
Human Mitochondrial RNA Processing and Modifications: Overview


Abstract
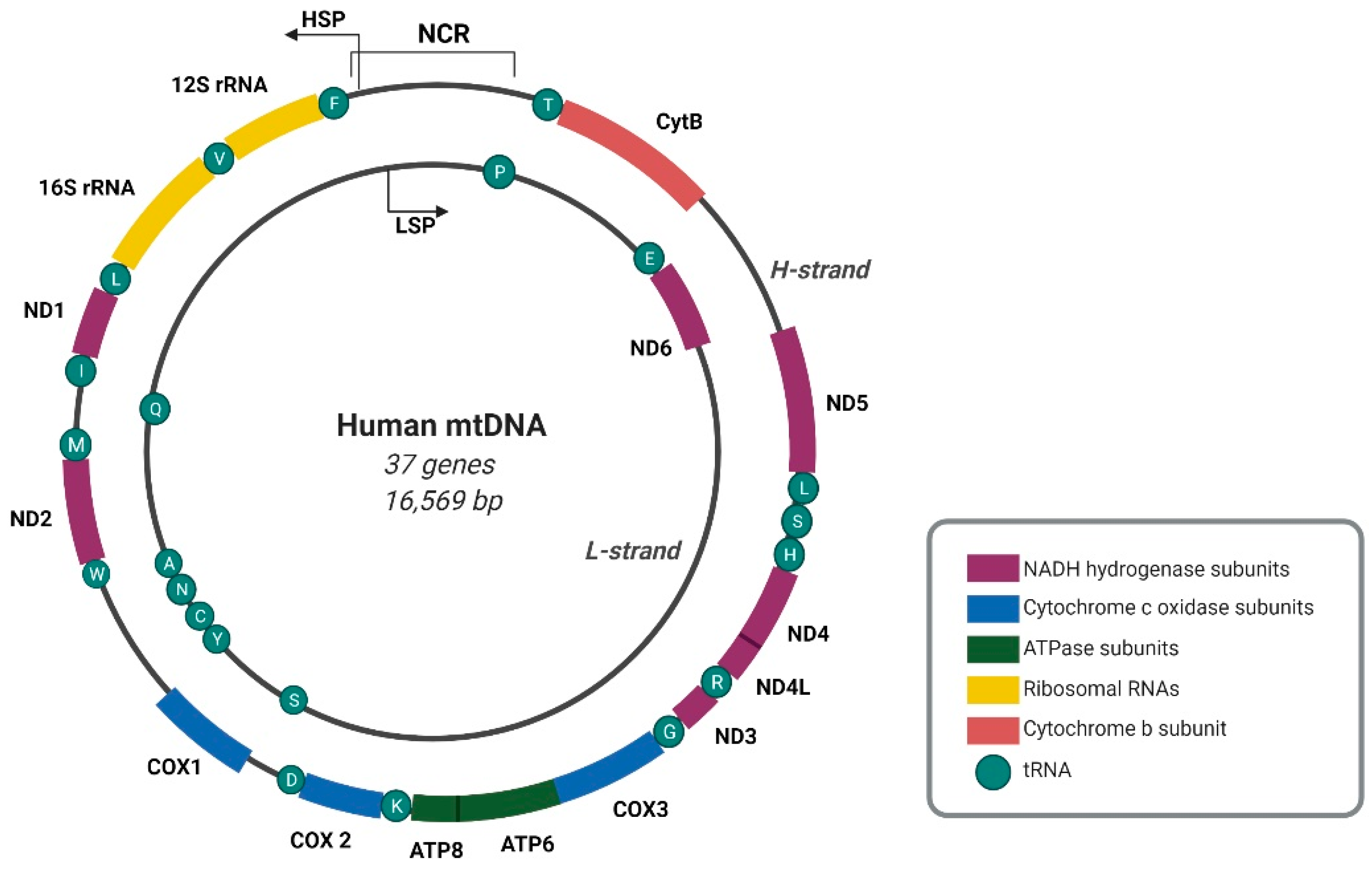
:1. Mitochondrial Genome Organization and Mitochondrial RNA Processing Compartmentalization
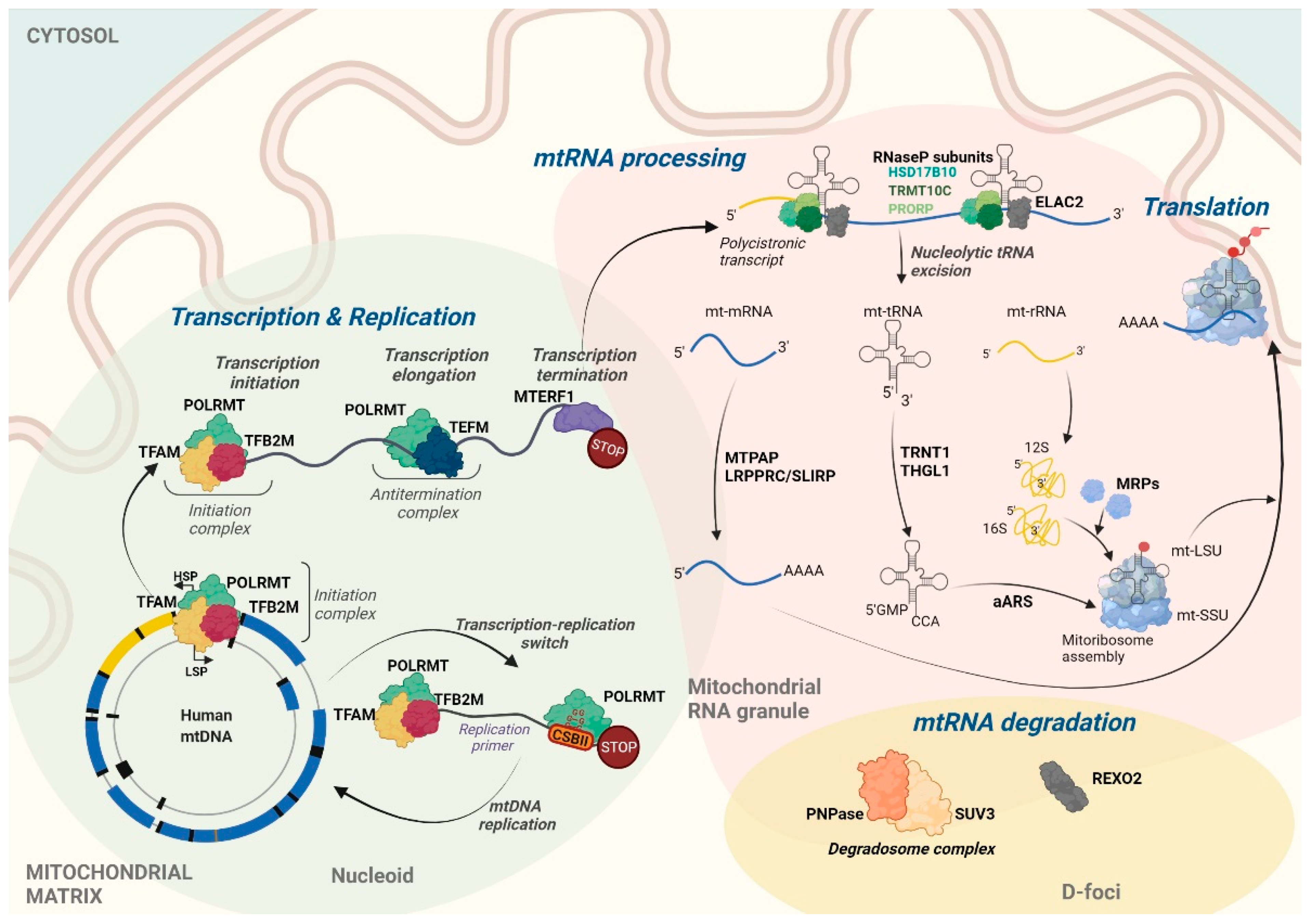
2. Transcription of the Human Mitochondrial Genome
3. Maturation and Post-transcriptional Processing of RNA in Human Mitochondria
3.1. Processing of Primary Polycistronic Transcripts
3.2. Polyadenylation and Aminoacetylation
3.3. Post-transcriptional Chemical Modifications of mRNAs
3.4. Post-transcriptional Chemical Modifications of tRNAs
3.5. Post-transcriptional Chemical Modifications of rRNAs

{kind=link}
{kind=link}
{kind=link}
| Transcript | Position | Modification | Modifying Enzyme | Ref. |
|---|---|---|---|---|
| mRNA | ||||
| MT-CO1 | 1472 | m1A | TRMT6/61A, TRMT61B | [66,67] |
| 391 | Ψ | RPUSD3, TRUB2 | [68] | |
| MT-CO2 | 297 | m1A | TRMT6/61A, TRMT61B | [66,67] |
| MT-CO3 | 707 | m1A | TRMT6/61A, TRMT61B | [66,67] |
| 698–700 | Ψ | RPUSD3, TRUB2 | [68] | |
| MT-ND5 | 1374 | m1A | TRMT10C | [66] |
| tRNA | ||||
| Asn, Arg, Asp, Ala, His, Gly Glu, Leu(CUN), Lys, Pro, Phe, Val, Trp, Thr | 9 | m1A | MRPP1/MRPP2 | [69] |
| Gln, Cys, Ile, Leu(UUR), Tyr | m1G | MRPP1/MRPP2 | [69] | |
| Ala, Asp, Glu, Phe, Gly, His, Lys, Leu(UUR), Leu(CUN), Tyr, Trp, Val, Asn, Thr, Val | 10 | m2G | Unknown | [70] |
| Arg | 16 | m1A | Predicted: TRMT61B | [70] |
| Leu(UUR), Asn, Gln | 20 | D | Predicted: DUS2 | [70,103] |
| Ala, Glu, Arg | 26 | m2G | Unknown | [70] |
| Ile | m22G | Unknown | [70] | |
| Asn, Asp, Cys, His, Ile, Leu(UUR), Leu(CUN), Met, Pro, Val, Ser(UCN), Tyr, Lys | 27 | Ψ | PUS1 | [70,87] |
| Asn, Cys, Ala, Leu(CUN), Ser(UCN), Lys, Glu, Tyr, Phe, Gly, Ile | 28 | Ψ | PUS1 | [70,87] |
| Leu(CUN) | 31 | Ψ | Unknown | [70] |
| Cys, Pro, Arg | 32 | Ψ | Unknown | [70] |
| Ser(UCN), Thr | m3C | Predicted METTL2A, METTL2B, METTL6, or METTL8 | [70] | |
| Gln | 33 | Ψ | Unknown | [70] |
| Lys, Glu, Gln, Leu(UUR), Trp | wobble position 34 | τm5U | GTPBP3, MTO1 | [74] |
| Lys, Glu, Gln | τm5s2U | GTPBP3, MTO1, MTU1, NFS1 | [74,76] | |
| Met | f5C | NSUN3, ALKBH1 | [71,72] | |
| Asp, His, Asn, and Tyr | Q | QTRT1, QTRT2 | [70] | |
| His | 35 | Ψ | Predicted: PUS7 | [70] |
| Phe, Ser(UCN), Trp, Tyr, Cys | 37 | i6A | TRIT1 | [70,77] |
| Phe, Ser(UCN), Trp, Tyr | ms2i6A | TRIT1, CDK5RAP1 | [79] | |
| Gln, Leu(CUN), Pro, Ala | m1G | TRMT5 | [78] | |
| Ser(AGY), Thr, Asn, Ile, Lys | t6A | YRDC, OSGEPL1 | [70,80,81] | |
| Ala, Pro | 38 | Ψ | Predicted: PUS3 | [70] |
| Ala, Pro, Cys, Gly, His, Gln, Arg, Val, Tyr | 39 | Ψ | Unknown | [70] |
| Phe | RPUSD4 | [88] | ||
| Glu, Gly Asn, Gln | 40 | Ψ | Unknown | [70] |
| Phe, His Leu(UUR), Ser(AGY), Tyr | 48 | m5C | NSUN2 | [70,82,83] |
| Glu, Ser(AGY) | 49 | m5C | NSUN2 | [70,82,83] |
| Ser(AGY) | 50 | m5C | NSUN2 | [70,82,83] |
| Met | Ψ | Unknown | [70] | |
| Pro, Asn, Leu(UUR), Ser(UCN), Gln | 54 | m5U | TRMT2B | [84,102] |
| Glu, Met, Leu(UUR), Ser(UCN), Asn, Pro, Gln | 55 | Ψ | Predicted: TRUB2 | [70,104] |
| Leu(UUR), Lys, Ser(UCN) Cys, Glu, Ile | 58 | m1A | TRMT61B | [85] |
| Pro | 66 | Ψ | Predicted: PUS1 | [70] |
| Pro | 67 | Ψ | PUS1 | [70] |
| Ala | 68 | Ψ | Predicted: PUS1 | [70] |
| rRNA | ||||
| 12S | 429 | m5U | TRMT2B | [102] |
| 839 | m4C | METTL15 | [100] | |
| 841 | m5C | NSUN4 | [99] | |
| 936 | m62A | TFB1M | [95,96,97] | |
| 937 | m62A | TFB1M | [95,96,97] | |
| 16S | 947 | m1A | TRMT61B | [91] |
| 1145 | Gm | MRM1 | [93] | |
| 1369 | Um | MRM2 | [92] | |
| 1370 | Gm | MRM3 | [92] | |
| 1397 | Ψ | RPUSD4 | [68] | |
4. Mitochondrial RNA Surveillance and Decay
5. Unresolved Issues
5.1. Processing of Non-Canonical Sites in Primary Polycistronic Transcripts
5.2. Post-Transcriptional RNA Uridylation in Human Mitochondria
5.3. Mitochondrial RNA Editing
5.4. Links between mtDNA Transcription and Mitoribosome Assembly
5.5. Regulation of Mitochondrial Gene Expression
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| aARS | aminoacyl-tRNA synthetases |
| ATP6 | ATP synthase membrane subunit 6 |
| ATP8 | ATP synthase membrane subunit 8 |
| CO1 | cytochrome c oxidase subunit 1 |
| CO2 | cytochrome c oxidase subunit 2 |
| CO3 | cytochrome c oxidase subunit 3 |
| CSBII | G-rich sequence block II |
| CYB | cytochrome b |
| ELAC2 | zinc phosphodiesterase ELAC protein 2 |
| FASTK | Fas-activated serine/threonine kinase |
| G4 | G-quadruplex |
| GRSF1 | G-rich sequence factor 1 |
| HSD17B10 | hydroxysteroid 17-b dehydrogenase 10 |
| HSP | H-strand promoter |
| LRPPRC | leucine-rich pentatricopeptide repeat-containing protein |
| LSP | L-strand promoter |
| MRG | mitochondrial RNA granules |
| MRPL12 | mitochondrial ribosomal protein L7/L12 |
| MRPs | mitoribosomal proteins |
| MTERF1 | mitochondrial transcription termination factor 1 |
| mt-LSU | mitochondrial ribosome large subunit |
| MTPAP | mitochondrial poly(A) polymerase |
| MTRES1 | mitochondrial transcription rescue factor 1 |
| MTS | mitochondrial targeting sequence |
| mtSSB (SSBP1) | single-stranded DNA-binding protein |
| mt-SSU | mitochondrial ribosome small subunit |
| NCR | non-coding regulatory region |
| ND1 | NADH-ubiquinone oxidoreductase chain 1 |
| ND2 | NADH-ubiquinone oxidoreductase chain 2 |
| ND3 | NADH-ubiquinone oxidoreductase chain 3 |
| ND4 | NADH-ubiquinone oxidoreductase chain 4 |
| ND4L | NADH-ubiquinone oxidoreductase chain 4L |
| ND5 | NADH-ubiquinone oxidoreductase chain 5 |
| ND6 | NADH-ubiquinone oxidoreductase chain 6 |
| OXPHOS | oxidative phosphorylation system |
| PDE12 | 3′ exonuclease phosphodiesterase 12 |
| PNPase (PNPT1) | polynucleotide phosphorylase |
| POLG | polymerase γ |
| POLRMT | mitochondrial RNA polymerase |
