
SERR Spectroelectrochemistry as a Guide for Rational Design of DyP-Based Bioelectronics Devices

 , , ,
, , ,  and
and 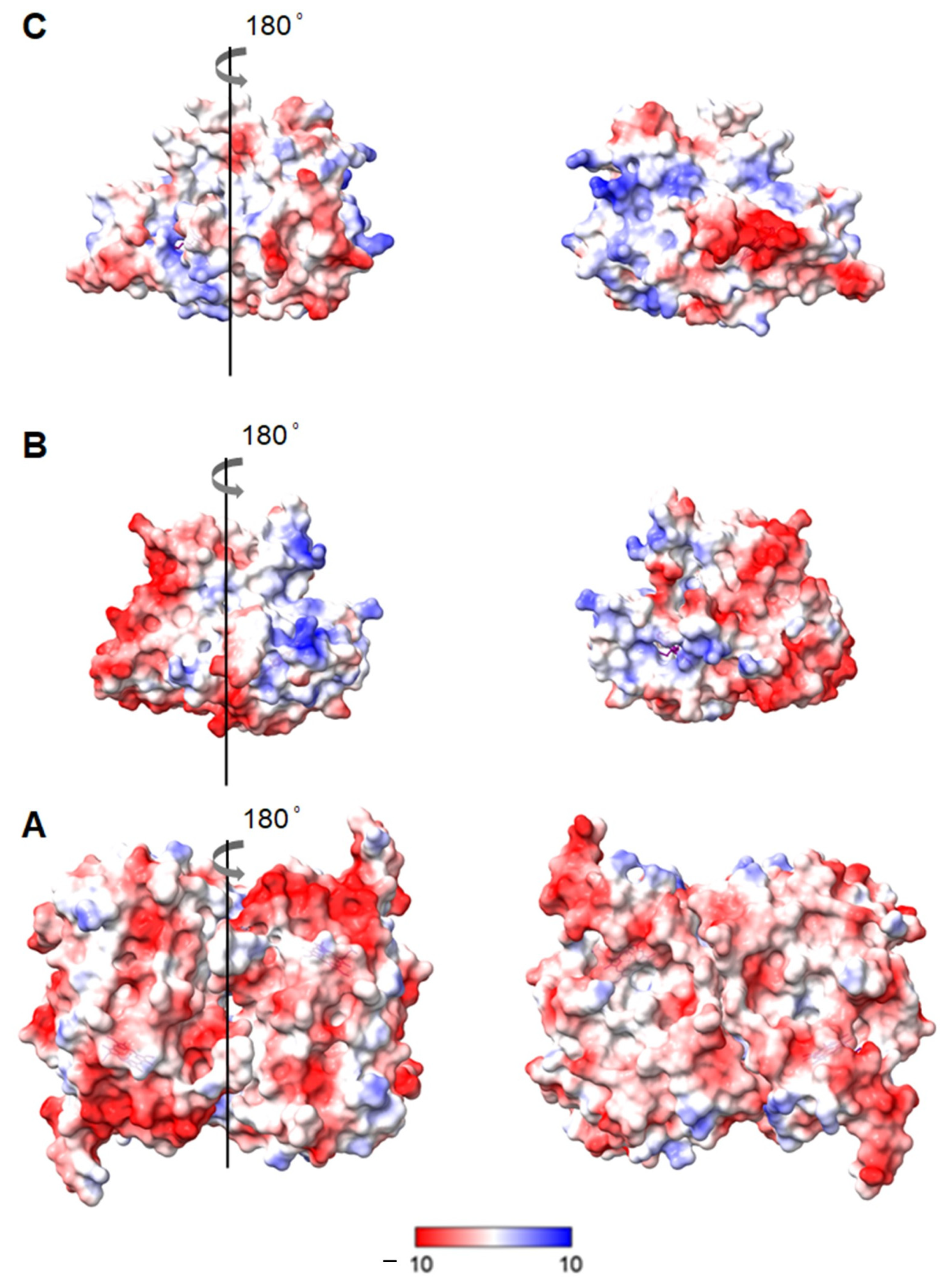{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
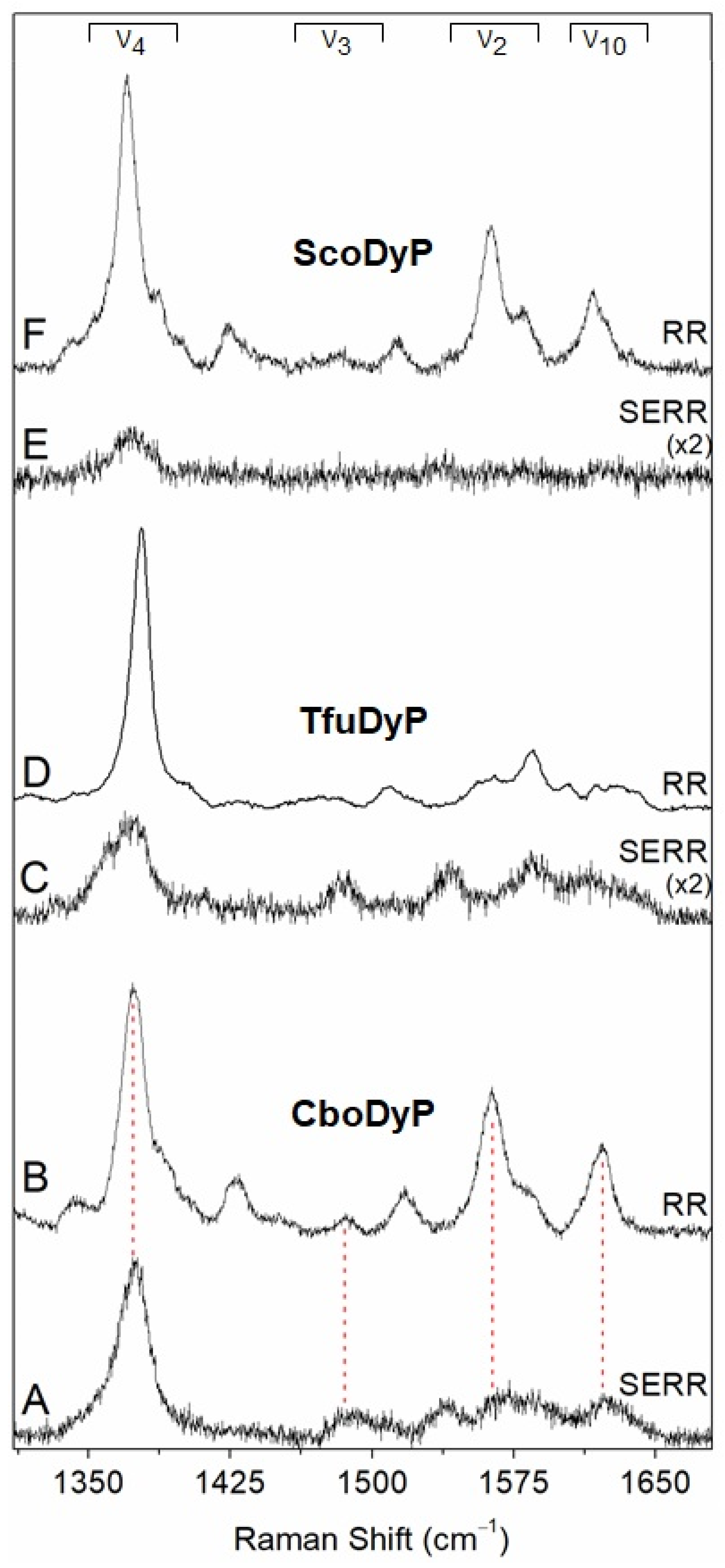
2.1. Evaluation of Structural Integrity of DyPs upon Immobilisation
2.2. Probing Redox Activity, Reversibility and Electronic Coupling of the Immobilised CboDyP
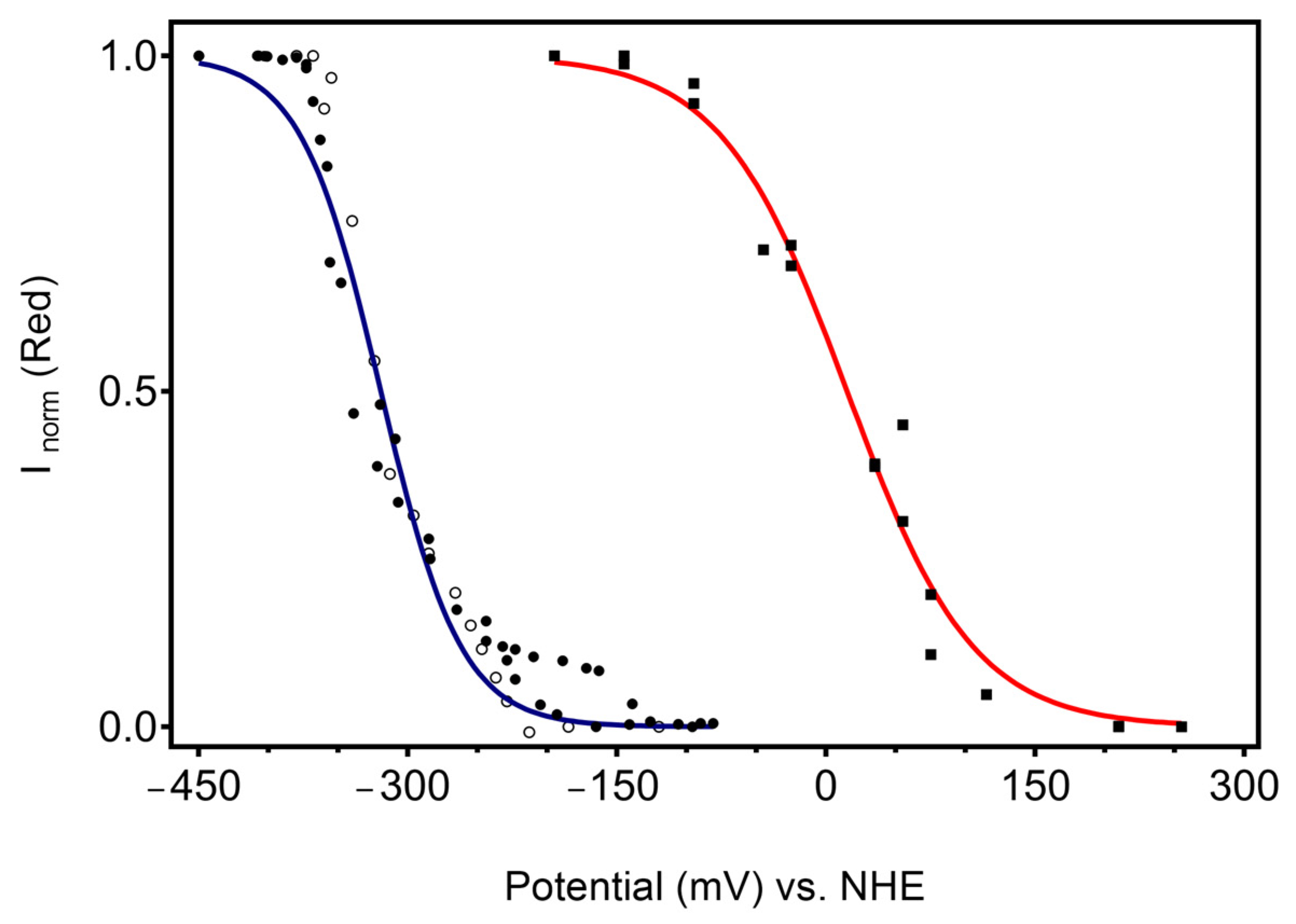
2.3. Assessement of the Redox Potential and Catalytic Efficiency of the Immobilised CboDyP
3. Discussion
3.1. Structural Integrity upon Immobilisation
3.2. Redox Properties upon Immobilisation and Electronic Communication
3.3. Probing Electrocatalytic Activity upon Immobilisation
4. Materials and Methods
4.1. Enzymes and Reagents
4.2. Enzyme Immobilisation
4.3. RR Spectroscopy
4.4. Redox Potential Determination in the Immobilised State
4.5. Redox Potential Determination in Solution
4.6. Electrochemistry Assays
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sugano, Y. DyP-Type Peroxidases Comprise a Novel Heme Peroxidase Family. Cell. Mol. Life Sci. 2009, 66, 1387–1403. [Google Scholar] [CrossRef]
- Colpa, D.I.; Fraaije, M.W.; Van Bloois, E. DyP-Type Peroxidases: A Promising and Versatile Class of Enzymes. J. Ind. Microbiol. Biotechnol. 2014, 41, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Chen, W. Recent Advances in Graphene-Based Nanomaterials for Fabricating Electrochemical Hydrogen Peroxide Sensors. Biosens. Bioelectron. 2017, 89, 249–268. [Google Scholar] [CrossRef]
- Lippert, A.R.; Van de Bittner, G.C.; Chang, C.J. Boronate Oxidation as a Bioorthogonal Reaction Approach for Studying the Chemistry of Hydrogen Peroxide in Living Systems. Acc. Chem. Res. 2011, 44, 793–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, P.; Das, M.; Chinnadayyala, S.R.; Singha, I.M.; Goswami, P. Recent Advances on Developing 3rd Generation Enzyme Electrode for Biosensor Applications. Biosens. Bioelectron. 2016, 79, 386–397. [Google Scholar] [CrossRef]
