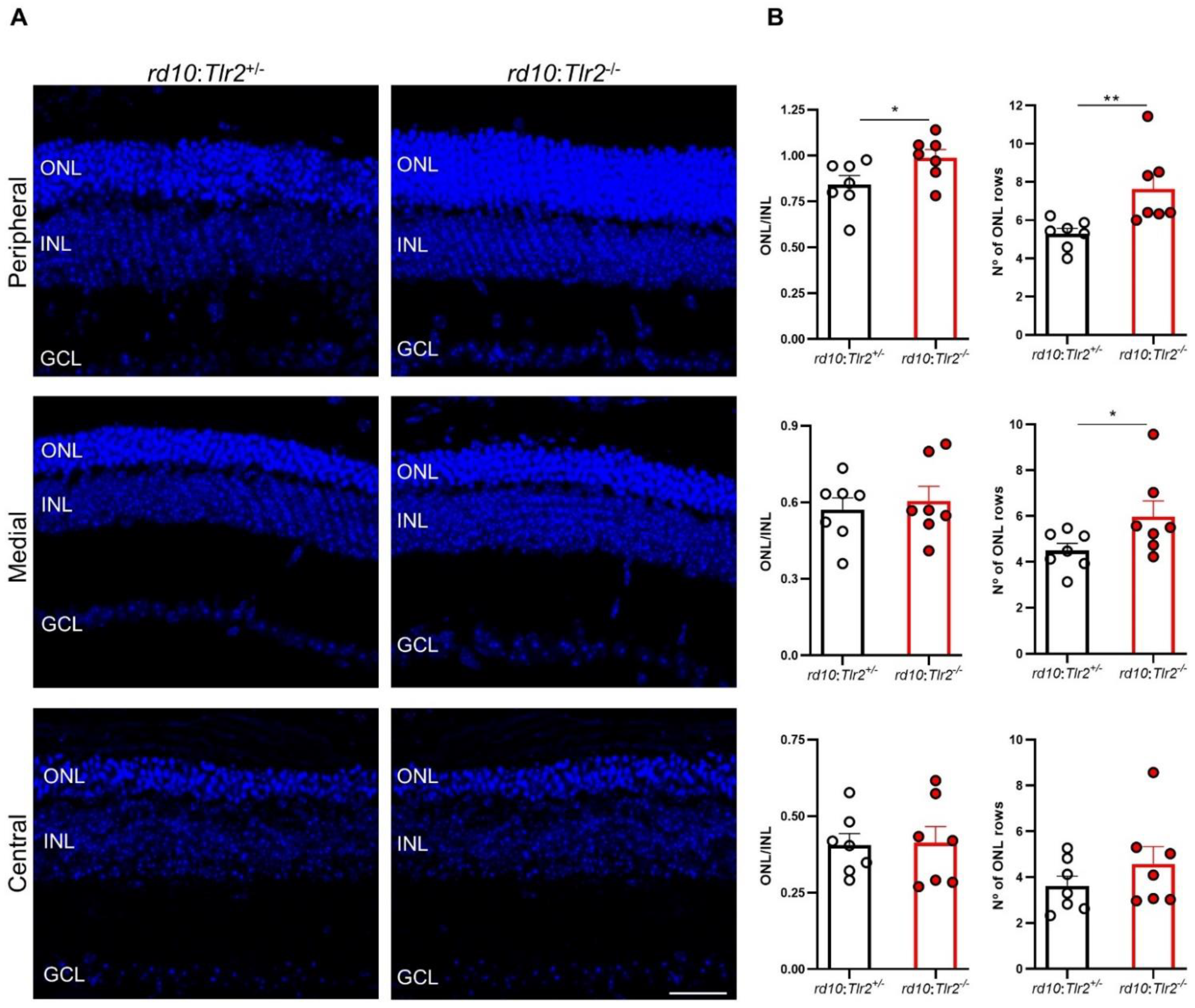
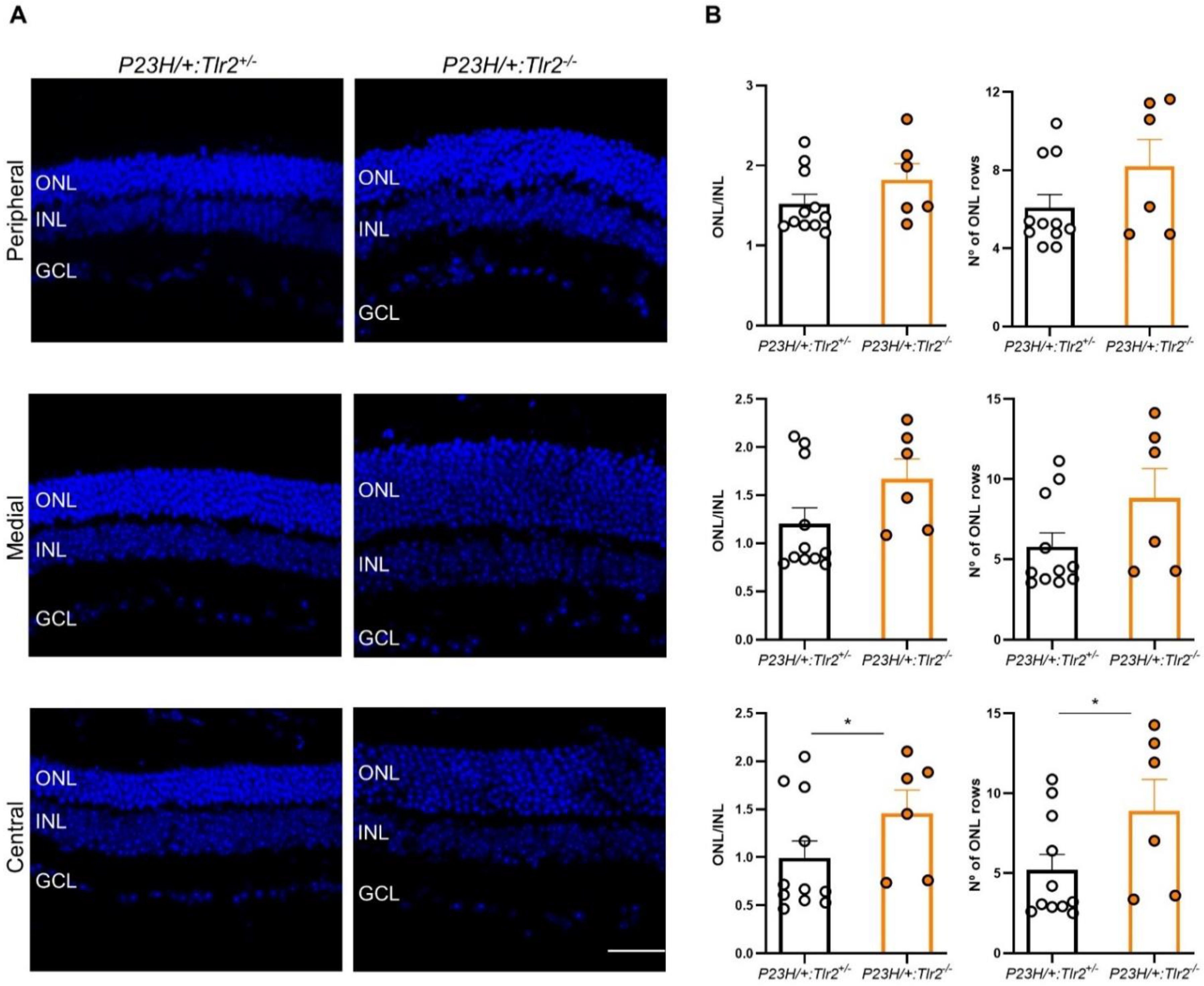
2.3. Effect of Tlr2 Deletion on Photoreceptor Preservation
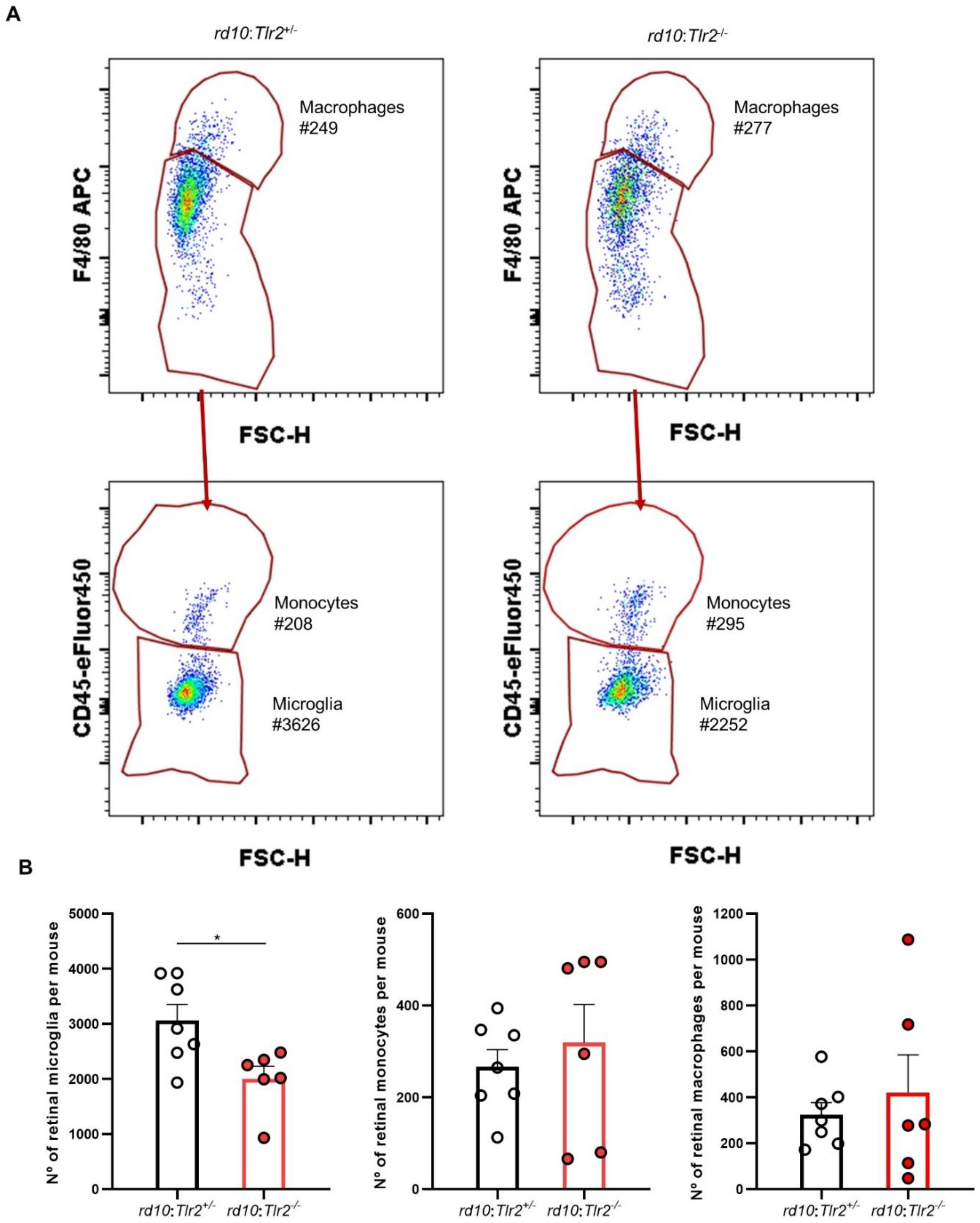
2.4. Effect of Tlr2 Deletion on the Myeloid Cell Population
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. RNA Isolation and Quantitative PCR
4.3. Histological and Myeloid Cell Analysis of Retinal Sections
4.4. Microglia Quantification in Wholemount Retinas
4.5. Flow Cytometry
4.6. Electroretinography (ERG) Recordings
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Verbakel, S.K.; van Huet, R.A.; Boon, C.; Hollander, A.I.D.; Collin, R.W.; Klaver, C.C.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Silverman, S.M.; Wong, W.T. Microglia in the Retina: Roles in Development, Maturity, and Disease. Annu. Rev. Vis. Sci. 2018, 4, 45–77. [Google Scholar] [CrossRef]
- Murakami, Y.; Ishikawa, K.; Nakao, S.; Sonoda, K.-H. Innate immune response in retinal homeostasis and inflammatory disorders. Prog. Retin. Eye Res. 2020, 74, 100778. [Google Scholar] [CrossRef]
- Wubben, T.; Zacks, D.; Besirli, C. Retinal neuroprotection: Current strategies and future directions. Curr. Opin. Ophthalmol. 2019, 30, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Marigo, V.; Kutluer, M.; Huang, L. Targeting molecular pathways for the treatment of inherited retinal degeneration. Neural Regen. Res. 2020, 15, 1784–1791. [Google Scholar] [CrossRef]
- Franz, K.M.; Kagan, J.C. Innate Immune Receptors as Competitive Determinants of Cell Fate. Mol. Cell 2017, 66, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Hanke, M.L.; Kielian, T. Toll-like receptors in health and disease in the brain: Mechanisms and therapeutic potential. Clin. Sci. 2011, 121, 367–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heneka, M.T.; Kummer, M.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Bsibsi, M.; Ravid, R.; Gveric, D.; van Noort, J. Broad Expression of Toll-Like Receptors in the Human Central Nervous System. J. Neuropathol. Exp. Neurol. 2002, 61, 1013–1021. [Google Scholar] [CrossRef] [Green Version]