| PPR | pentatricopeptide repeat |
| PRORP | protein-only RNase P catalytic subunit |
| RAP | RNA-binding domain abundant in apicomplexans |
| RBP | RNA-binding protein |
| REXO2 | RNA exonuclease 2 |
| RNase P | endoribonuclease P |
| RRM | RNA recognition motif |
| SIRT1 | protein deacetylase sirtuin-1 |
| SLIRP | stem-loop-interacting RNA-binding protein |
| SUV3 (SUPV3L1) | ATP-dependent RNA helicase SUPV3L1 |
| TEFM | mitochondrial transcription elongation factor |
| TFAM | mitochondrial transcription factor A |
| TFB2M | mitochondrial transcription factor B2 |
| TRMT10C | tRNA methyltransferase 10C |
References
- Robberson, D.L.; Clayton, D.A. Replication of Mitochondrial DNA in Mouse L Cells and Their Thymidine Kinase- Derivatives: Displacement Replication on a Covalently-Closed Circular Template. Proc. Natl. Acad. Sci. USA 1972, 69, 3810–3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.L.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Istiaq Alam, T.; Kanki, T.; Muta, T.; Ukaji, K.; Abe, Y.; Nakayama, H.; Takio, K.; Hamasaki, N.; Kang, D. Human mitochondrial DNA is packaged with TFAM. Nucleic Acids Res. 2003, 31, 1640–1645. [Google Scholar] [CrossRef]
- Garrido, N.; Griparic, L.; Jokitalo, E.; Wartiovaara, J.; van der Bliek, A.M.; Spelbrink, J.N. Composition and dynamics of human mitochondrial nucleoids. Mol. Biol. Cell 2003, 14, 1583–1596. [Google Scholar] [CrossRef] [Green Version]
- Bogenhagen, D.F.; Wang, Y.; Shen, E.L.; Kobayashi, R. Protein components of mitochondrial DNA nucleoids in higher eukaryotes. Mol. Cell. Proteom. 2003, 2, 1205–1216. [Google Scholar] [CrossRef] [Green Version]
- Bogenhagen, D.F. Mitochondrial DNA nucleoid structure. Biochim. Biophys. Acta 2012, 1819, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Antonicka, H.; Shoubridge, E.A. Mitochondrial RNA Granules Are Centers for Posttranscriptional RNA Processing and Ribosome Biogenesis. Cell Rep. 2015, 10, 920–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jourdain, A.A.; Boehm, E.; Maundrell, K.; Martinou, J.-C. Mitochondrial RNA granules: Compartmentalizing mitochondrial gene expression. J. Cell Biol. 2016, 212, 611–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hensen, F.; Potter, A.; van Esveld, S.L.; Tarrés-Solé, A.; Chakraborty, A.; Solà, M.; Spelbrink, J.N. Mitochondrial RNA granules are critically dependent on mtDNA replication factors Twinkle and mtSSB. Nucleic Acids Res. 2019, 47, 3680–3698. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, B.A.; Durisic, N.; Mativetsky, J.M.; Costantino, S.; Hancock, M.A.; Grutter, P.; Shoubridge, E.A. The Mitochondrial Transcription Factor TFAM Coordinates the Assembly of Multiple DNA Molecules into Nucleoid-like Structures. Mol. Biol. Cell 2007, 18, 3225–3236. [Google Scholar] [CrossRef] [PubMed]
- Kukat, C.; Davies, K.M.; Wurm, C.A.; Spåhr, H.; Bonekamp, N.A.; Kühl, I.; Joos, F.; Polosa, P.L.; Park, C.B.; Posse, V.; et al. Cross-strand binding of TFAM to a single mtDNA molecule forms the mitochondrial nucleoid. Proc. Natl. Acad. Sci. USA 2015, 112, 11288–11293. [Google Scholar] [CrossRef] [Green Version]
- Ježek, P.; Špaček, T.; Tauber, J.; Pavluch, V. Mitochondrial Nucleoids: Superresolution microscopy analysis. Int. J. Biochem. Cell Biol. 2019, 106, 21–25. [Google Scholar] [CrossRef]
- Jourdain, A.A.; Koppen, M.; Wydro, M.; Rodley, C.D.; Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M.; Martinou, J.-C. GRSF1 Regulates RNA Processing in Mitochondrial RNA Granules. Cell Metab. 2013, 17, 399–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, Y.-T.; Barrientos, A. The Human Mitochondrial DEAD-Box Protein DDX28 Resides in RNA Granules and Functions in Mitoribosome Assembly. Cell Rep. 2015, 10, 854–864. [Google Scholar] [CrossRef] [Green Version]
- Litonin, D.; Sologub, M.; Shi, Y.; Savkina, M.; Anikin, M.; Falkenberg, M.; Gustafsson, C.M.; Temiakov, D. Human mitochondrial transcription revisited: Only TFAM and TFB2M are required for transcription of the mitochondrial genes in vitro. J. Biol. Chem. 2010, 285, 18129–18133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minczuk, M.; He, J.; Duch, A.M.; Ettema, T.J.; Chlebowski, A.; Dzionek, K.; Nijtmans, L.G.J.; Huynen, M.A.; Holt, I.J. TEFM (c17orf42) is necessary for transcription of human mtDNA. Nucleic Acids Res. 2011, 39, 4284–4299. [Google Scholar] [CrossRef] [Green Version]
- Hillen, H.S.; Temiakov, D.; Cramer, P. Structural basis of mitochondrial transcription. Nat. Struct. Mol. Biol. 2018, 25, 754–765. [Google Scholar] [CrossRef] [PubMed]
- Hillen, H.S.; Morozov, Y.I.; Sarfallah, A.; Temiakov, D.; Cramer, P. Structural Basis of Mitochondrial Transcription Initiation. Cell 2017, 171, 1072–1081.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yakubovskaya, E.; Guja, K.E.; Eng, E.T.; Choi, W.S.; Mejia, E.; Beglov, D.; Lukin, M.; Kozakov, D.; Garcia-Diaz, M. Organization of the human mitochondrial transcription initiation complex. Nucleic Acids Res. 2014, 42, 4100–4112. [Google Scholar] [CrossRef]
- Morozov, Y.I.; Parshin, A.V.; Agaronyan, K.; Cheung, A.C.M.; Anikin, M.; Cramer, P.; Temiakov, D. A model for transcription initiation in human mitochondria. Nucleic Acids Res. 2015, 43, 3726–3735. [Google Scholar] [CrossRef] [Green Version]
- Ringel, R.; Sologub, M.; Morozov, Y.I.; Litonin, D.; Cramer, P.; Temiakov, D. Structure of human mitochondrial RNA polymerase. Nature 2011, 478, 269–273. [Google Scholar] [CrossRef] [Green Version]
- Cermakian, N.; Ikeda, T.M.; Miramontes, P.; Lang, B.F.; Gray, M.W.; Cedergren, R. On the evolution of the single-subunit RNA polymerases. J. Mol. Evol. 1997, 45, 671–681. [Google Scholar] [CrossRef] [Green Version]