- Bollella, P.; Medici, L.; Tessema, M.; Poloznikov, A.A.; Hushpulian, D.M.; Tishkov, V.I.; Andreu, R.; Leech, D.; Megersa, N.; Marcaccio, M.; et al. Highly Sensitive, Stable and Selective Hydrogen Peroxide Amperometric Biosensors Based on Peroxidases from Different Sources Wired by Os-Polymer: A Comparative Study. Solid State Ion. 2018, 314, 178–186. [Google Scholar] [CrossRef]
- Zuccarello, L.; Barbosa, C.; Todorovic, S.; Silveira, C.M. Electrocatalysis by Heme Enzymes—Applications in Biosensing. Catalysts 2021, 11, 218. [Google Scholar] [CrossRef]
- Murgida, D.H.; Hildebrandt, P. Electron-Transfer Processes of Cytochrome c at Interfaces. New Insights by Surface-Enhanced Resonance Raman Spectroscopy. Acc. Chem. Res. 2004, 37, 854–861. [Google Scholar] [CrossRef]
- Murgida, D.H.; Hildebrandt, P. Heterogeneous Electron Transfer of Cytochrome c on Coated Silver Electrodes. Electric Field Effects on Structure and Redox Potential. J. Phys. Chem. B 2001, 105, 1578–1586. [Google Scholar] [CrossRef]
- Sezer, M.; Millo, D.; Weidinger, I.M.; Zebger, I.; Hildebrandt, P. Analyzing the Catalytic Processes of Immobilized Redox Enzymes by Vibrational Spectroscopies. IUBMB Life 2012, 64, 455–464. [Google Scholar] [CrossRef] [Green Version]
- Siebert, F.; Hildebrandt, P. Vibrational Spectroscopy in Life Science; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Khoa Ly, H.; Sezer, M.; Wisitruangsakul, N.; Feng, J.-J.; Kranich, A.; Millo, D.; Weidinger, I.M.; Zebger, I.; Murgida, D.H.; Hildebrandt, P. Surface-Enhanced Vibrational Spectroscopy for Probing Transient Interactions of Proteins with Biomimetic Interfaces: Electric Field Effects on Structure, Dynamics and Function of Cytochrome c. FEBS J. 2011, 27, 1382–1390. [Google Scholar] [CrossRef] [Green Version]