- Olson, J.K.; Miller, S.D. Microglia Initiate Central Nervous System Innate and Adaptive Immune Responses through Multiple TLRs. J. Immunol. 2004, 173, 3916–3924. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, I.; Said, D.G.; Dua, H.S. Human antimicrobial peptides in ocular surface defense. Prog. Retin. Eye Res. 2017, 61, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.; Hawes, N.; Hurd, R.; Davisson, M.; Nusinowitz, S.; Heckenlively, J. Retinal degeneration mutants in the mouse. Vis. Res. 2002, 42, 517–525. [Google Scholar] [CrossRef] [Green Version]
- Sakami, S.; Maeda, T.; Bereta, G.; Okano, K.; Golczak, M.; Sumaroka, A.; Roman, A.J.; Cideciyan, A.V.; Jacobson, S.G.; Palczewski, K. Probing Mechanisms of Photoreceptor Degeneration in a New Mouse Model of the Common Form of Autosomal Dominant Retinitis Pigmentosa due to P23H Opsin Mutations. J. Biol. Chem. 2011, 286, 10551–10567. [Google Scholar] [CrossRef] [Green Version]
- Pang, J.-J.; Boye, S.L.; Kumar, A.; Dinculescu, A.; Deng, W.; Li, J.; Li, Q.; Rani, A.; Foster, T.C.; Chang, B.; et al. AAV-Mediated Gene Therapy for Retinal Degeneration in the rd10 Mouse Containing a Recessive PDEβ Mutation. Investig. Opthalmol. Vis. Sci. 2008, 49, 4278–4283. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Cruz, A.; Villarejo-Zori, B.; Marchena, M.; Zaldivar-Diez, J.; Palomo, V.; Gil, C.; Lizasoain, I.; De La Villa, P.; Martínez, A.; De La Rosa, E.J.; et al. Modulation of GSK-3 provides cellular and functional neuroprotection in the rd10 mouse model of retinitis pigmentosa. Mol. Neurodegener. 2018, 13, 19. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, O.; Hoshino, K.; Kawai, T.; Sanjo, H.; Takada, H.; Ogawa, T.; Takeda, K.; Akira, S. Differential Roles of TLR2 and TLR4 in Recognition of Gram-Negative and Gram-Positive Bacterial Cell Wall Components. Immunity 1999, 11, 443–451. [Google Scholar] [CrossRef] [Green Version]
- Siegert, S.; Scherf, B.G.; Del Punta, K.; Didkovsky, N.; Heintz, N.; Roska, B. Genetic address book for retinal cell types. Nat. Neurosci. 2009, 12, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Hooper, M.J.; Wang, J.; Browning, R.; Ash, J.D. Damage-associated molecular pattern recognition is required for induction of retinal neuroprotective pathways in a sex-dependent manner. Sci. Rep. 2018, 8, 9115. [Google Scholar] [CrossRef] [PubMed]