- Posse, V.; Gustafsson, C.M. Human Mitochondrial Transcription Factor B2 Is Required for Promoter Melting during Initiation of Transcription. J. Biol. Chem. 2017, 292, 2637–2645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Cotney, J.; Shadel, G.S. Human mitochondrial ribosomal protein MRPL12 interacts directly with mitochondrial RNA polymerase to modulate mitochondrial gene expression. J. Biol. Chem. 2007, 282, 12610–12618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surovtseva, Y.V.; Shutt, T.E.; Cotney, J.; Cimen, H.; Chen, S.Y.; Koc, E.C.; Shadel, G.S. Mitochondrial Ribosomal Protein L12 selectively associates with human mitochondrial RNA polymerase to activate transcription. Proc. Natl. Acad. Sci. USA 2011, 108, 17921–17926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotrys, A.V.; Cysewski, D.; Czarnomska, S.D.; Pietras, Z.; Borowski, L.S.; Dziembowski, A.; Szczesny, R.J. Quantitative proteomics revealed C6orf203/MTRES1 as a factor preventing stress-induced transcription deficiency in human mitochondria. Nucleic Acids Res. 2019, 47, 7502–7517. [Google Scholar] [CrossRef] [PubMed]
- Agaronyan, K.; Morozov, Y.I.; Anikin, M.; Temiakov, D. Mitochondrial biology. Replication-transcription switch in human mitochondria. Science 2015, 347, 548–551. [Google Scholar] [CrossRef] [Green Version]
- Chang, D.D.; Clayton, D.A. Priming of human mitochondrial DNA replication occurs at the light-strand promoter. Proc. Natl. Acad. Sci. USA 1985, 82, 351–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanrooij, P.H.; Uhler, J.P.; Simonsson, T.; Falkenberg, M.; Gustafsson, C.M. G-quadruplex structures in RNA stimulate mitochondrial transcription termination and primer formation. Proc. Natl. Acad. Sci. USA 2010, 107, 16072–16077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanrooij, P.H.; Uhler, J.P.; Shi, Y.; Westerlund, F.; Falkenberg, M.; Gustafsson, C.M. A hybrid G-quadruplex structure formed between RNA and DNA explains the extraordinary stability of the mitochondrial R-loop. Nucleic Acids Res. 2012, 40, 10334–10344. [Google Scholar] [CrossRef] [Green Version]
- Pham, X.H.; Farge, G.; Shi, Y.; Gaspari, M.; Gustafsson, C.M.; Falkenberg, M. Conserved sequence box II directs transcription termination and primer formation in mitochondria. J. Biol. Chem. 2006, 281, 24647–24652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terzioglu, M.; Ruzzenente, B.; Harmel, J.; Mourier, A.; Jemt, E.; López, M.D.; Kukat, C.; Stewart, J.B.; Wibom, R.; Meharg, C.; et al. MTERF1 binds mtDNA to prevent transcriptional interference at the light-strand promoter but is dispensable for rRNA gene transcription regulation. Cell Metab. 2013, 17, 618–626. [Google Scholar] [CrossRef] [Green Version]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Holzmann, J.; Frank, P.; Löffler, E.; Bennett, K.L.; Gerner, C.; Rossmanith, W. RNase P without RNA: Identification and Functional Reconstitution of the Human Mitochondrial tRNA Processing Enzyme. Cell 2008, 135, 462–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brzezniak, L.K.; Bijata, M.; Szczesny, R.J.; Stepien, P.P. Involvement of human ELAC2 gene product in 3’ end processing of mitochondrial tRNAs. RNA Biol. 2011, 8, 616–626. [Google Scholar] [CrossRef] [Green Version]
- Jarrous, N.; Reiner, R.; Wesolowski, D.; Mann, H.; Guerrier-Takada, C.; Altman, S. Function and subnuclear distribution of Rpp21, a protein subunit of the human ribonucleoprotein ribonuclease P. RNA 2001, 7, 1153–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossmanith, W. Localization of Human RNase Z Isoforms: Dual Nuclear/Mitochondrial Targeting of the ELAC2 Gene Product by Alternative Translation Initiation. PLoS ONE 2011, 6, e0019152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jourdain, A.A.; Popow, J.; de la Fuente, M.A.; Martinou, J.-C.; Anderson, P.; Simarro, M. The FASTK family of proteins: Emerging regulators of mitochondrial RNA biology. Nucleic Acids Res. 2017, 45, 10941–10947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehm, E.; Zaganelli, S.; Maundrell, K.; Jourdain, A.A.; Thore, S.; Martinou, J.-C. FASTKD1 and FASTKD4 have opposite effects on expression of specific mitochondrial RNAs, depending upon their endonuclease-like RAP domain. Nucleic Acids Res. 2017, 45, 6135–6146. [Google Scholar] [CrossRef]
- Jourdain, A.A.; Koppen, M.; Rodley, C.D.; Maundrell, K.; Gueguen, N.; Reynier, P.; Guaras, A.M.; Enriquez, J.A.; Anderson, P.; Simarro, M.; et al. A Mitochondria-Specific Isoform of FASTK Is Present In Mitochondrial RNA Granules and Regulates Gene Expression and Function. Cell Rep. 2015, 10, 1110–1121. [Google Scholar] [CrossRef] [Green Version]
- Popow, J.; Alleaume, A.-M.; Curk, T.; Schwarzl, T.; Sauer, S.; Hentze, M.W. FASTKD2 is an RNA-binding protein required for mitochondrial RNA processing and translation. RNA 2015, 21, 1873–1884. [Google Scholar] [CrossRef] [Green Version]
- Ghezzi, D.; Saada, A.; D’Adamo, P.; Fernandez-Vizarra, E.; Gasparini, P.; Tiranti, V.; Elpeleg, O.; Zeviani, M. FASTKD2 Nonsense Mutation in an Infantile Mitochondrial Encephalomyopathy Associated with Cytochrome C Oxidase Deficiency. Am. J. Hum. Genet. 2008, 83, 415–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, D.H.; Choi, Y.-C.; Nam, D.E.; Choi, S.S.; Kim, J.W.; Choi, B.-O.; Chung, K.W. Identification of FASTKD2 compound heterozygous mutations as the underlying cause of autosomal recessive MELAS-like syndrome. Mitochondrion 2017, 35, 54–58. [Google Scholar] [CrossRef]
