
The Slow-Releasing and Mitochondria-Targeted Hydrogen Sulfide (H2S) Delivery Molecule AP39 Induces Brain Tolerance to Ischemia

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
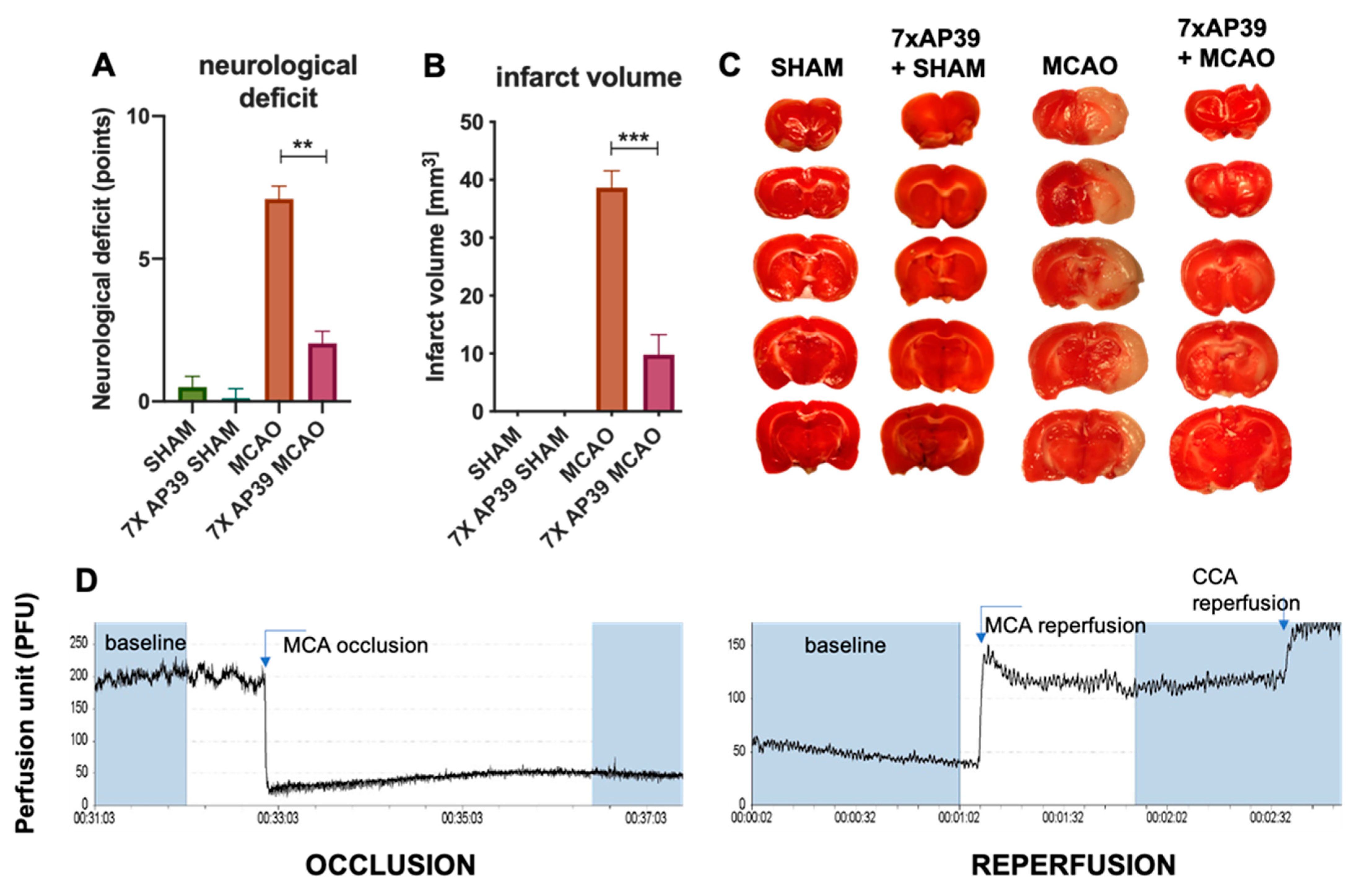
2.1. AP39 Treatment Reduced the Neurological Deficit and Infarct Volume
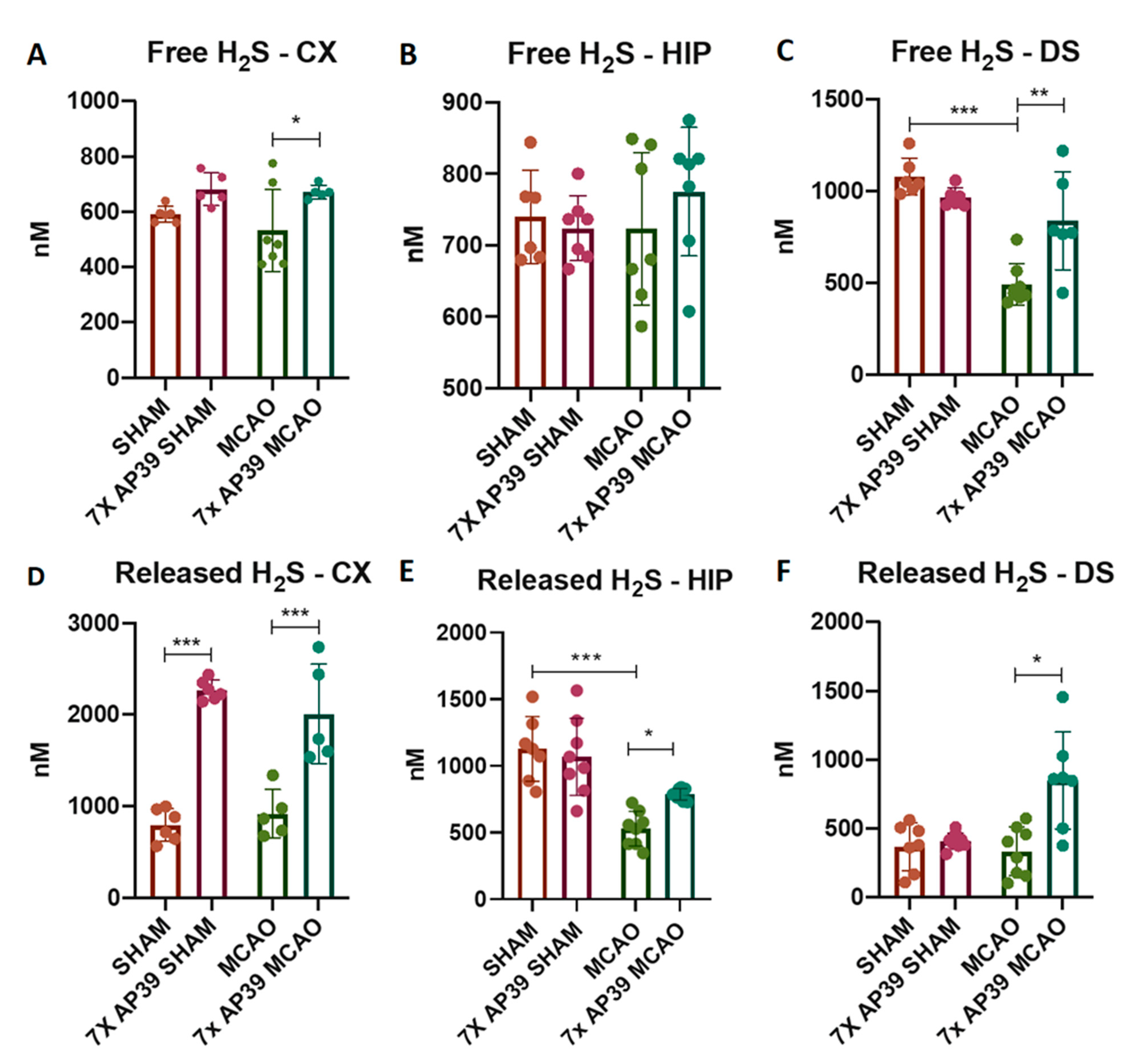
2.2. AP39 Prevented the MCAO-Induced Depletion of Free and DTT-Releasable H2S Levels
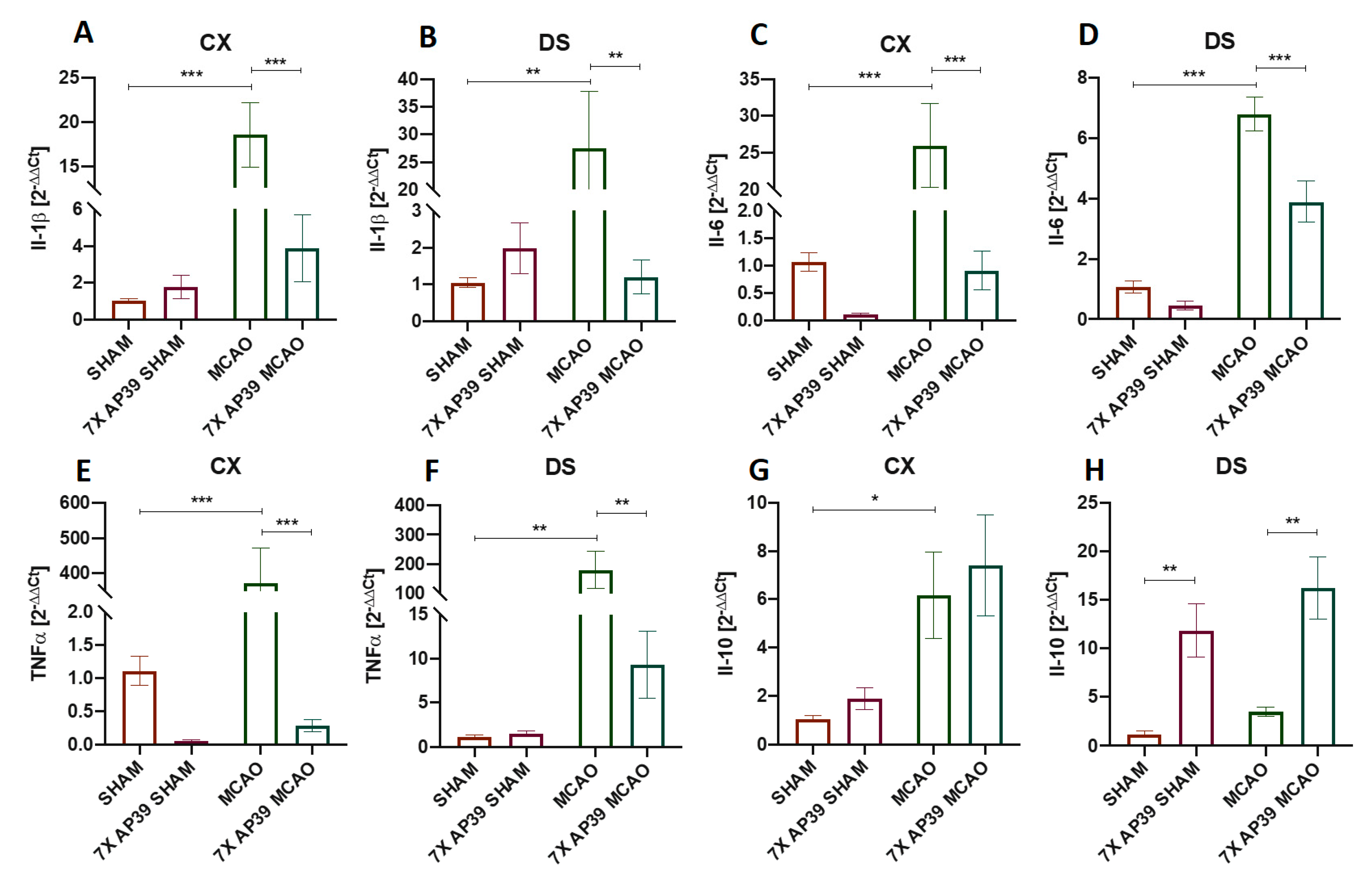
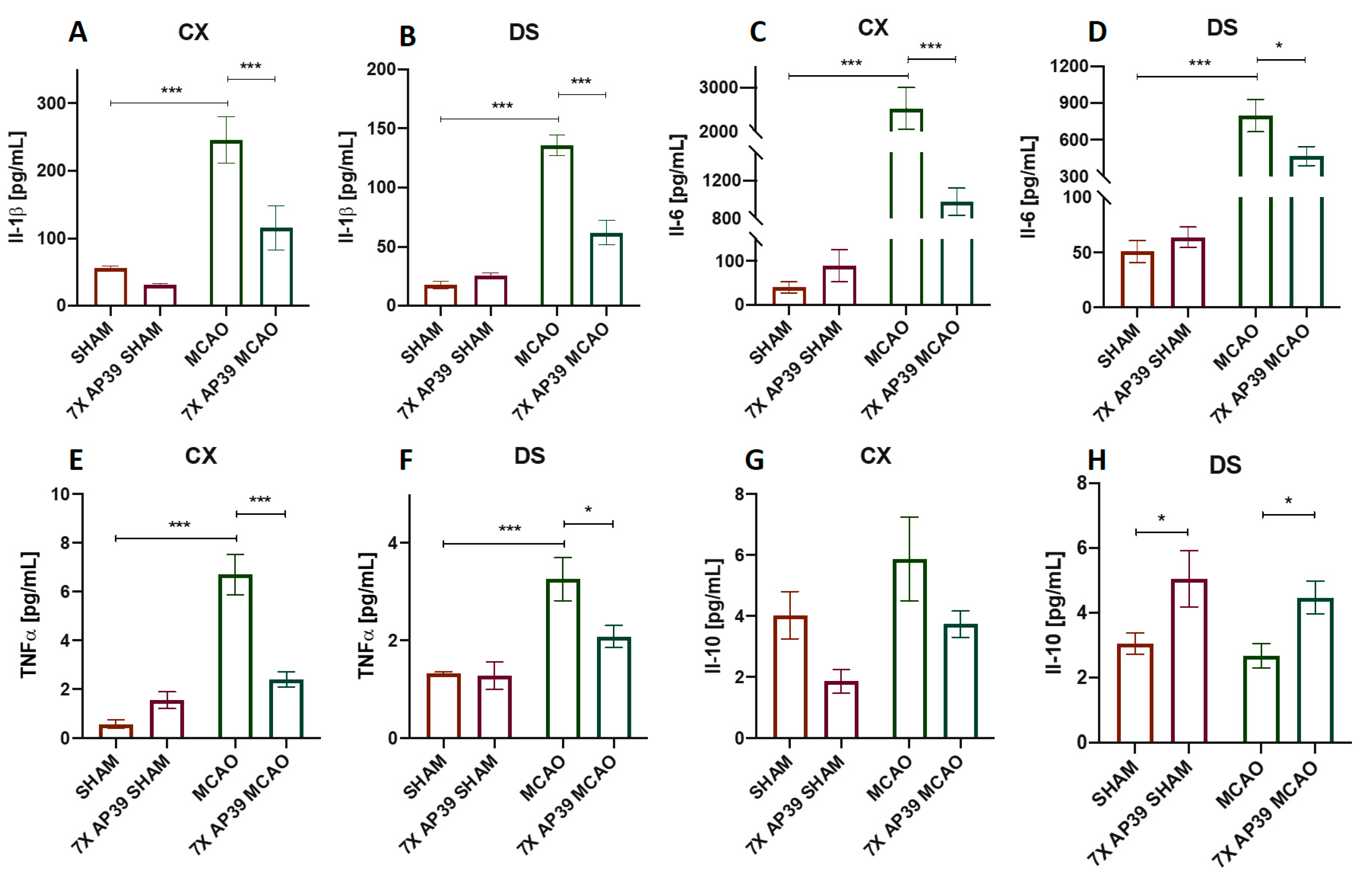
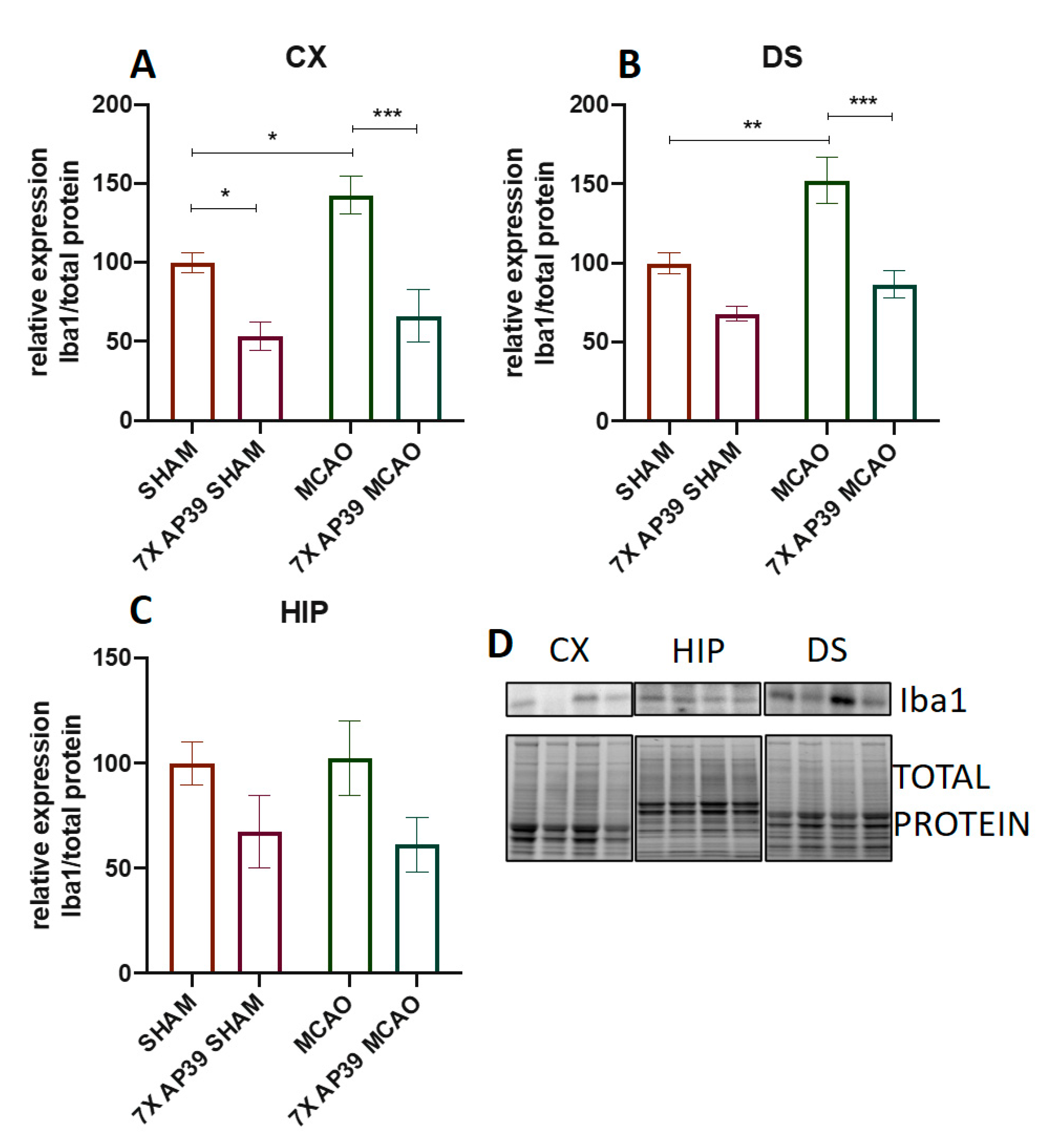
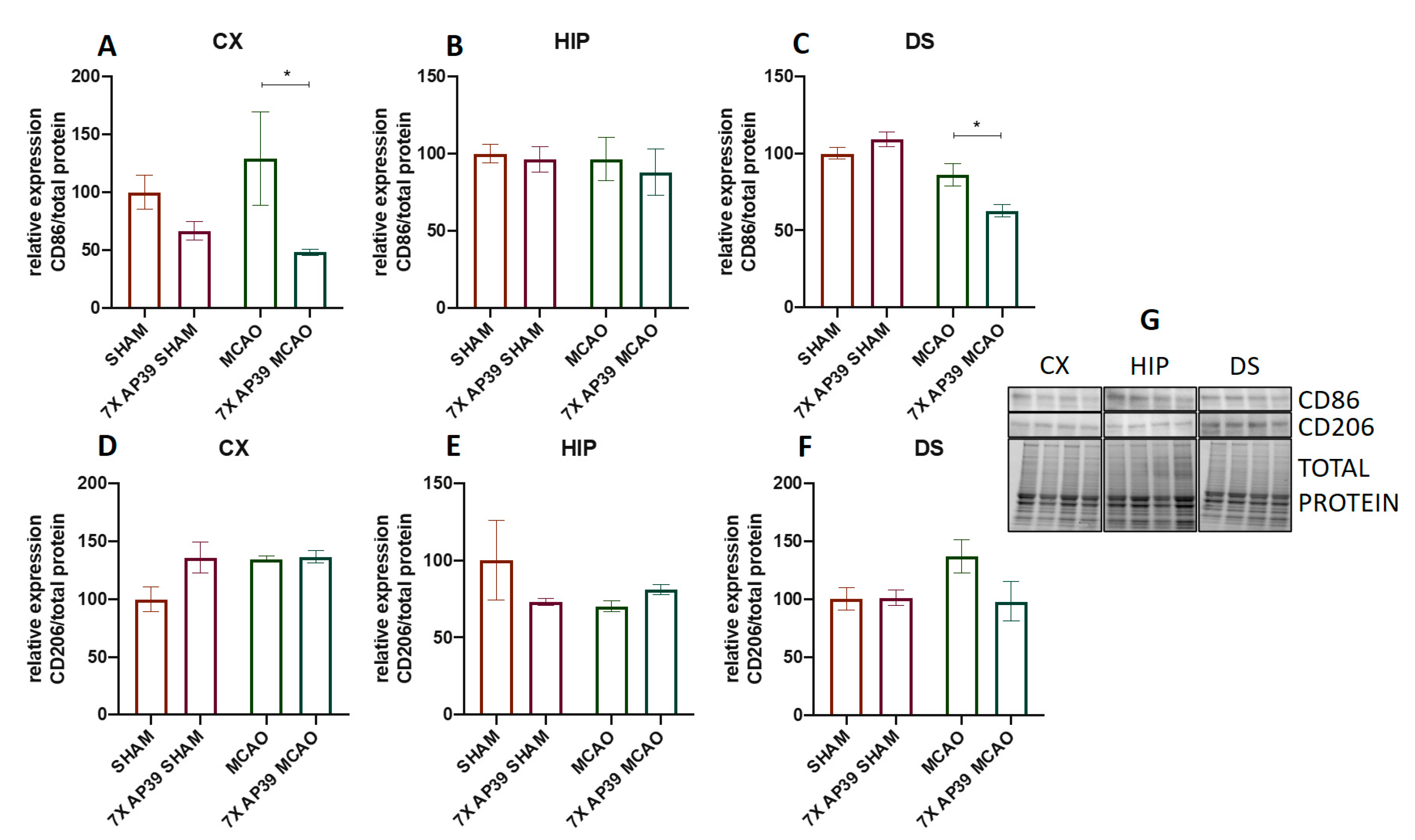
2.3. AP39 Mitigated the Neuroinflammation after MCAO
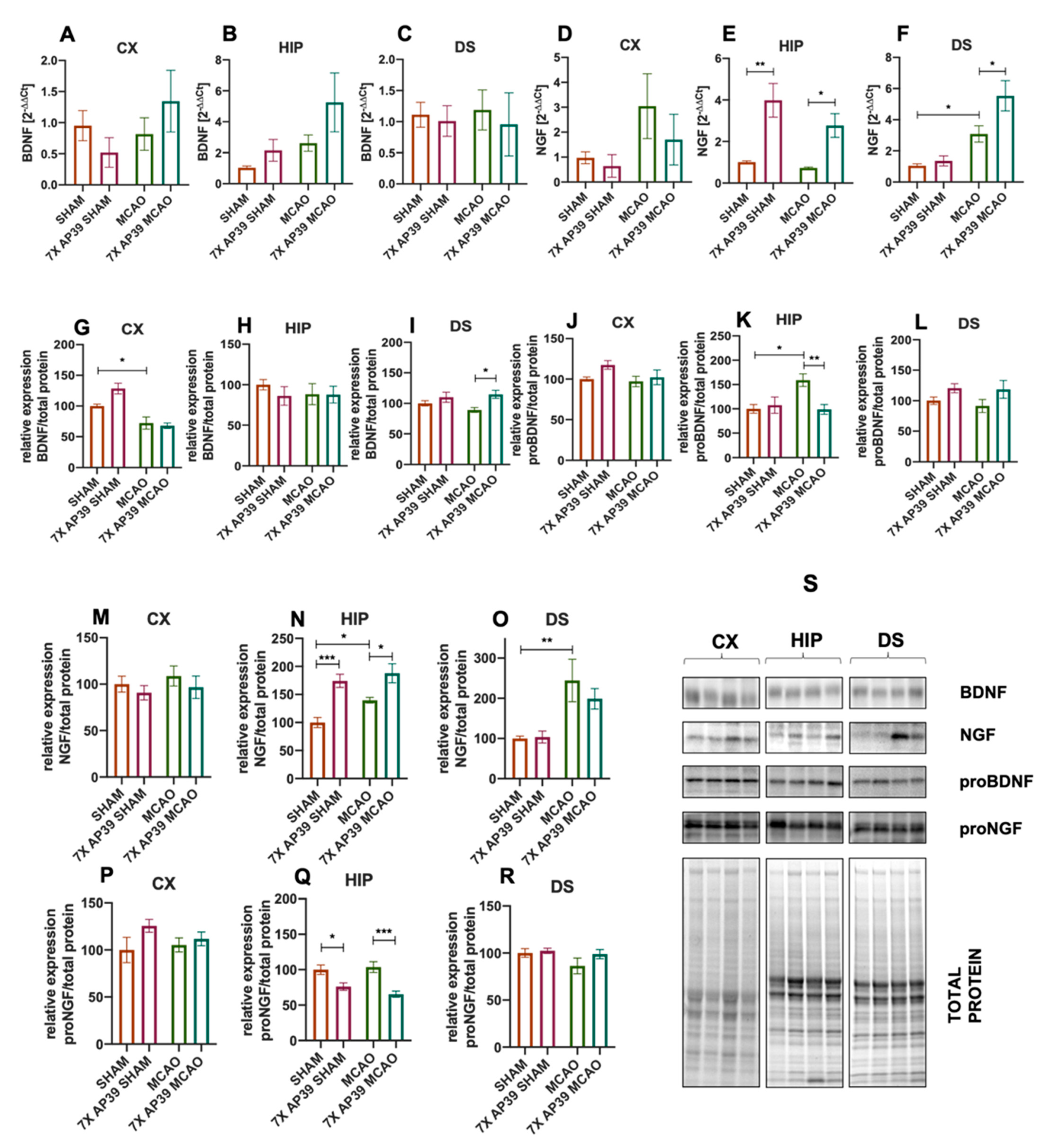
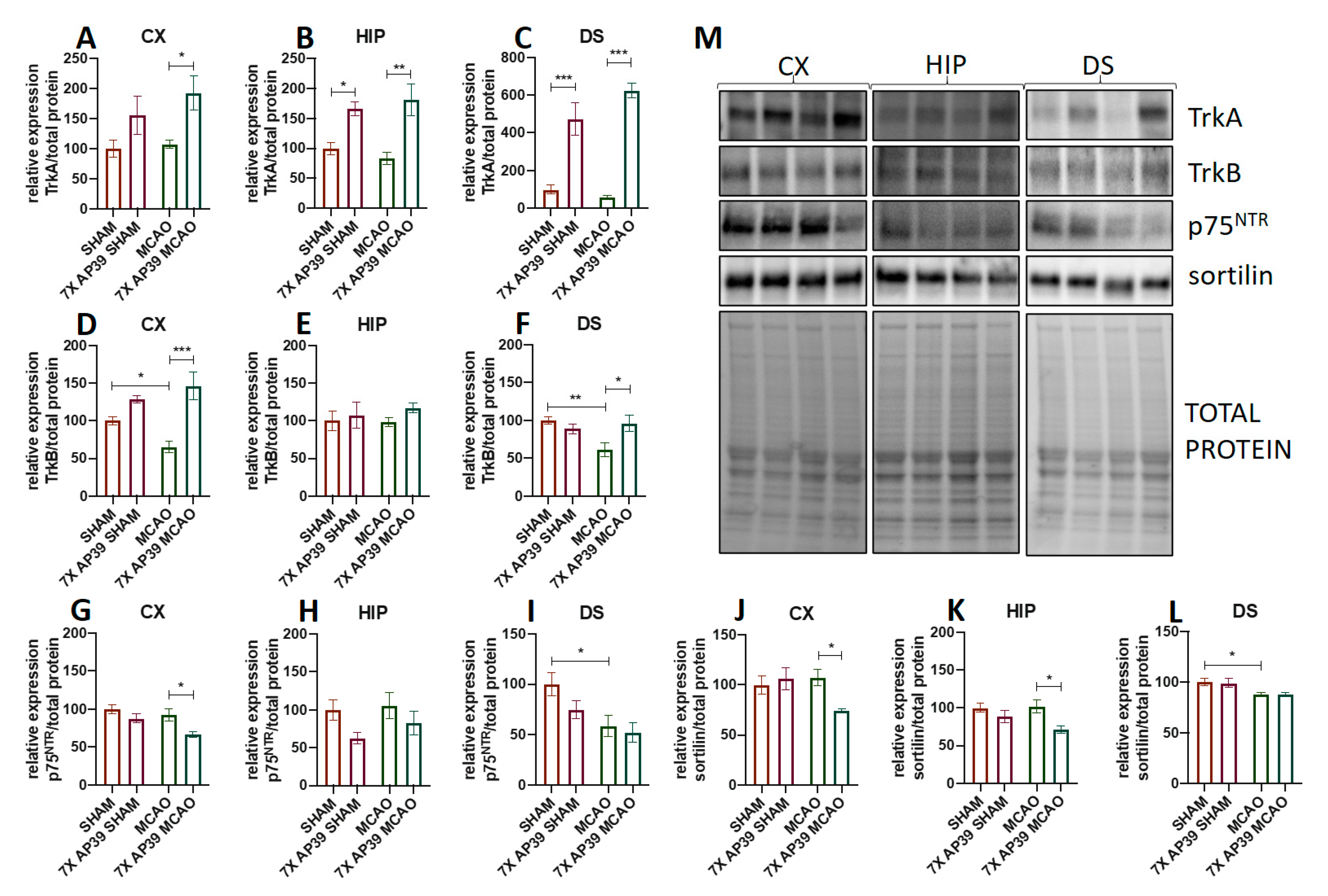
2.4. AP39 Modulates the Expression of Neurotrophic Factors and Their Receptors
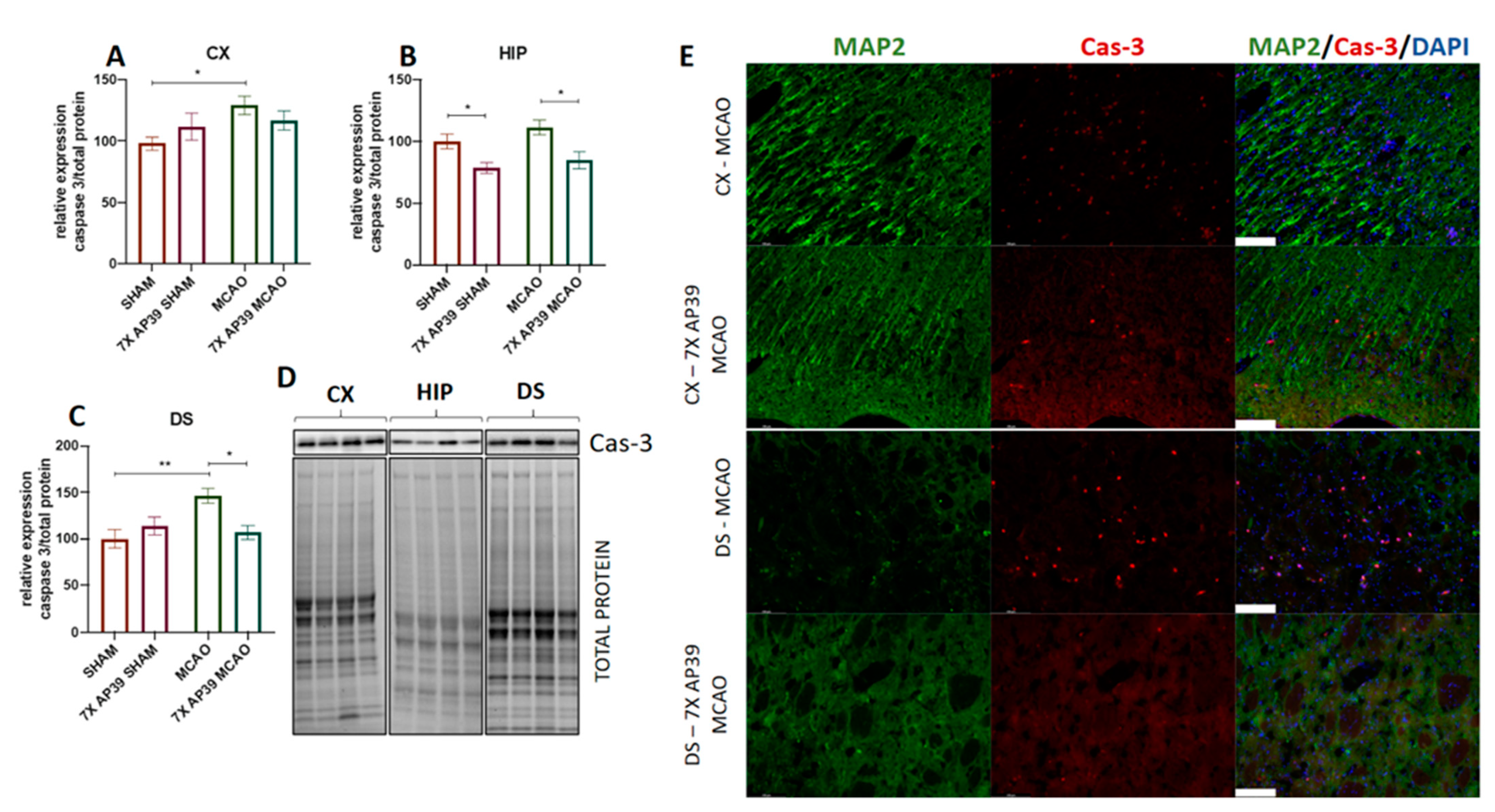
2.5. AP39 Mitigated MCAO-Induced Caspase 3 Activation
3. Discussion
4. Materials and Methods
4.1. Animals and Experimental Design
4.2. Animal Treatment
4.3. Focal Cerebral Ischemia Model
4.4. Neurological Deficit
4.5. TTC Staining—Measurement of the Infarct Volume
4.6. Luminex Assay
4.7. Western Blot
4.8. Real-Time qPCR
4.9. Tissue Fixation for the Immunofluorescent Double-Staining
4.10. Immunofluorescent Double-Staining
4.11. Measurement of H2S Content
4.12. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Feigin, V.L.; Lawes, C.M.; Bennett, D.A.; Barker-Collo, S.L.; Parag, V. Worldwide stroke incidence and early case fatality reported in 56 population-based studies: A systematic review. Lancet Neurol. 2009, 8, 355–369. [Google Scholar] [CrossRef]
- Berge, E.; Whiteley, W.; Audebert, H.; Marchis, G.M.D.; Fonseca, A.C.; Padiglioni, C.; Ossa, N.P.d.l.; Strbian, D.; Tsivgoulis, G.; Turc, G. European Stroke Organisation (ESO) guidelines on intravenous thrombolysis for acute ischaemic stroke. Eur. Stroke J. 2021, 6, I–LXII. [Google Scholar] [CrossRef]
- Wang, R. Physiological implications of hydrogen sulfide: A whiff exploration that blossomed. Physiol. Rev. 2012, 92, 791–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabõ, C. Hydrogen sulphide and its therapeutic potential. Nat. Rev. Drug Discov. 2007, 6, 917–935. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.K.; Bhatia, M.; Moochhala, S. Hydrogen sulfide: From the smell of the past to the mediator of the future? Trends Pharmacol. Sci. 2003, 24, 609–611. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Kimura, H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 1996, 16, 1066–1071. [Google Scholar] [CrossRef] [Green Version]
- Kimura, H. Physiological role of hydrogen sulfide and polysulfide in the central nervous system. Neurochem. Int. 2013, 63, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Marutani, E.; Morita, M.; Hirai, S.; Kai, S.; Grange, R.M.H.; Miyazaki, Y.; Nagashima, F.; Traeger, L.; Magliocca, A.; Ida, T.; et al. Sulfide catabolism ameliorates hypoxic brain injury. Nat. Commun. 2021, 12, 3108. [Google Scholar] [CrossRef]