- Todorovic, S.; Jung, C.; Hildebrandt, P.; Murgida, D.H. Conformational Transitions and Redox Potential Shifts of Cytochrome P450 Induced by Immobilization. J. Biol. Inorg. Chem. 2006, 1, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Todorovic, S.; Verissimo, A.; Wisitruangsakul, N.; Zebger, I.; Hildebrandt, P.; Pereira, M.M.; Teixeira, M.; Murgida, D.H. SERR-Spectroelectrochemical Study of a Cbb3 Oxygen Reductase in a Biomimetic Construct. J. Phys. Chem. B 2008, 11, 16952–16959. [Google Scholar] [CrossRef] [PubMed]
- Sezer, M.; Genebra, T.; Mendes, S.; Martins, L.O.; Todorovic, S. A DyP-Type Peroxidase at a Bio-Compatible Interface: Structural and Mechanistic Insights. Soft Matter 2012, 8, 10314–10321. [Google Scholar] [CrossRef]
- Sezer, M.; Santos, A.; Kielb, P.; Pinto, T.; Martins, L.O.; Todorovic, S. Distinct Structural and Redox Properties of the Heme Active Site in Bacterial Dye Decolorizing Peroxidase-Type Peroxidases from Two Subfamilies: Resonance Raman and Electrochemical Study. Biochemistry 2013, 5, 3074–3084. [Google Scholar] [CrossRef]
- Silveira, C.M.; Quintas, P.O.; Moura, I.; Moura, J.J.G.; Hildebrandt, P.; Almeida, M.G.; Todorovic, S. SERR Spectroelectrochemical Study of Cytochrome Cd1 Nitrite Reductase Coimmobilized with Physiological Redox Partner Cytochrome C552 on Biocompatible Metal Electrodes. PLoS ONE 2015, 1, e0129940. [Google Scholar] [CrossRef] [Green Version]
- Silveira, C.M.; Castro, M.A.; Dantas, J.M.; Salgueiro, C.; Murgida, D.H.; Todorovic, S. Structure, Electrocatalysis and Dynamics of Immobilized Cytochrome PccH and Its Microperoxidase. Phys. Chem. Chem. Phys. 2017, 1, 8908–8918. [Google Scholar] [CrossRef]