- Silverman, S.M.; Ma, W.; Wang, X.; Zhao, L.; Wong, W.T. C3- and CR3-dependent microglial clearance protects photoreceptors in retinitis pigmentosa. J. Exp. Med. 2019, 216, 1925–1943. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Roubeix, C.; Sennlaub, F.; Saban, D.R. Microglia versus Monocytes: Distinct Roles in Degenerative Diseases of the Retina. Trends Neurosci. 2020, 43, 433–449. [Google Scholar] [CrossRef] [PubMed]
- Platón-Corchado, M.; Barcelona, P.; Jmaeff, S.; Marchena, M.; Hernández-Pinto, A.M.; Sánchez, C.H.; Saragovi, H.U.; De La Rosa, E.J. p75NTR antagonists attenuate photoreceptor cell loss in murine models of retinitis pigmentosa. Cell Death Dis. 2017, 8, e2922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Koren, E.G.; Mathew, R.; Saban, D.R. Fate mapping reveals that microglia and recruited monocyte-derived macrophages are definitively distinguishable by phenotype in the retina. Sci. Rep. 2016, 6, 20636. [Google Scholar] [CrossRef] [PubMed]
- Zeiss, C.J.; Johnson, E.A. Proliferation of Microglia, but not Photoreceptors, in the Outer Nuclear Layer of the rd-1 Mouse. Investig. Opthalmol. Vis. Sci. 2004, 45, 971–976. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Zabel, M.; Wang, X.; Ma, W.; Shah, P.; Fariss, R.; Qian, H.; Parkhurst, C.N.; Gan, W.; Wong, W.T. Microglial phagocytosis of living photoreceptors contributes to inherited retinal degeneration. EMBO Mol. Med. 2015, 7, 1179–1197. [Google Scholar] [CrossRef]
- Akhtar-Schäfer, I.; Wang, L.; Krohne, T.U.; Xu, H.; Langmann, T. Modulation of three key innate immune pathways for the most common retinal degenerative diseases. EMBO Mol. Med. 2018, 10, e8259. [Google Scholar] [CrossRef] [PubMed]
- Sudharsan, R.; Beiting, D.P.; Aguirre, G.D.; Beltran, W.A. Involvement of Innate Immune System in Late Stages of Inherited Photoreceptor Degeneration. Sci. Rep. 2017, 7, 17897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-J.; Wang, P.-W.; Yang, I.-H.; Huang, H.-M.; Chang, C.-S.; Wu, C.-L.; Chuang, J.-H. High-Fat Diet Induces Toll-Like Receptor 4-Dependent Macrophage/Microglial Cell Activation and Retinal Impairment. Investig. Opthalmol. Vis. Sci. 2015, 56, 3041–3050. [Google Scholar] [CrossRef] [Green Version]