- Goubern, M.; Andriamihaja, M.; Nübel, T.; Blachier, F.; Bouillaud, F. Sulfide, the first inorganic substrate for human cells. FASEB J. 2007, 21, 1699–1706. [Google Scholar] [CrossRef]
- Theissen, U.; Martin, W. Sulfide:quinone oxidoreductase (SQR) from the lugworm Arenicola marina shows cyanide- and thioredoxin-dependent activity. FEBS J. 2008, 275, 1131–1139. [Google Scholar] [CrossRef]
- Gerő, D.; Torregrossa, R.; Perry, A.; Waters, A.; Le-Trionnaire, S.; Whatmore, J.L.; Wood, M.; Whiteman, M. The novel mitochondria-targeted hydrogen sulfide (H2S) donors AP123 and AP39 protect against hyperglycemic injury in microvascular endothelial cells in vitro. Pharmacol. Res. 2016, 113, 186–198. [Google Scholar] [CrossRef]
- Libiad, M.; Vitvitsky, V.; Bostelaar, T.; Bak, D.W.; Lee, H.J.; Sakamoto, N.; Fearon, E.; Lyssiotis, C.A.; Weerapana, E.; Banerjee, R. Hydrogen sulfide perturbs mitochondrial bioenergetics and triggers metabolic reprogramming in colon cells. J. Biol. Chem. 2019, 294, 12077–12090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, H. Hydrogen sulfide induces cyclic AMP and modulates the NMDA receptor. Biochem. Biophys. Res. Commun. 2000, 267, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Qin, J.; Chang, X.; Yang, Z.; Bu, D.; Du, J. Modulating effect of hydrogen sulfide on gamma-aminobutyric acid B receptor in recurrent febrile seizures in rats. Neurosci. Res. 2005, 53, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Cheung, N.S.; Zhu, Y.Z.; Chu, S.H.; Siau, J.L.; Wong, B.S.; Armstrong, J.S.; Moore, P.K. Hydrogen sulphide: A novel inhibitor of hypochlorous acid-mediated oxidative damage in the brain? Biochem. Biophys. Res. Commun. 2005, 326, 794–798. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Kimura, H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J. 2004, 18, 1165–1167. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Goto, Y.I.; Kimura, H. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid. Redox Signal. 2010, 12, 1–13. [Google Scholar] [CrossRef]
- Hu, L.F.; Lu, M.; Hon Wong, P.T.; Bian, J.S. Hydrogen sulfide: Neurophysiology and neuropathology. Antioxid. Redox Signal. 2011, 15, 405–419. [Google Scholar] [CrossRef]
- Seifert, H.A.; Pennypacker, K.R. Molecular and Cellular Immune Responses to Ischemic Brain Injury. Transl. Stroke Res. 2014, 5, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.F.; Li, Y.; Song, J.N.; Pang, H.G. Role of hydrogen sulfide in secondary neuronal injury. Neurochem. Int. 2014, 64, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Winyard, P.G. Hydrogen sulfide and inflammation: The good, the bad, the ugly and the promising. Expert Rev. Clin. Pharmacol. 2011, 4, 13–32. [Google Scholar] [CrossRef]