- Todorovic, S.; Pereira, M.M.; Bandeiras, T.M.; Teixeira, M.; Hildebrandt, P.; Murgida, D.H. Midpoint Potentials of Hemes a and a3 in the Quinol Oxidase from Acidianus ambivalens Are Inverted. J. Am. Chem. Soc. 2005, 12, 13561–13566. [Google Scholar] [CrossRef]
- Todorovic, S.; Rodrigues, M.L.; Matos, D.; Pereira, I.A.C. Redox Properties of Lysine- and Methionine-Coordinated Hemes Ensure Downhill Electron Transfer in NrfH2A4 Nitrite Reductase. J. Phys. Chem. B 2012, 11, 5637–5643. [Google Scholar] [CrossRef]
- Barbosa, C.; Silveira, C.M.; Silva, D.; Brissos, V.; Hildebrandt, P.; Martins, L.O.; Todorovic, S. Immobilized Dye-Decolorizing Peroxidase (DyP) and Directed Evolution Variants for Hydrogen Peroxide Biosensing. Biosens. Bioelectron. 2020, 153, 112055. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Z.; Rui, Y.; Li, M. Horseradish Peroxidase Immobilization on Carbon Nanodots/CoFe Layered Double Hydroxides: Direct Electrochemistry and Hydrogen Peroxide Sensing. Biosens. Bioelectron. 2015, 64, 57–62. [Google Scholar] [CrossRef]
- Zhang, D.; Zhao, H.; Fan, Z.; Li, M.; Du, P.; Liu, C.; Li, Y.; Li, H.; Cao, H. A Highly Sensitive and Selective Hydrogen Peroxide Biosensor Based on Gold Nanoparticles and Three-Dimensional Porous Carbonized Chicken Eggshell Membrane. PLoS ONE 2015, 1, e0130156. [Google Scholar] [CrossRef]
- Villalonga, R.; Díez, P.; Yáñez-Sedeño, P.; Pingarrón, J.M. Wiring Horseradish Peroxidase on Gold Nanoparticles-Based Nanostructured Polymeric Network for the Construction of Mediatorless Hydrogen Peroxide Biosensor. Electrochim. Acta 2011, 5, 4672–4677. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure Visualization for Researchers, Educators, and Developers. Protein Sci. 2021, 3, 70–82. [Google Scholar] [CrossRef]
- Silveira, C.M.; Moe, E.; Fraaije, M.; Martins, L.O.; Todorovic, S. Resonance Raman View of the Active Site Architecture in Bacterial DyP-Type Peroxidases. RSC Adv. 2020, 1, 11095–11104. [Google Scholar] [CrossRef]