- Kohno, H.; Chen, Y.; Kevany, B.M.; Pearlman, E.; Miyagi, M.; Maeda, T.; Palczewski, K.; Maeda, A. Photoreceptor Proteins Initiate Microglial Activation via Toll-like Receptor 4 in Retinal Degeneration Mediated by All-trans-retinal. J. Biol. Chem. 2013, 288, 15326–15341. [Google Scholar] [CrossRef] [Green Version]
- Gargini, C.; Terzibasi, E.; Mazzoni, F.; Strettoi, E. Retinal organization in the retinal degeneration 10 (rd10) mutant mouse: A morphological and ERG study. J. Comp. Neurol. 2006, 500, 222–238. [Google Scholar] [CrossRef] [Green Version]
- Barhoum, R.; Navarrete, G.M.; Corrochano, S.; Germain, F.; Fernandez-Sanchez, L.; de la Rosa, E.; de la Villa, P.; Cuenca, N. Functional and structural modifications during retinal degeneration in the rd10 mouse. Neuroscience 2008, 155, 698–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulfaul, K.; Ozaki, E.; Fernando, N.; Brennan, K.; Chirco, K.R.; Connolly, E.; Greene, C.; Maminishkis, A.; Salomon, R.G.; Linetsky, M.; et al. Toll-like Receptor 2 Facilitates Oxidative Damage-Induced Retinal Degeneration. Cell Rep. 2020, 30, 2209–2224.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syeda, S.; Patel, A.K.; Lee, T.; Hackam, A.S. Reduced photoreceptor death and improved retinal function during retinal degeneration in mice lacking innate immunity adaptor protein MyD88. Exp. Neurol. 2015, 267, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garces, K.; Carmy, T.; Illiano, P.; Brambilla, R.; Hackam, A.S. Increased Neuroprotective Microglia and Photoreceptor Survival in the Retina from a Peptide Inhibitor of Myeloid Differentiation Factor 88 (MyD88). J. Mol. Neurosci. 2020, 70, 968–980. [Google Scholar] [CrossRef]
- McDonald, C.; Hennessy, E.; Rubio-Araiz, A.; Keogh, B.; McCormack, W.; McGuirk, P.; Reilly, M.; Lynch, M.A. Inhibiting TLR2 activation attenuates amyloid accumulation and glial activation in a mouse model of Alzheimer’s disease. Brain Behav. Immun. 2016, 58, 191–200. [Google Scholar] [CrossRef]