- Zhao, F.L.; Fang, F.; Qiao, P.F.; Yan, N.; Gao, D.; Yan, Y. AP39, a Mitochondria-Targeted Hydrogen Sulfide Donor, Supports Cellular Bioenergetics and Protects against Alzheimer’s Disease by Preserving Mitochondrial Function in APP/PS1 Mice and Neurons. Oxid. Med. Cell. Longev. 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.; Yuan, Y.; Sheng, Y.; Yuan, B.; Wang, Y.; Zheng, J.; Liu, C.F.; Zhang, X.; Hu, L.F. GYY4137, an H2S slow-releasing donor, prevents nitrative stress and α-synuclein nitration in an MPTP mouse model of parkinson’s disease. Front. Pharmacol. 2017, 8, 741. [Google Scholar] [CrossRef] [Green Version]
- Woo, C.W.; Kwon, J.I.; Kim, K.W.; Kim, J.K.; Jeon, S.B.; Jung, S.C.; Choi, C.G.; Kim, S.T.; Kim, J.; Ham, S.J.; et al. The administration of hydrogen sulphide prior to ischemic reperfusion has neuroprotective effects in an acute stroke model. PLoS ONE 2017, 12, e0187910. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Yang, X.; Zhao, S.; Wei, C.; Yin, Y.; Liu, T.; Jiang, S.; Xie, J.; Wan, X.; Mao, M.; et al. Hydrogen sulfide prevents OGD/R-induced apoptosis via improving mitochondrial dysfunction and suppressing an ROS-mediated caspase-3 pathway in cortical neurons. Neurochem. Int. 2013, 63, 826–831. [Google Scholar] [CrossRef] [PubMed]
- Tay, A.S.; Hu, L.F.; Lu, M.; Wong, P.T.H.; Bian, J.S. Hydrogen sulfide protects neurons against hypoxic injury via stimulation of ATP-sensitive potassium channel/protein kinase C/extracellular signal-regulated kinase/heat shock protein90 pathway. Neuroscience 2010, 167, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Li, G.F.; Luo, H.K.; Li, L.F.; Zhang, Q.Z.; Xie, L.J.; Jiang, H.; Li, L.P.; Hao, N.; Wang, W.W.; Zhang, J.X. Dual effects of hydrogen sulphide on focal cerebral ischaemic injury via modulation of oxidative stress-induced apoptosis. Clin. Exp. Pharmacol. Physiol. 2012, 39, 765–771. [Google Scholar] [CrossRef]
- Florian, B.; Vintilescu, R.; Balseanu, A.T.; Buga, A.M.; Grisk, O.; Walker, L.C.; Kessler, C.; Popa-Wagner, A. Long-term hypothermia reduces infarct volume in aged rats after focal ischemia. Neurosci. Lett. 2008, 438, 180–185. [Google Scholar] [CrossRef]
- Ren, C.; Du, A.; Li, D.; Sui, J.; Mayhan, W.G.; Zhao, H. Dynamic change of hydrogen sulfide during global cerebral ischemia-reperfusion and its effect in rats. Brain Res. 2010, 1345, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Tu, C.; Zhao, J.; Ou, D.; Chen, G.; Liu, Y.; Xiao, X. Exogenous hydrogen sulfide protects against global cerebral ischemia/reperfusion injury via its anti-oxidative, anti-inflammatory and anti-apoptotic effects in rats. Brain Res. 2013, 1491, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Gheibi, S.; Aboutaleb, N.; Khaksari, M.; Kalalian-Moghaddam, H.; Vakili, A.; Asadi, Y.; Mehrjerdi, F.Z.; Gheibi, A. Hydrogen Sulfide Protects the Brain Against Ischemic Reperfusion Injury in a Transient Model of Focal Cerebral Ischemia. J. Mol. Neurosci. 2014, 54, 264–270. [Google Scholar] [CrossRef]