- Van Bloois, E.; Torres Pazmiño, D.E.; Winter, R.T.; Fraaije, M.W. A Robust and Extracellular Heme-Containing Peroxidase from Thermobifida fusca as Prototype of a Bacterial Peroxidase Superfamily. Appl. Microbiol. Biotechnol. 2010, 8, 1419–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habib, M.H.; Rozeboom, H.J.; Fraaije, M.W. Characterization of a New DyP-Peroxidase from the alkaliphilic cellulomonad, Cellulomonas bogoriensis. Molecules 2019, 24, 1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollella, P.; Gorton, L. Enzyme Based Amperometric Biosensors. Curr. Opin. Electrochem. 2018, 10, 157–173. [Google Scholar] [CrossRef]
- Monteiro, T.; Almeida, M.G. Electrochemical Enzyme Biosensors Revisited: Old Solutions for New Problems. Crit. Rev. Anal. Chem. 2019, 4, 44–66. [Google Scholar] [CrossRef]
- Brissos, V.; Tavares, D.; Sousa, A.C.; Robalo, M.P.; Martins, L.O. Engineering a Bacterial DyP-Type Peroxidase for Enhanced Oxidation of Lignin-Related Phenolics at Alkaline PH. ACS Catal. 2017, 5, 3454–3465. [Google Scholar] [CrossRef]
- Santos, A.; Mendes, S.; Brissos, V. New Dye-Decolorizing Peroxidases from Bacillus subtilis and Pseudomonas putida MET94: Towards Biotechnological Applications. Appl. Microbiol. Biotechnol. 2014, 2053–2065. [Google Scholar] [CrossRef] [PubMed]
- Mendes, S.; Catarino, T.; Silveira, C.; Todorovic, S.; Martins, L.O. The Catalytic Mechanism of A-Type Dye-Decolourising Peroxidase BsDyP: Neither Aspartate nor Arginine Is Individually Essential for Peroxidase Activity. Catal. Sci. Technol. 2015, 5, 5196–5207. [Google Scholar] [CrossRef]
- Mendes, S.; Brissos, V.; Gabriel, A.; Catarino, T.; Turner, D.L.; Todorovic, S.; Martins, L.O. An Integrated View of Redox and Catalytic Properties of B-Type PpDyP from Pseudomonas putida MET94 and Its Distal Variants. Arch. Biochem. Biophys. 2015, 574, 99–107. [Google Scholar] [CrossRef]