- Kim, C.; Spencer, B.; Rockenstein, E.; Yamakado, H.; Mante, M.; Adame, A.; Fields, J.A.; Masliah, D.; Iba, M.; Lee, H.-J.; et al. Immunotherapy targeting toll-like receptor 2 alleviates neurodegeneration in models of synucleinopathy by modulating α-synuclein transmission and neuroinflammation. Mol. Neurodegener. 2018, 13. [Google Scholar] [CrossRef]
- Karlen, S.J.; Miller, E.B.; Burns, M.E. Microglia Activation and Inflammation During the Death of Mammalian Photoreceptors. Annu. Rev. Vis. Sci. 2020, 6, 149–169. [Google Scholar] [CrossRef]
- Babcock, A.A.; Wirenfeldt, M.; Holm, T.; Nielsen, H.H.; Dissing-Olesen, L.; Toft-Hansen, H.; Millward, J.M.; Landmann, R.; Rivest, S.; Finsen, B.; et al. Toll-Like Receptor 2 Signaling in Response to Brain Injury: An Innate Bridge to Neuroinflammation. J. Neurosci. 2006, 26, 12826–12837. [Google Scholar] [CrossRef] [Green Version]
- Bohacek, I.; Cordeau, P.; Lalancette–Hébert, M.; Gorup, D.; Weng, Y.-C.; Gajovic, S.; Kriz, J. Toll-like receptor 2 deficiency leads to delayed exacerbation of ischemic injury. J. Neuroinflamm. 2012, 9, 191. [Google Scholar] [CrossRef] [Green Version]
- Zabel, M.; Zhao, L.; Zhang, Y.; Gonzalez, S.R.; Ma, W.; Wang, X.; Fariss, R.; Wong, W.T. Microglial phagocytosis and activation underlying photoreceptor degeneration is regulated by CX3CL1-CX3CR1 signaling in a mouse model of retinitis pigmentosa. Glia 2016, 64, 1479–1491. [Google Scholar] [CrossRef]
- Guo, C.; Otani, A.; Oishi, A.; Kojima, H.; Makiyama, Y.; Nakagawa, S.; Yoshimura, N. Knockout of Ccr2 alleviates photoreceptor cell death in a model of retinitis pigmentosa. Exp. Eye Res. 2012, 104, 39–47. [Google Scholar] [CrossRef] [Green Version]
- Kohno, H.; Maeda, T.; Perusek, L.; Pearlman, E.; Maeda, A. CCL3 Production by Microglial Cells Modulates Disease Severity in Murine Models of Retinal Degeneration. J. Immunol. 2014, 192, 3816–3827. [Google Scholar] [CrossRef] [PubMed]