- Wu, D.; Wang, J.; Li, H.; Xue, M.; Ji, A.; Li, Y. Role of hydrogen sulfide in ischemia-reperfusion injury. Oxid. Med. Cell. Longev. 2015, 2015, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, B.; Ootani, A.; Iwakiri, R.; Fujise, T.; Tsunada, S.; Toda, S.; Fujimoto, K. Ischemic preconditioning attenuates ischemia-reperfusion-induced mucosal apoptosis by inhibiting the mitochondria-dependent pathway in rat small intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G580–G587. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Hydrogen sulfide: The third gasotransmitter in biology and medicine. Antioxid. Redox Signal. 2010, 12, 1061–1064. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.T.; Neo, K.L.; Hu, L.F.; Yong, Q.C.; Bian, J.S. H2S preconditioning-induced PKC activation regulates intracellular calcium handling in rat cardiomyocytes. Am. J. Physiol. Cell Physiol. 2008, 294, C169–C177. [Google Scholar] [CrossRef]
- Du, J.; Jin, H.; Yang, L. Role of hydrogen sulfide in retinal diseases. Front. Pharmacol. 2017, 8, 588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Y.; Li, Y.; Zhao, Z.; Li, P.; Xie, Y. Hydrogen Sulfide Overproduction Is Involved in Acute Ischemic Cerebral Injury Under Hyperhomocysteinemia. Front. Neurosci. 2020, 14, 1224. [Google Scholar] [CrossRef] [PubMed]
- Qu, K.; Chen, C.P.L.H.; Halliwell, B.; Moore, P.K.; Wong, P.T.H. Hydrogen sulfide is a mediator of cerebral ischemic damage. Stroke 2006, 37, 889–893. [Google Scholar] [CrossRef] [Green Version]
- Le Trionnaire, S.; Perry, A.; Szczesny, B.; Szabo, C.; Winyard, P.G.; Whatmore, J.L.; Wood, M.E.; Whiteman, M. The synthesis and functional evaluation of a mitochondria-targeted hydrogen sulfide donor, (10-oxo-10-(4-(3-thioxo-3H-1,2-dithiol-5-yl)phenoxy)decyl) triphenylphosphonium bromide (AP39). Medchemcomm 2014, 5, 728–736. [Google Scholar] [CrossRef] [Green Version]
- Zemke, D.; Smith, J.L.; Reeves, M.J.; Majid, A. Ischemia and ischemic tolerance in the brain: An overview. Neurotoxicology 2004, 25, 895–904. [Google Scholar] [CrossRef]
- Ji, K.; Xue, L.; Cheng, J.; Bai, Y. Preconditioning of H2S inhalation protects against cerebral ischemia/reperfusion injury by induction of HSP70 through PI3K/Akt/Nrf2 pathway. Brain Res. Bull. 2016, 121, 68–74. [Google Scholar] [CrossRef]
- Krzyzanowska, W.; Pomierny, B.; Budziszewska, B.; Filip, M.; Pera, J. N-Acetylcysteine and Ceftriaxone as Preconditioning Strategies in Focal Brain Ischemia: Influence on Glutamate Transporters Expression. Neurotox. Res. 2016, 29, 539–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krzyżanowska, W.; Pomierny, B.; Bystrowska, B.; Pomierny-Chamioło, L.; Filip, M.; Budziszewska, B.; Pera, J. Ceftriaxone- and N-acetylcysteine-induced brain tolerance to ischemia: Influence on glutamate levels in focal cerebral ischemia. PLoS ONE 2017, 12, e0186243. [Google Scholar] [CrossRef] [Green Version]
- Shah, F.A.; Li, T.; Kury, L.T.A.; Zeb, A.; Khatoon, S.; Liu, G.; Yang, X.; Liu, F.; Yao, H.; Khan, A.U.; et al. Pathological Comparisons of the Hippocampal Changes in the Transient and Permanent Middle Cerebral Artery Occlusion Rat Models. Front. Neurol. 2019, 10, 1178. [Google Scholar] [CrossRef]