- Smulevich, G.; Feis, A.; Howes, B.D.; Ivancich, A. Structure-Function Relationships among Heme Peroxidases: New Insights from Electronic Absorption, Resonance Raman and Multifrequency Electron Paramagnetic Resonance Spectroscopies. In Handbook of Porphyrin Science; World Scientific: Singapore, 2010; pp. 367–453. [Google Scholar] [CrossRef]
- Smulevich, G.; Feis, A.; Howes, B.D. Fifteen Years of Raman Spectroscopy of Engineered Heme Containing Peroxidases: What Have We Learned? Acc. Chem. Res. 2005, 3, 433–440. [Google Scholar] [CrossRef]
- Battistuzzi, G.; Bellei, M.; Bortolotti, C.A.; Sola, M. Redox Properties of Heme Peroxidases. Arch. Biochem. Biophys. 2010, 50, 21–36. [Google Scholar] [CrossRef]
- Brown, M.E.; Barros, T.; Chang, M.C.Y. Identification and Characterization of a Multifunctional Dye Peroxidase from a Lignin-Reactive Bacterium. ACS Chem. Biol. 2012, 7, 2074–2081. [Google Scholar] [CrossRef]
- Chen, C.; Shrestha, R.; Jia, K.; Gao, P.F.; Geisbrecht, B.V.; Bossmann, S.H.; Shi, J.; Li, P. Characterization of Dye-Decolorizing Peroxidase (DyP) from Thermomonospora curvata Reveals Unique Catalytic Properties of A-Type DyPs. J. Biol. Chem. 2015, 29, 23447–23463. [Google Scholar] [CrossRef] [Green Version]
- Pfanzagl, V.; Nys, K.; Bellei, M.; Michlits, H.; Mlynek, G.; Battistuzzi, G.; Djinovic-Carugo, K.; Van Doorslaer, S.; Furtmüller, P.G.; Hofbauer, S.; et al. Roles of Distal Aspartate and Arginine of B-Class Dye-Decolorizing Peroxidase in Heterolytic Hydrogen Peroxide Cleavage. J. Biol. Chem. 2018, 29, 14823–14838. [Google Scholar] [CrossRef] [Green Version]