- Arroba, A.I.; Alvarez-Lindo, N.L.; Van Rooijen, N.; De La Rosa, E.J. Microglia-Mediated IGF-I Neuroprotection in the rd10 Mouse Model of Retinitis Pigmentosa. Investig. Opthalmol. Vis. Sci. 2011, 52, 9124–9130. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Svetkauskaite, D.; He, Q.; Kim, J.-Y.; Strassheim, D.; Ishizaka, A.; Abraham, E. Involvement of Toll-like Receptors 2 and 4 in Cellular Activation by High Mobility Group Box 1 Protein. J. Biol. Chem. 2004, 279, 7370–7377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, N.; Dewan, V.; Grace, P.; Gunn, R.J.; Tamura, R.; Tzarum, N.; Watkins, L.R.; Wilson, I.A.; Yin, H. HMGB1 Activates Proinflammatory Signaling via TLR5 Leading to Allodynia. Cell Rep. 2016, 17, 1128–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinle, J.J. Role of HMGB1 signaling in the inflammatory process in diabetic retinopathy. Cell. Signal. 2020, 73, 109687. [Google Scholar] [CrossRef]
- Murakami, Y.; Ikeda, Y.; Nakatake, S.; Tachibana, T.; Fujiwara, K.; Yoshida, N.; Notomi, S.; Nakao, S.; Hisatomi, T.; Miller, J.W.; et al. Necrotic enlargement of cone photoreceptor cells and the release of high-mobility group box-1 in retinitis pigmentosa. Cell Death Discov. 2015, 1, 15058. [Google Scholar] [CrossRef] [Green Version]
- Corrochano, S.; Barhoum, R.; Boya, P.; Arroba, A.I.; Rodríguez-Muela, N.; Gómez-Vicente, V.; Bosch, F.; De Pablo, F.; De La Villa, P.; De La Rosa, E.J. Attenuation of Vision Loss and Delay in Apoptosis of Photoreceptors Induced by Proinsulin in a Mouse Model of Retinitis Pigmentosa. Investig. Opthalmol. Vis. Sci. 2008, 49, 4188–4194. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Sánchez, L.; Lax, P.; Isiegas, C.; Ayuso, E.; Ruiz, J.M.; De La Villa, P.; Bosch, F.; De La Rosa, E.J.; Cuenca, N. Proinsulin Slows Retinal Degeneration and Vision Loss in the P23H Rat Model of Retinitis Pigmentosa. Hum. Gene Ther. 2012, 23, 1290–1300. [Google Scholar] [CrossRef] [Green Version]
- Isiegas, C.; Marinich-Madzarevich, J.A.; Marchena, M.; Ruiz, J.M.; Cano, M.J.; De La Villa, P.; Hernández-Sánchez, C.; De La Rosa, E.J.; De Pablo, F. Intravitreal Injection of Proinsulin-Loaded Microspheres Delays Photoreceptor Cell Death and Vision Loss in the rd10 Mouse Model of Retinitis Pigmentosa. Investig. Opthalmol. Vis. Sci. 2016, 57, 3610. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Cruz, A.; Hernández-Pinto, A.; Lillo, C.; Isiegas, C.; Marchena, M.; Lizasoain, I.; Bosch, F.; de la Villa, P.; Hernández-Sánchez, C.; de la Rosa, E.J. Insulin receptor activation by proinsulin preserves synapses and vision in retinitis pigmento-sa. bioRxiv 2020. [Google Scholar] [CrossRef]