- Weinstein, P.R.; Hong, S.; Sharp, F.R. Molecular identification of the ischemic penumbra. Stroke 2004, 35, 2666–2670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahul, A.B.; Joshi, P.C.; Kumar, A.; Chakravarty, S. Transient global cerebral ischemia differentially affects cortex, striatum and hippocampus in Bilateral Common Carotid Arterial occlusion (BCCAo) mouse model. J. Chem. Neuroanat. 2018, 92, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Pawluk, H.; Woźniak, A.; Grześk, G.; Kołodziejska, R.; Kozakiewicz, M.; Kopkowska, E.; Grzechowiak, E.; Kozera, G. The role of selected pro-inflammatory cytokines in pathogenesis of ischemic stroke. Clin. Interv. Aging 2020, 15, 469–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouda, A.Y.; Kozak, A.; Alhusban, A.; Switzer, J.A.; Fagan, S.C. Anti-inflammatory IL-10 is upregulated in both hemispheres after experimental ischemic stroke: Hypertension blunts the response. Exp. Transl. Stroke Med. 2013, 5, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Baron, J.C.; Yamauchi, H.; Fujioka, M.; Endres, M. Selective neuronal loss in ischemic stroke and cerebrovascular disease. J. Cereb. Blood Flow Metab. 2014, 34, 2–18. [Google Scholar] [CrossRef] [Green Version]
- Szczesny, B.; Módis, K.; Yanagi, K.; Coletta, C.; Le Trionnaire, S.; Perry, A.; Wood, M.E.; Whiteman, M.; Szabo, C. AP39, a novel mitochondria-targeted hydrogen sulfide donor, stimulates cellular bioenergetics, exerts cytoprotective effects and protects against the loss of mitochondrial DNA integrity in oxidatively stressed endothelial cells in vitro. Nitric Oxide Biol. Chem. 2014, 41, 120–130. [Google Scholar] [CrossRef] [Green Version]
- Szabo, C.; Ransy, C.; Módis, K.; Andriamihaja, M.; Murghes, B.; Coletta, C.; Olah, G.; Yanagi, K.; Bouillaud, F. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. Br. J. Pharmacol. 2014, 171, 2099–2122. [Google Scholar] [CrossRef] [Green Version]
- Yong, R.; Searcy, D.G. Sulfide oxidation coupled to ATP synthesis in chicken liver mitochondria. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2001, 129, 129–137. [Google Scholar] [CrossRef]
- Zivanovic, J.; Kouroussis, E.; Kohl, J.B.; Adhikari, B.; Bursac, B.; Schott-Roux, S.; Petrovic, D.; Miljkovic, J.L.; Thomas-Lopez, D.; Jung, Y.; et al. Selective Persulfide Detection Reveals Evolutionarily Conserved Antiaging Effects of S-Sulfhydration. Cell Metab. 2019, 30, 1152–1170.e13. [Google Scholar] [CrossRef]
- Wedmann, R.; Onderka, C.; Wei, S.; Szijártó, I.A.; Miljkovic, J.L.; Mitrovic, A.; Lange, M.; Savitsky, S.; Yadav, P.K.; Torregrossa, R.; et al. Improved tag-switch method reveals that thioredoxin acts as depersulfidase and controls the intracellular levels of protein persulfidation. Chem. Sci. 2016, 7, 3414–3426. [Google Scholar] [CrossRef] [Green Version]
- Aroca, A.; Benito, J.M.; Gotor, C.; Romero, L.C. Persulfidation proteome reveals the regulation of protein function by hydrogen sulfide in diverse biological processes in Arabidopsis. J. Exp. Bot. 2017, 68, 4915–4927. [Google Scholar] [CrossRef] [Green Version]