- Boehm, E.; Zornoza, M.; Jourdain, A.A.; Delmiro Magdalena, A.; García-Consuegra, I.; Torres Merino, R.; Orduña, A.; Martín, M.A.; Martinou, J.-C.; De la Fuente, M.A.; et al. Role of FAST Kinase Domains 3 (FASTKD3) in Post-transcriptional Regulation of Mitochondrial Gene Expression. J. Biol. Chem. 2016, 291, 25877–25887. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.; Sripada, L.; Lipatova, A.; Roy, M.; Prajapati, P.; Gohel, D.; Bhatelia, K.; Chumakov, P.M.; Singh, R. NLRX1 resides in mitochondrial RNA granules and regulates mitochondrial RNA processing and bioenergetic adaptation. Biochim. Biophys. Acta BBA Mol. Cell Res. 2018, 1865, 1260–1276. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Neph, S.; Dinger, M.E.; Crawford, J.; Smith, M.A.; Shearwood, A.-M.J.; Haugen, E.; Bracken, C.P.; Rackham, O.; Stamatoyannopoulos, J.A.; et al. The human mitochondrial transcriptome. Cell 2011, 146, 645–658. [Google Scholar] [CrossRef] [Green Version]
- Tomecki, R.; Dmochowska, A.; Gewartowski, K.; Dziembowski, A.; Stepien, P.P. Identification of a novel human nuclear-encoded mitochondrial poly(A) polymerase. Nucleic Acids Res. 2004, 32, 6001–6014. [Google Scholar] [CrossRef] [Green Version]
- Nagaike, T.; Suzuki, T.; Katoh, T.; Ueda, T. Human Mitochondrial mRNAs Are Stabilized with Polyadenylation Regulated by Mitochondria-specific Poly(A) Polymerase and Polynucleotide Phosphorylase. J. Biol. Chem. 2005, 280, 19721–19727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, W.C.; Hornig-Do, H.-T.; Bruni, F.; Chang, J.H.; Jourdain, A.A.; Martinou, J.-C.; Falkenberg, M.; Spåhr, H.; Larsson, N.-G.; Lewis, R.J.; et al. A human mitochondrial poly(A) polymerase mutation reveals the complexities of post-transcriptional mitochondrial gene expression. Hum. Mol. Genet. 2014, 23, 6345–6355. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.H.; Tong, L. Mitochondrial poly(A) polymerase and polyadenylation. Biochim. Biophys. Acta 2012, 1819, 992–997. [Google Scholar] [CrossRef] [Green Version]
- Fiedler, M.; Rossmanith, W.; Wahle, E.; Rammelt, C. Mitochondrial poly(A) polymerase is involved in tRNA repair. Nucleic Acids Res. 2015, 43, 9937–9949. [Google Scholar] [CrossRef] [Green Version]
- Wende, S.; Bonin, S.; Götze, O.; Betat, H.; Mörl, M. The identity of the discriminator base has an impact on CCA addition. Nucleic Acids Res. 2015, 43, 5617–5629. [Google Scholar] [CrossRef] [Green Version]
- Pearce, S.F.; Rorbach, J.; Haute, L.V.; D’Souza, A.R.; Rebelo-Guiomar, P.; Powell, C.A.; Brierley, I.; Firth, A.E.; Minczuk, M. Maturation of selected human mitochondrial tRNAs requires deadenylation. eLife 2021, 6. [Google Scholar] [CrossRef] [Green Version]
- Toompuu, M.; Tuomela, T.; Laine, P.; Paulin, L.; Dufour, E.; Jacobs, H.T. Polyadenylation and degradation of structurally abnormal mitochondrial tRNAs in human cells. Nucleic Acids Res. 2018, 46, 5209–5226. [Google Scholar] [CrossRef] [Green Version]
- Slomovic, S.; Laufer, D.; Geiger, D.; Schuster, G. Polyadenylation and Degradation of Human Mitochondrial RNA: The Prokaryotic Past Leaves Its Mark. Mol. Cell. Biol. 2005, 25, 6427–6435. [Google Scholar] [CrossRef] [Green Version]
- Szczesny, R.J.; Borowski, L.S.; Brzezniak, L.K.; Dmochowska, A.; Gewartowski, K.; Bartnik, E.; Stepien, P.P. Human mitochondrial RNA turnover caught in flagranti: Involvement of hSuv3p helicase in RNA surveillance. Nucleic Acids Res. 2010, 38, 279–298. [Google Scholar] [CrossRef]
- Hajnsdorf, E.; Kaberdin, V.R. RNA polyadenylation and its consequences in prokaryotes. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373. [Google Scholar] [CrossRef] [Green Version]
- Tudek, A.; Lloret-Llinares, M.; Jensen, T.H. The multitasking polyA tail: Nuclear RNA maturation, degradation and export. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bratic, A.; Clemente, P.; Calvo-Garrido, J.; Maffezzini, C.; Felser, A.; Wibom, R.; Wedell, A.; Freyer, C.; Wredenberg, A. Mitochondrial Polyadenylation Is a One-Step Process Required for mRNA Integrity and tRNA Maturation. PLoS Genet. 2016, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Temperley, R.J.; Wydro, M.; Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M. Human mitochondrial mRNAs—like members of all families, similar but different. Biochim. Biophys. Acta 2010, 1797, 1081–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wydro, M.; Bobrowicz, A.; Temperley, R.J.; Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M. Targeting of the cytosolic poly(A) binding protein PABPC1 to mitochondria causes mitochondrial translation inhibition. Nucleic Acids Res. 2010, 38, 3732–3742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudge, S.J.; Williams, J.H.; Eyre, H.J.; Sutherland, G.R.; Cowan, P.J.; Power, D.A. Complex organisation of the 5′-end of the human glycine tRNA synthetase gene. Gene 1998, 209, 45–50. [Google Scholar] [CrossRef]
- Tolkunova, E.; Park, H.; Xia, J.; King, M.P.; Davidson, E. The human lysyl-tRNA synthetase gene encodes both the cytoplasmic and mitochondrial enzymes by means of an unusual alternative splicing of the primary transcript. J. Biol. Chem. 2000, 275, 35063–35069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sissler, M.; González-Serrano, L.E.; Westhof, E. Recent Advances in Mitochondrial Aminoacyl-tRNA Synthetases and Disease. Trends Mol. Med. 2017, 23, 693–708. [Google Scholar] [CrossRef] [Green Version]
- Nagao, A.; Suzuki, T.; Katoh, T.; Sakaguchi, Y.; Suzuki, T. Biogenesis of glutaminyl-mt tRNAGln in human mitochondria. Proc. Natl. Acad. Sci. USA 2009, 106, 16209–16214. [Google Scholar] [CrossRef] [Green Version]