- Jung, S.; Aliberti, J.; Graemmel, P.; Sunshine, M.J.; Kreutzberg, G.W.; Sher, A.; Littman, D.R. Analysis of Fractalkine Receptor CX 3 CR1 Function by Targeted Deletion and Green Fluorescent Protein Reporter Gene Insertion. Mol. Cell. Biol. 2000, 20, 4106–4114. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Probe |
|---|---|
| Cd68 | Mm03047343_m1 |
| Cx3cr1 | Mm02620111_s1 |
| Iba1 | Mm00479862_g1 |
| P2ry12 | Mm01950543_s1 |
| Tbp | Mm01277042_m1 |
| Tmem119 | Mm00525305_m1 |
| Trem2 | Mm04209424_g1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Cruz, A.; Méndez, A.C.; Lizasoain, I.; de la Villa, P.; de la Rosa, E.J.; Hernández-Sánchez, C. Tlr2 Gene Deletion Delays Retinal Degeneration in Two Genetically Distinct Mouse Models of Retinitis Pigmentosa. Int. J. Mol. Sci. 2021, 22, 7815. https://doi.org/10.3390/ijms22157815
Sánchez-Cruz A, Méndez AC, Lizasoain I, de la Villa P, de la Rosa EJ, Hernández-Sánchez C. Tlr2 Gene Deletion Delays Retinal Degeneration in Two Genetically Distinct Mouse Models of Retinitis Pigmentosa. International Journal of Molecular Sciences. 2021; 22(15):7815. https://doi.org/10.3390/ijms22157815
Chicago/Turabian StyleSánchez-Cruz, Alonso, Andrea C. Méndez, Ignacio Lizasoain, Pedro de la Villa, Enrique J. de la Rosa, and Catalina Hernández-Sánchez. 2021. "Tlr2 Gene Deletion Delays Retinal Degeneration in Two Genetically Distinct Mouse Models of Retinitis Pigmentosa" International Journal of Molecular Sciences 22, no. 15: 7815. https://doi.org/10.3390/ijms22157815
APA StyleSánchez-Cruz, A., Méndez, A. C., Lizasoain, I., de la Villa, P., de la Rosa, E. J., & Hernández-Sánchez, C. (2021). Tlr2 Gene Deletion Delays Retinal Degeneration in Two Genetically Distinct Mouse Models of Retinitis Pigmentosa. International Journal of Molecular Sciences, 22(15), 7815. https://doi.org/10.3390/ijms22157815