- Linden, D.R.; Furne, J.; Stoltz, G.J.; Abdel-Rehim, M.S.; Levitt, M.D.; Szurszewski, J.H. Sulphide quinone reductase contributes to hydrogen sulphide metabolism in murine peripheral tissues but not in the CNS. Br. J. Pharmacol. 2012, 165, 2178–2190. [Google Scholar] [CrossRef] [Green Version]
- Kanazawa, M.; Ninomiya, I.; Hatakeyama, M.; Takahashi, T.; Shimohata, T. Microglia and monocytes/macrophages polarization reveal novel therapeutic mechanism against stroke. Int. J. Mol. Sci. 2017, 18, 2135. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Tian, T.; Gong, S.X.; Huang, W.Q.; Zhou, Q.Y.; Wang, A.P.; Tian, Y. Microglia-associated neuroinflammation is a potential therapeutic target for ischemic stroke. Neural Regen. Res. 2021, 16, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yang, J.; Zhang, C.; Geng, X.; Zhao, H. Remote ischemic conditioning reduced cerebral ischemic injury by modulating inflammatory responses and ERK activity in type 2 diabetic mice. Neurochem. Int. 2020, 135, 104690. [Google Scholar] [CrossRef] [PubMed]
- Pera, J.; Zawadzka, M.; Kaminska, B.; Szczudlik, A. Influence of chemical and ischemic preconditioning on cytokine expression after focal brain ischemia. J. Neurosci. Res. 2004, 78, 132–140. [Google Scholar] [CrossRef]
- Spera, P.A.; Ellison, J.A.; Feuerstein, G.Z.; Barone, F.C. IL-10 reduces rat brain injury following focal stroke. Neurosci. Lett. 1998, 251, 189–192. [Google Scholar] [CrossRef]
- Sabat, R.; Grütz, G.; Warszawska, K.; Kirsch, S.; Witte, E.; Wolk, K.; Geginat, J. Biology of interleukin-10. Cytokine Growth Factor Rev. 2010, 21, 331–344. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.T.; Wu, W.F.; Deng, Y.H.; Ge, J.W. Modulators of microglia activation and polarization in ischemic stroke (Review). Mol. Med. Rep. 2020, 21, 2006–2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yenari, M.A.; Kauppinen, T.M.; Swanson, R.A. Microglial Activation in Stroke: Therapeutic Targets. Neurotherapeutics 2010, 7, 378–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, L.F.; Wong, P.T.H.; Moore, P.K.; Bian, J.S. Hydrogen sulfide attenuates lipopolysaccharide-induced inflammation by inhibition of p38 mitogen-activated protein kinase in microglia. J. Neurochem. 2007, 100, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Castrén, E.; Antila, H. Neuronal plasticity and neurotrophic factors in drug responses. Mol. Psychiatry 2017, 22, 1085–1095. [Google Scholar] [CrossRef]
- Lee, R.; Kermani, P.; Teng, K.K.; Hempstead, B.L. Regulation of cell survival by secreted proneurotrophins. Science (80-) 2001, 294, 1945–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gehler, S.; Gallo, G.; Veien, E.; Letourneau, P.C. p75 Neurotrophin Receptor Signaling Regulates Growth Cone Filopodial Dynamics through Modulating RhoA Activity. J. Neurosci. 2004, 24, 4363–4372. [Google Scholar] [CrossRef]