- Safra, M.; Sas-Chen, A.; Nir, R.; Winkler, R.; Nachshon, A.; Bar-Yaacov, D.; Erlacher, M.; Rossmanith, W.; Stern-Ginossar, N.; Schwartz, S. The m 1 A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nature 2017, 551, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xiong, X.; Zhang, M.; Wang, K.; Chen, Y.; Zhou, J.; Mao, Y.; Lv, J.; Yi, D.; Chen, X.-W.; et al. Base-resolution mapping reveals distinct m1A methylome in nuclear- and mitochondrial-encoded transcripts. Mol. Cell 2017, 68, 993–1005.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonicka, H.; Choquet, K.; Lin, Z.; Gingras, A.; Kleinman, C.L.; Shoubridge, E.A. A pseudouridine synthase module is essential for mitochondrial protein synthesis and cell viability. EMBO Rep. 2017, 18, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Vilardo, E.; Nachbagauer, C.; Buzet, A.; Taschner, A.; Holzmann, J.; Rossmanith, W. A subcomplex of human mitochondrial RNase P is a bifunctional methyltransferase—Extensive moonlighting in mitochondrial tRNA biogenesis. Nucleic Acids Res. 2012, 40, 11583–11593. [Google Scholar] [CrossRef]
- Suzuki, T.; Yashiro, Y.; Kikuchi, I.; Ishigami, Y.; Saito, H.; Matsuzawa, I.; Okada, S.; Mito, M.; Iwasaki, S.; Ma, D.; et al. Complete chemical structures of human mitochondrial tRNAs. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Nakano, S.; Suzuki, T.; Kawarada, L.; Iwata, H.; Asano, K.; Suzuki, T. NSUN3 methylase initiates 5-formylcytidine biogenesis in human mitochondrial tRNA Met. Nat. Chem. Biol. 2016, 12, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Kawarada, L.; Suzuki, T.; Ohira, T.; Hirata, S.; Miyauchi, K.; Suzuki, T. ALKBH1 is an RNA dioxygenase responsible for cytoplasmic and mitochondrial tRNA modifications. Nucleic Acids Res. 2017, 45, 7401–7415. [Google Scholar] [CrossRef]
- Haag, S.; Sloan, K.E.; Ranjan, N.; Warda, A.S.; Kretschmer, J.; Blessing, C.; Hübner, B.; Seikowski, J.; Dennerlein, S.; Rehling, P.; et al. NSUN3 and ABH1 modify the wobble position of mt-tRNA Met to expand codon recognition in mitochondrial translation. EMBO J. 2016, 35, 2104–2119. [Google Scholar] [CrossRef] [PubMed]
- Umeda, N.; Suzuki, T.; Yukawa, M.; Ohya, Y.; Shindo, H.; Watanabe, K.; Suzuki, T. Mitochondria-specific RNA-modifying Enzymes Responsible for the Biosynthesis of the Wobble Base in Mitochondrial tRNAs: Implications for the Molecular Pathogenesis of Human Mitochondrial Diseases. J. Biol. Chem. 2005, 280, 1613–1624. [Google Scholar] [CrossRef] [Green Version]
- Boutoual, R.; Meseguer, S.; Villarroya, M.; Martín-Hernández, E.; Errami, M.; Martín, M.A.; Casado, M.; Armengod, M.-E. Defects in the mitochondrial-tRNA modification enzymes MTO1 and GTPBP3 promote different metabolic reprogramming through a HIF-PPARγ-UCP2-AMPK axis. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Sasarman, F.; Antonicka, H.; Horvath, R.; Shoubridge, E.A. The 2-thiouridylase function of the human MTU1 (TRMU) enzyme is dispensable for mitochondrial translation. Hum. Mol. Genet. 2011, 20, 4634–4643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarham, J.W.; Lamichhane, T.N.; Pyle, A.; Mattijssen, S.; Baruffini, E.; Bruni, F.; Donnini, C.; Vassilev, A.; He, L.; Blakely, E.L.; et al. Defective i6A37 Modification of Mitochondrial and Cytosolic tRNAs Results from Pathogenic Mutations in TRIT1 and Its Substrate tRNA. PLoS Genet. 2014, 10. [Google Scholar] [CrossRef] [Green Version]
- Powell, C.A.; Kopajtich, R.; D’Souza, A.R.; Rorbach, J.; Kremer, L.S.; Husain, R.A.; Dallabona, C.; Donnini, C.; Alston, C.L.; Griffin, H.; et al. TRMT5 Mutations Cause a Defect in Post-transcriptional Modification of Mitochondrial tRNA Associated with Multiple Respiratory-Chain Deficiencies. Am. J. Hum. Genet. 2015, 97, 319–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiter, V.; Matschkal, D.M.S.; Wagner, M.; Globisch, D.; Kneuttinger, A.C.; Müller, M.; Carell, T. The CDK5 repressor CDK5RAP1 is a methylthiotransferase acting on nuclear and mitochondrial RNA. Nucleic Acids Res. 2012, 40, 6235–6240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.; Miyauchi, K.; Harada, T.; Okita, R.; Takeshita, E.; Komaki, H.; Fujioka, K.; Yagasaki, H.; Goto, Y.; Yanaka, K.; et al. CO2-sensitive tRNA modification associated with human mitochondrial disease. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Zhou, J.-B.; Wang, Y.; Zeng, Q.-Y.; Meng, S.-X.; Wang, E.-D.; Zhou, X.-L. Molecular basis for t6A modification in human mitochondria. Nucleic Acids Res. 2020, 48, 3181–3194. [Google Scholar] [CrossRef] [PubMed]
- Van Haute, L.; Lee, S.-Y.; McCann, B.J.; Powell, C.A.; Bansal, D.; Vasiliauskaitė, L.; Garone, C.; Shin, S.; Kim, J.-S.; Frye, M.; et al. NSUN2 introduces 5-methylcytosines in mammalian mitochondrial tRNAs. Nucleic Acids Res. 2019, 47, 8720–8733. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, S.; Kitagawa, S.; Nakagawa, S.; Wei, F.-Y.; Tomizawa, K.; Araki, K.; Araki, M.; Suzuki, T.; Suzuki, T. Mammalian NSUN2 introduces 5-methylcytidines into mitochondrial tRNAs. Nucleic Acids Res. 2019, 47, 8734–8745. [Google Scholar] [CrossRef] [Green Version]
- Laptev, I.; Shvetsova, E.; Levitskii, S.; Serebryakova, M.; Rubtsova, M.; Bogdanov, A.; Kamenski, P.; Sergiev, P.; Dontsova, O. Mouse Trmt2B protein is a dual specific mitochondrial metyltransferase responsible for m5U formation in both tRNA and rRNA. RNA Biol. 2019, 17, 441–450. [Google Scholar] [CrossRef]
- Chujo, T.; Suzuki, T. Trmt61B is a methyltransferase responsible for 1-methyladenosine at position 58 of human mitochondrial tRNAs. RNA 2012, 18, 2269–2276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, D.R. Stabilization of RNA stacking by pseudouridine. Nucleic Acids Res. 1995, 23, 5020–5026. [Google Scholar] [CrossRef]