- Pfanzagl, V.; Bellei, M.; Hofbauer, S.; Laurent, C.V.F.P.; Furtmüller, P.G.; Oostenbrink, C.; Battistuzzi, G.; Obinger, C. Redox Thermodynamics of B-Class Dye-Decolorizing Peroxidases. J. Inorg. Biochem. 2019, 199, 110761. [Google Scholar] [CrossRef]
- Kassner, R.J. Theoretical Model for the Effects of Local Nonpolar Heme Environments on the Redox Potentials in Cytochromes. J. Am. Chem. Soc. 1973, 9, 2674–2677. [Google Scholar] [CrossRef]
- Döpner, S.; Hildebrandt, P.; Mauk, A.G.; Lenk, H.; Stempfle, W. Analysis of Vibrational Spectra of Multicomponent Systems. Application to PH-Dependent Resonance Raman Spectra of Ferricytochrome C. Spectrochim. Acta Part. A Mol. Spectrosc. 1996, 52, 573–584. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zuccarello, L.; Barbosa, C.; Galdino, E.; Lončar, N.; Silveira, C.M.; Fraaije, M.W.; Todorovic, S. SERR Spectroelectrochemistry as a Guide for Rational Design of DyP-Based Bioelectronics Devices. Int. J. Mol. Sci. 2021, 22, 7998. https://doi.org/10.3390/ijms22157998
Zuccarello L, Barbosa C, Galdino E, Lončar N, Silveira CM, Fraaije MW, Todorovic S. SERR Spectroelectrochemistry as a Guide for Rational Design of DyP-Based Bioelectronics Devices. International Journal of Molecular Sciences. 2021; 22(15):7998. https://doi.org/10.3390/ijms22157998
Chicago/Turabian StyleZuccarello, Lidia, Catarina Barbosa, Edilson Galdino, Nikola Lončar, Célia M. Silveira, Marco W. Fraaije, and Smilja Todorovic. 2021. "SERR Spectroelectrochemistry as a Guide for Rational Design of DyP-Based Bioelectronics Devices" International Journal of Molecular Sciences 22, no. 15: 7998. https://doi.org/10.3390/ijms22157998
APA StyleZuccarello, L., Barbosa, C., Galdino, E., Lončar, N., Silveira, C. M., Fraaije, M. W., & Todorovic, S. (2021). SERR Spectroelectrochemistry as a Guide for Rational Design of DyP-Based Bioelectronics Devices. International Journal of Molecular Sciences, 22(15), 7998. https://doi.org/10.3390/ijms22157998