- Jiang, J.M.; Zhou, C.F.; Gao, S.L.; Tian, Y.; Wang, C.Y.; Wang, L.; Gu, H.F.; Tang, X.Q. BDNF-TrkB pathway mediates neuroprotection of hydrogen sulfide against formaldehyde-induced toxicity to PC12 cells. PLoS ONE 2015, 10, e0119478. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, F.; Sakakima, H.; Yoshida, Y. The effects of early exercise on brain damage and recovery after focal cerebral infarction in rats. Acta Physiol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.P.; Liu, H.J.; Yang, H.; Feng, P.Y. Therapeutic time window for the neuroprotective effects of NGF when administered after focal cerebral ischemia. Neurol. Sci. 2011, 32, 433–441. [Google Scholar] [CrossRef]
- Nykjaer, A.; Lee, R.; Teng, K.K.; Jansen, P.; Madsen, P.; Nielsen, M.S.; Jacobsen, C.; Kliemannel, M.; Schwarz, E.; Willnow, T.E.; et al. Sortilin is essential for proNGF-induced neuronal cell death. Nature 2004, 427, 843–848. [Google Scholar] [CrossRef] [PubMed]
- McAllister, A.K.; Katz, L.C.; Lo, D.C. Neurotrophin regulation of cortical dendritic growth requires activity. Neuron 1996, 17, 1057–1064. [Google Scholar] [CrossRef] [Green Version]
- Cohen-Cory, S.; Fraser, S.E. Effects of brain-derived neurotrophic factor on optic axon branching and remodelling in vivo. Nature 1995, 378, 192–196. [Google Scholar] [CrossRef]
- Datta, A.; Sarmah, D.; Mounica, L.; Kaur, H.; Kesharwani, R.; Verma, G.; Veeresh, P.; Kotian, V.; Kalia, K.; Borah, A.; et al. Cell Death Pathways in Ischemic Stroke and Targeted Pharmacotherapy. Transl. Stroke Res. 2020, 11, 1185–1202. [Google Scholar] [CrossRef]
- Nitatori, T.; Sato, N.; Waguri, S.; Karasawa, Y.; Araki, H.; Shibanai, K.; Kominami, E.; Uchiyama, Y. Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. J. Neurosci. 1995, 15, 1001–1011. [Google Scholar] [CrossRef] [Green Version]
- Longa, E.Z.; Weinstein, P.R.; Carlson, S.; Cummins, R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989, 20, 84–91. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.B.; Williams, A.J.; Adams, J.; Elliott, P.J.; Tortella, F.C. Proteasome inhibitor PS519 reduces infarction and attenuates leukocyte infiltration in a rat model of focal cerebral ischemia. Stroke 2000, 31, 1686–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, B.; Jin, S.; Sun, J.; Gu, Z.; Sun, X.; Zhu, Y.; Huo, K.; Cao, Z.; Yang, P.; Xin, X.; et al. New method for quantification of gasotransmitter hydrogen sulfide in biological matrices by LC-MS/MS. Sci. Rep. 2017, 7, 46278. [Google Scholar] [CrossRef]
- Ditrói, T.; Nagy, A.; Martinelli, D.; Rosta, A.; Kožich, V.; Nagy, P. Comprehensive analysis of how experimental parameters affect H 2 S measurements by the monobromobimane method. Free Radic. Biol. Med. 2019, 136, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Wintner, E.A.; Deckwerth, T.L.; Langston, W.; Bengtsson, A.; Leviten, D.; Hill, P.; Insko, M.A.; Dumpit, R.; Vandenekart, E.; Toombs, C.F.; et al. A monobromobimane-based assay to measure the pharmacokinetic profile of reactive sulphide species in blood. Br. J. Pharmacol. 2010, 160, 941–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Manufacturer | Catalog Number | Dilution Used |