- Patton, J.R.; Bykhovskaya, Y.; Mengesha, E.; Bertolotto, C.; Fischel-Ghodsian, N. Mitochondrial Myopathy and Sideroblastic Anemia (MLASA): Missense Mutation in the Pseudouridine Synthase 1 (PUS1) Gene Is Associated with the Loss of tRNA Pseudouridylation. J. Biol. Chem. 2005, 280, 19823–19828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaganelli, S.; Rebelo-Guiomar, P.; Maundrell, K.; Rozanska, A.; Pierredon, S.; Powell, C.A.; Jourdain, A.A.; Hulo, N.; Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M.; et al. The Pseudouridine Synthase RPUSD4 Is an Essential Component of Mitochondrial RNA Granules. J. Biol. Chem. 2017, 292, 4519–4532. [Google Scholar] [CrossRef] [Green Version]
- Nagaike, T.; Suzuki, T.; Tomari, Y.; Takemoto-Hori, C.; Negayama, F.; Watanabe, K.; Ueda, T. Identification and Characterization of Mammalian Mitochondrial tRNA nucleotidyltransferases. J. Biol. Chem. 2001, 276, 40041–40049. [Google Scholar] [CrossRef] [Green Version]
- Hyde, S.J.; Eckenroth, B.E.; Smith, B.A.; Eberley, W.A.; Heintz, N.H.; Jackman, J.E.; Doublié, S. tRNAHis guanylyltransferase (THG1), a unique 3′-5′ nucleotidyl transferase, shares unexpected structural homology with canonical 5′-3′ DNA polymerases. Proc. Natl. Acad. Sci. USA 2010, 107, 20305–20310. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.-W.; Okot-Kotber, C.; LaComb, J.F.; Bogenhagen, D.F. Mitochondrial Ribosomal RNA (rRNA) Methyltransferase Family Members Are Positioned to Modify Nascent rRNA in Foci near the Mitochondrial DNA Nucleoid. J. Biol. Chem. 2013, 288, 31386–31399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rorbach, J.; Boesch, P.; Gammage, P.A.; Nicholls, T.J.J.; Pearce, S.F.; Patel, D.; Hauser, A.; Perocchi, F.; Minczuk, M. MRM2 and MRM3 are involved in biogenesis of the large subunit of the mitochondrial ribosome. Mol. Biol. Cell 2014, 25, 2542–2555. [Google Scholar] [CrossRef]
- Lee, K.-W.; Bogenhagen, D.F. Assignment of 2′-O-Methyltransferases to Modification Sites on the Mammalian Mitochondrial Large Subunit 16 S Ribosomal RNA (rRNA). J. Biol. Chem. 2014, 289, 24936–24942. [Google Scholar] [CrossRef] [Green Version]
- Bar-Yaacov, D.; Frumkin, I.; Yashiro, Y.; Chujo, T.; Ishigami, Y.; Chemla, Y.; Blumberg, A.; Schlesinger, O.; Bieri, P.; Greber, B.; et al. Mitochondrial 16S rRNA Is Methylated by tRNA Methyltransferase TRMT61B in All Vertebrates. PLoS Biol. 2016, 14. [Google Scholar] [CrossRef] [PubMed]
- Seidel-Rogol, B.L.; McCulloch, V.; Shadel, G.S. Human mitochondrial transcription factor B1 methylates ribosomal RNA at a conserved stem-loop. Nat. Genet. 2003, 33, 23–24. [Google Scholar] [CrossRef]
- Metodiev, M.D.; Lesko, N.; Park, C.B.; Cámara, Y.; Shi, Y.; Wibom, R.; Hultenby, K.; Gustafsson, C.M.; Larsson, N.-G. Methylation of 12S rRNA Is Necessary for In Vivo Stability of the Small Subunit of the Mammalian Mitochondrial Ribosome. Cell Metab. 2009, 9, 386–397. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Shen, S.; Wu, P.; Li, F.; Liu, X.; Wang, C.; Gong, Q.; Wu, J.; Yao, X.; Zhang, H.; et al. Structural insights into dimethylation of 12S rRNA by TFB1M: Indispensable role in translation of mitochondrial genes and mitochondrial function. Nucleic Acids Res. 2019, 47, 7648–7665. [Google Scholar] [CrossRef] [Green Version]
- Rozanska, A.; Richter-Dennerlein, R.; Rorbach, J.; Gao, F.; Lewis, R.J.; Chrzanowska-Lightowlers, Z.M.; Lightowlers, R.N. The human RNA-binding protein RBFA promotes the maturation of the mitochondrial ribosome. Biochem. J. 2017, 474, 2145–2158. [Google Scholar] [CrossRef] [Green Version]
- Metodiev, M.D.; Spåhr, H.; Loguercio Polosa, P.; Meharg, C.; Becker, C.; Altmueller, J.; Habermann, B.; Larsson, N.-G.; Ruzzenente, B. NSUN4 Is a Dual Function Mitochondrial Protein Required for Both Methylation of 12S rRNA and Coordination of Mitoribosomal Assembly. PLoS Genet. 2014, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haute, L.V.; Hendrick, A.G.; D’Souza, A.R.; Powell, C.A.; Rebelo-Guiomar, P.; Harbour, M.E.; Ding, S.; Fearnley, I.M.; Andrews, B.; Minczuk, M. METTL15 introduces N4-methylcytidine into human mitochondrial 12S rRNA and is required for mitoribosome biogenesis. Nucleic Acids Res. 2019, 47, 10267–10281. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Shi, Z.; Guo, J.; Chang, K.; Chen, Q.; Yao, C.; Haigis, M.C.; Shi, Y. The human mitochondrial 12S rRNA m4C methyltransferase METTL15 is required for proper mitochondrial function. bioRxiv 2019, 809756. [Google Scholar] [CrossRef] [Green Version]
- Powell, C.A.; Minczuk, M. TRMT2B is responsible for both tRNA and rRNA m5U-methylation in human mitochondria. RNA Biol. 2020, 17, 451–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Suzuki, T. A complete landscape of post-transcriptional modifications in mammalian mitochondrial tRNAs. Nucleic Acids Res. 2014, 42, 7346–7357. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Deogharia, M.; Gupta, R. Mammalian nuclear TRUB1, mitochondrial TRUB2, and cytoplasmic PUS10 produce conserved pseudouridine 55 in different sets of tRNA. RNA 2021, 27, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Siira, S.J.; Spåhr, H.; Shearwood, A.-M.J.; Ruzzenente, B.; Larsson, N.-G.; Rackham, O.; Filipovska, A. LRPPRC-mediated folding of the mitochondrial transcriptome. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruzzenente, B.; Metodiev, M.D.; Wredenberg, A.; Bratic, A.; Park, C.B.; Cámara, Y.; Milenkovic, D.; Zickermann, V.; Wibom, R.; Hultenby, K.; et al. LRPPRC is necessary for polyadenylation and coordination of translation of mitochondrial mRNAs. EMBO J. 2012, 31, 443–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagouge, M.; Mourier, A.; Lee, H.J.; Spåhr, H.; Wai, T.; Kukat, C.; Silva Ramos, E.; Motori, E.; Busch, J.D.; Siira, S.; et al. SLIRP Regulates the Rate of Mitochondrial Protein Synthesis and Protects LRPPRC from Degradation. PLoS Genet. 2015, 11. [Google Scholar] [CrossRef]
- Spåhr, H.; Rozanska, A.; Li, X.; Atanassov, I.; Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M.A.; Rackham, O.; Larsson, N.-G. SLIRP stabilizes LRPPRC via an RRM–PPR protein interface. Nucleic Acids Res. 2016, 44, 6868–6882. [Google Scholar] [CrossRef]
- Aibara, S.; Singh, V.; Modelska, A.; Amunts, A. Structural basis of mitochondrial translation. eLife 2020, 9, e58362. [Google Scholar] [CrossRef]
- Chujo, T.; Ohira, T.; Sakaguchi, Y.; Goshima, N.; Nomura, N.; Nagao, A.; Suzuki, T. LRPPRC/SLIRP suppresses PNPase-mediated mRNA decay and promotes polyadenylation in human mitochondria. Nucleic Acids Res. 2012, 40, 8033–8047. [Google Scholar] [CrossRef] [Green Version]
- Bruni, F.; Proctor-Kent, Y.; Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M. Messenger RNA delivery to mitoribosomes—hints from a bacterial toxin. FEBS J. 2021, 288, 437–451. [Google Scholar] [CrossRef] [Green Version]
- Borowski, L.S.; Dziembowski, A.; Hejnowicz, M.S.; Stepien, P.P.; Szczesny, R.J. Human mitochondrial RNA decay mediated by PNPase–hSuv3 complex takes place in distinct foci. Nucleic Acids Res. 2013, 41, 1223–1240. [Google Scholar] [CrossRef]
- Lin, C.L.; Wang, Y.-T.; Yang, W.-Z.; Hsiao, Y.-Y.; Yuan, H.S. Crystal structure of human polynucleotide phosphorylase: Insights into its domain function in RNA binding and degradation. Nucleic Acids Res. 2012, 40, 4146–4157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietras, Z.; Wojcik, M.A.; Borowski, L.S.; Szewczyk, M.; Kulinski, T.M.; Cysewski, D.; Stepien, P.P.; Dziembowski, A.; Szczesny, R.J. Controlling the mitochondrial antisense—Role of the SUV3-PNPase complex and its co-factor GRSF1 in mitochondrial RNA surveillance. Mol. Cell. Oncol. 2018, 5, e1516452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, S.; Camino, L.P.; Aguilera, A. Human mitochondrial degradosome prevents harmful mitochondrial R loops and mitochondrial genome instability. Proc. Natl. Acad. Sci. USA 2018, 115, 11024–11029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhir, A.; Dhir, S.; Borowski, L.S.; Jimenez, L.; Teitell, M.; Rötig, A.; Crow, Y.J.; Rice, G.I.; Duffy, D.; Tamby, C.; et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 2018, 560, 238–242. [Google Scholar] [CrossRef]
- Minczuk, M.; Piwowarski, J.; Papworth, M.A.; Awiszus, K.; Schalinski, S.; Dziembowski, A.; Dmochowska, A.; Bartnik, E.; Tokatlidis, K.; Stepien, P.P.; et al. Localisation of the human hSuv3p helicase in the mitochondrial matrix and its preferential unwinding of dsDNA. Nucleic Acids Res. 2002, 30, 5074–5086. [Google Scholar] [CrossRef] [Green Version]
- Bruni, F.; Gramegna, P.; Oliveira, J.M.A.; Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M.A. REXO2 Is an Oligoribonuclease Active in Human Mitochondria. PLoS ONE 2013, 8, e0064670. [Google Scholar] [CrossRef] [Green Version]
- Szewczyk, M.; Malik, D.; Borowski, L.S.; Czarnomska, S.D.; Kotrys, A.V.; Klosowska-Kosicka, K.; Nowotny, M.; Szczesny, R.J. Human REXO2 controls short mitochondrial RNAs generated by mtRNA processing and decay machinery to prevent accumulation of double-stranded RNA. Nucleic Acids Res. 2020, 48, 5572–5590. [Google Scholar] [CrossRef]
- Chu, L.-Y.; Agrawal, S.; Chen, Y.-P.; Yang, W.-Z.; Yuan, H.S. Structural insights into nanoRNA degradation by human Rexo2. RNA 2019, 25, 737–746. [Google Scholar] [CrossRef]
- Nicholls, T.J.; Spåhr, H.; Jiang, S.; Siira, S.J.; Koolmeister, C.; Sharma, S.; Kauppila, J.H.K.; Jiang, M.; Kaever, V.; Rackham, O.; et al. Dinucleotide Degradation by REXO2 Maintains Promoter Specificity in Mammalian Mitochondria. Mol. Cell 2019, 76, 784–796.e6. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, C.; Fisette, J.-F.; Chabot, B.; Allain, F.H.-T. Structural basis of G-tract recognition and encaging by hnRNP F quasi-RRMs. Nat. Struct. Mol. Biol. 2010, 17, 853–861. [Google Scholar] [CrossRef] [Green Version]
- Antonicka, H.; Sasarman, F.; Nishimura, T.; Paupe, V.; Shoubridge, E.A. The Mitochondrial RNA-Binding Protein GRSF1 Localizes to RNA Granules and Is Required for Posttranscriptional Mitochondrial Gene Expression. Cell Metab. 2013, 17, 386–398. [Google Scholar] [CrossRef] [Green Version]
- Pietras, Z.; Wojcik, M.A.; Borowski, L.S.; Szewczyk, M.; Kulinski, T.M.; Cysewski, D.; Stepien, P.P.; Dziembowski, A.; Szczesny, R.J. Dedicated surveillance mechanism controls G-quadruplex forming non-coding RNAs in human mitochondria. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Van Haute, L.; Pearce, S.F.; Powell, C.A.; D’Souza, A.R.; Nicholls, T.J.; Minczuk, M. Mitochondrial transcript maturation and its disorders. J. Inherit. Metab. Dis. 2015, 38, 655–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuznetsova, I.; Siira, S.J.; Shearwood, A.-M.J.; Ermer, J.A.; Filipovska, A.; Rackham, O. Simultaneous processing and degradation of mitochondrial RNAs revealed by circularized RNA sequencing. Nucleic Acids Res. 2017, 45, 5487–5500. [Google Scholar] [CrossRef] [Green Version]
- Malecki, M.; Viegas, S.C.; Carneiro, T.; Golik, P.; Dressaire, C.; Ferreira, M.G.; Arraiano, C.M. The exoribonuclease Dis3L2 defines a novel eukaryotic RNA degradation pathway. EMBO J. 2013, 32, 1842–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warkocki, Z.; Liudkovska, V.; Gewartowska, O.; Mroczek, S.; Dziembowski, A. Terminal nucleotidyl transferases (TENTs) in mammalian RNA metabolism. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattiacio, J.L.; Read, L.K. Roles for TbDSS-1 in RNA surveillance and decay of maturation by-products from the 12S rRNA locus. Nucleic Acids Res. 2008, 36, 319–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aphasizheva, I.; Aphasizhev, R. RET1-Catalyzed Uridylylation Shapes the Mitochondrial Transcriptome in Trypanosoma brucei. Mol. Cell. Biol. 2010, 30, 1555–1567. [Google Scholar] [CrossRef] [Green Version]