|---|---|---|---|
| pro-BDNF | Alomone | AGP-032 | 1:300 |
| mBDNF | Abcam | ab108319 | 1:1000 |
| pro-NGF | Alomone | AGP-031 | 1:300 |
| NGF | Alomone | AN-240 | 1:300 |
| TrkA | Alomone | ANT-018 | 1:300 |
| TrkB | Alomone | ANT-019 | 1:400 |
| p75NTR | Alomone | ANT-007 | 1:300 |
| Sortilin | Alomone | ANT-009 | 1:500 |
| Iba1 | Abcam | ab5076 | 1:1000 |
| Caspase 3 | Cell Signaling | 14220S | 1:1000 |
| CD206 | Abcam | ab125028 | 1:1000 |
| CD86 | Santa Cruz Biotechnology | sc-28347 | 1:300 |
| MAP2 (neuronal marker) | Abcam | ab5392 | 1:1000 |
| goat anti-guinea pig (HRP) | Abcam | ab97155 | 1:10,000 |
| goat anti-rabbit (HRP) | Abcam | ab6721 | 1:5000 |
| donkey anti-goat (HRP) | Abcam | ab97120 | 1:10,000 |
| goat anti-chicken (Alexa Fluor 488) | Abcam | ab150173 | 1:300 |
| donkey anti-rabbit (Texas Red) | Abcam | ab6800 | 1:1000 |
| Gene | Assay ID: |
|---|---|
| Ngf | Rn01533872_m1 |
| Bdnf | Rn01484924_m1 |
| Il-6 | Rn00561420_m1 |
| Il-1ß | Rn00580432_m1 |
| Tnfa | Rn99999017_m1 |
| Il-10 | Rn99999012_m1 |
| Ppia—HKG | Rn00690933_m1 |
| Ywhaz—HKG | Rn00755072_m1 |
| Hprt1—HKG | Rn01527840_m1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pomierny, B.; Krzyżanowska, W.; Jurczyk, J.; Skórkowska, A.; Strach, B.; Szafarz, M.; Przejczowska-Pomierny, K.; Torregrossa, R.; Whiteman, M.; Marcinkowska, M.; et al. The Slow-Releasing and Mitochondria-Targeted Hydrogen Sulfide (H2S) Delivery Molecule AP39 Induces Brain Tolerance to Ischemia. Int. J. Mol. Sci. 2021, 22, 7816. https://doi.org/10.3390/ijms22157816
Pomierny B, Krzyżanowska W, Jurczyk J, Skórkowska A, Strach B, Szafarz M, Przejczowska-Pomierny K, Torregrossa R, Whiteman M, Marcinkowska M, et al. The Slow-Releasing and Mitochondria-Targeted Hydrogen Sulfide (H2S) Delivery Molecule AP39 Induces Brain Tolerance to Ischemia. International Journal of Molecular Sciences. 2021; 22(15):7816. https://doi.org/10.3390/ijms22157816
Chicago/Turabian StylePomierny, Bartosz, Weronika Krzyżanowska, Jakub Jurczyk, Alicja Skórkowska, Beata Strach, Małgorzata Szafarz, Katarzyna Przejczowska-Pomierny, Roberta Torregrossa, Matthew Whiteman, Monika Marcinkowska, and et al. 2021. "The Slow-Releasing and Mitochondria-Targeted Hydrogen Sulfide (H2S) Delivery Molecule AP39 Induces Brain Tolerance to Ischemia" International Journal of Molecular Sciences 22, no. 15: 7816. https://doi.org/10.3390/ijms22157816
APA StylePomierny, B., Krzyżanowska, W., Jurczyk, J., Skórkowska, A., Strach, B., Szafarz, M., Przejczowska-Pomierny, K., Torregrossa, R., Whiteman, M., Marcinkowska, M., Pera, J., & Budziszewska, B. (2021). The Slow-Releasing and Mitochondria-Targeted Hydrogen Sulfide (H2S) Delivery Molecule AP39 Induces Brain Tolerance to Ischemia. International Journal of Molecular Sciences, 22(15), 7816. https://doi.org/10.3390/ijms22157816