- Slomovic, S.; Schuster, G. Stable PNPase RNAi silencing: Its effect on the processing and adenylation of human mitochondrial RNA. RNA 2008, 14, 310–323. [Google Scholar] [CrossRef] [Green Version]
- Bazak, L.; Haviv, A.; Barak, M.; Jacob-Hirsch, J.; Deng, P.; Zhang, R.; Isaacs, F.J.; Rechavi, G.; Li, J.B.; Eisenberg, E.; et al. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 2014, 24, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Sloan, D.B. Nuclear and mitochondrial RNA editing systems have opposite effects on protein diversity. Biol. Lett. 2017, 13. [Google Scholar] [CrossRef]
- Bar-Yaacov, D.; Avital, G.; Levin, L.; Richards, A.L.; Hachen, N.; Rebolledo Jaramillo, B.; Nekrutenko, A.; Zarivach, R.; Mishmar, D. RNA-DNA differences in human mitochondria restore ancestral form of 16S ribosomal RNA. Genome Res. 2013, 23, 1789–1796. [Google Scholar] [CrossRef] [Green Version]
- Rackham, O.; Busch, J.D.; Matic, S.; Siira, S.J.; Kuznetsova, I.; Atanassov, I.; Ermer, J.A.; Shearwood, A.-M.J.; Richman, T.R.; Stewart, J.B.; et al. Hierarchical RNA Processing Is Required for Mitochondrial Ribosome Assembly. Cell Rep. 2016, 16, 1874–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, N.; Yang, H.; Chandrasekaran, V.; Kazi, R.; Minczuk, M.; Ramakrishnan, V. Elongational Stalling Activates Mitoribosome-associated Quality Control. Science 2020, 370, 1105–1110. [Google Scholar] [CrossRef]
- Piechota, J.; Tomecki, R.; Gewartowski, K.; Szczesny, R.; Dmochowska, A.; Kudła, M.; Dybczyńska, L.; Stepien, P.P.; Bartnik, E. Differential stability of mitochondrial mRNA in HeLa cells. Acta Biochim. Pol. 2006, 53, 157–168. [Google Scholar] [CrossRef]
- Iborra, F.J.; Kimura, H.; Cook, P.R. The functional organization of mitochondrial genomes in human cells. BMC Biol. 2004, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quax, T.E.F.; Wolf, Y.I.; Koehorst, J.J.; Wurtzel, O.; van der Oost, R.; Ran, W.; Blombach, F.; Makarova, K.S.; Brouns, S.J.J.; Forster, A.C.; et al. Differential Translation Tunes Uneven Production of Operon-Encoded Proteins. Cell Rep. 2013, 4, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.; Tian, S.; Fujii, K.; Kladwang, W.; Das, R.; Barna, M. RNA regulons in Hox 5′UTRs confer ribosome specificity to gene regulation. Nature 2015, 517, 33–38. [Google Scholar] [CrossRef] [Green Version]
- Shi, Z.; Fujii, K.; Kovary, K.M.; Genuth, N.R.; Röst, H.L.; Teruel, M.N.; Barna, M. Heterogeneous ribosomes preferentially translate distinct subpools of mRNAs genome-wide. Mol. Cell 2017, 67, 71–83.e7. [Google Scholar] [CrossRef] [Green Version]
- Naithani, S.; Saracco, S.A.; Butler, C.A.; Fox, T.D. Interactions among COX1, COX2, andCOX3 mRNA-specific Translational Activator Proteins on the Inner Surface of the Mitochondrial Inner Membrane ofSaccharomyces cerevisiae. Mol. Biol. Cell 2002, 14, 324–333. [Google Scholar] [CrossRef] [Green Version]
- Weraarpachai, W.; Antonicka, H.; Sasarman, F.; Seeger, J.; Schrank, B.; Kolesar, J.E.; Lochmüller, H.; Chevrette, M.; Kaufman, B.A.; Horvath, R.; et al. Mutation in TACO1, encoding a translational activator of COX I, results in cytochrome c oxidase deficiency and late-onset Leigh syndrome. Nat. Genet. 2009, 41, 833–837. [Google Scholar] [CrossRef]
- Richman, T.R.; Spåhr, H.; Ermer, J.A.; Davies, S.M.K.; Viola, H.M.; Bates, K.A.; Papadimitriou, J.; Hool, L.C.; Rodger, J.; Larsson, N.-G.; et al. Loss of the RNA-binding protein TACO1 causes late-onset mitochondrial dysfunction in mice. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Kotrys, A.V.; Szczesny, R.J. Mitochondrial Gene Expression and Beyond-Novel Aspects of Cellular Physiology. Cells 2019, 9, 17. [Google Scholar] [CrossRef] [Green Version]
- Bandiera, S.; Rüberg, S.; Girard, M.; Cagnard, N.; Hanein, S.; Chrétien, D.; Munnich, A.; Lyonnet, S.; Henrion-Caude, A. Nuclear outsourcing of RNA interference components to human mitochondria. PLoS ONE 2011, 6, e20746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ro, S.; Ma, H.-Y.; Park, C.; Ortogero, N.; Song, R.; Hennig, G.W.; Zheng, H.; Lin, Y.-M.; Moro, L.; Hsieh, J.-T.; et al. The mitochondrial genome encodes abundant small noncoding RNAs. Cell Res. 2013, 23, 759–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zuo, X.; Yang, B.; Li, Z.; Xue, Y.; Zhou, Y.; Huang, J.; Zhao, X.; Zhou, J.; Yan, Y.; et al. MicroRNA directly enhances mitochondrial translation during muscle differentiation. Cell 2014, 158, 607–619. [Google Scholar] [CrossRef] [Green Version]
- Gao, K.; Cheng, M.; Zuo, X.; Lin, J.; Hoogewijs, K.; Murphy, M.P.; Fu, X.-D.; Zhang, X. Active RNA interference in mitochondria. Cell Res. 2021, 31, 219–228. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jedynak-Slyvka, M.; Jabczynska, A.; Szczesny, R.J. Human Mitochondrial RNA Processing and Modifications: Overview. Int. J. Mol. Sci. 2021, 22, 7999. https://doi.org/10.3390/ijms22157999
Jedynak-Slyvka M, Jabczynska A, Szczesny RJ. Human Mitochondrial RNA Processing and Modifications: Overview. International Journal of Molecular Sciences. 2021; 22(15):7999. https://doi.org/10.3390/ijms22157999
Chicago/Turabian StyleJedynak-Slyvka, Marta, Agata Jabczynska, and Roman J. Szczesny. 2021. "Human Mitochondrial RNA Processing and Modifications: Overview" International Journal of Molecular Sciences 22, no. 15: 7999. https://doi.org/10.3390/ijms22157999
APA StyleJedynak-Slyvka, M., Jabczynska, A., & Szczesny, R. J. (2021). Human Mitochondrial RNA Processing and Modifications: Overview. International Journal of Molecular Sciences, 22(15), 7999. https://doi.org/10.3390/ijms22157999