
Maize DNA Methylation in Response to Drought Stress Is Involved in Target Gene Expression and Alternative Splicing

 and
and
Abstract
:1. Introduction
2. Results
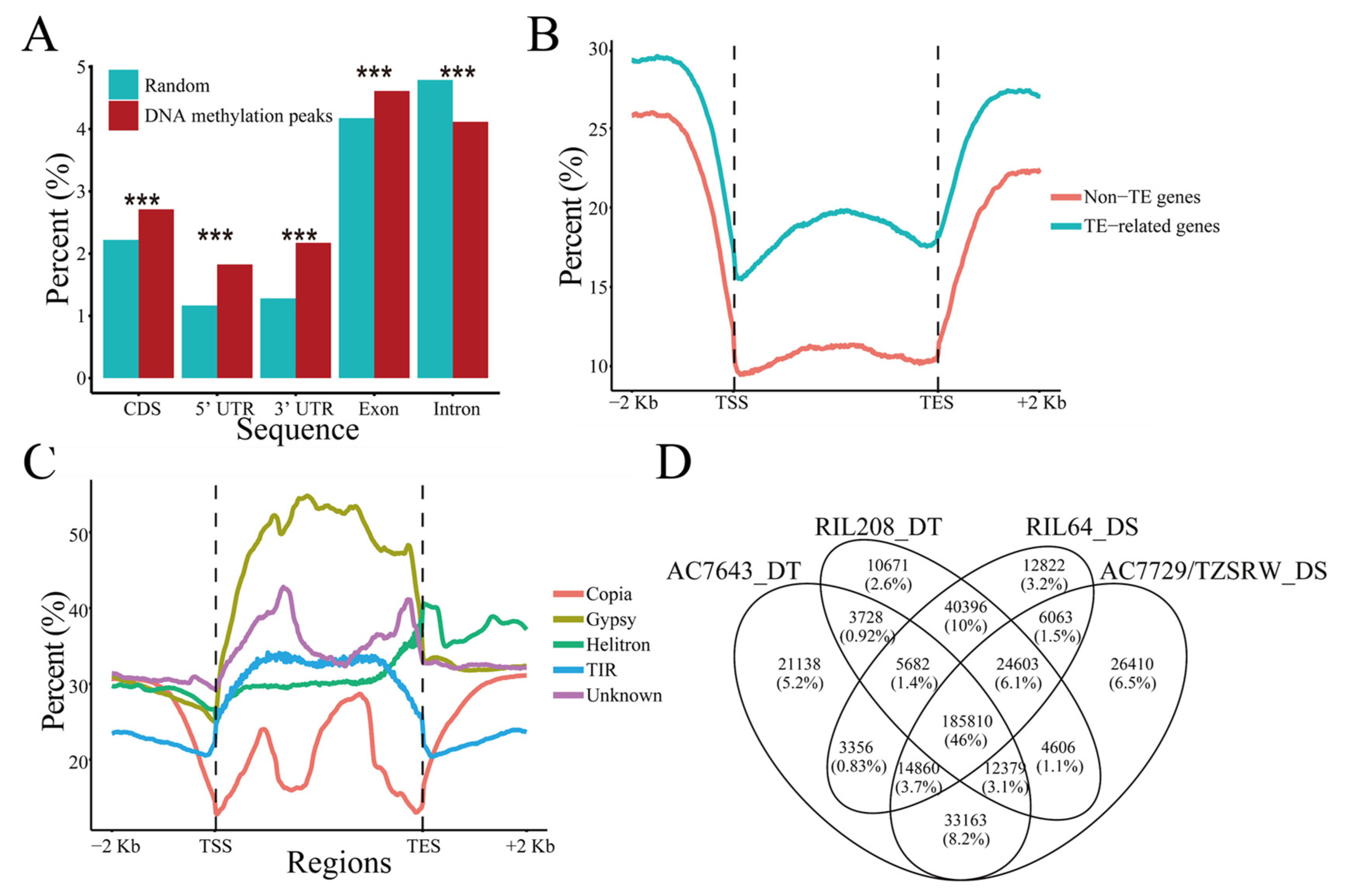
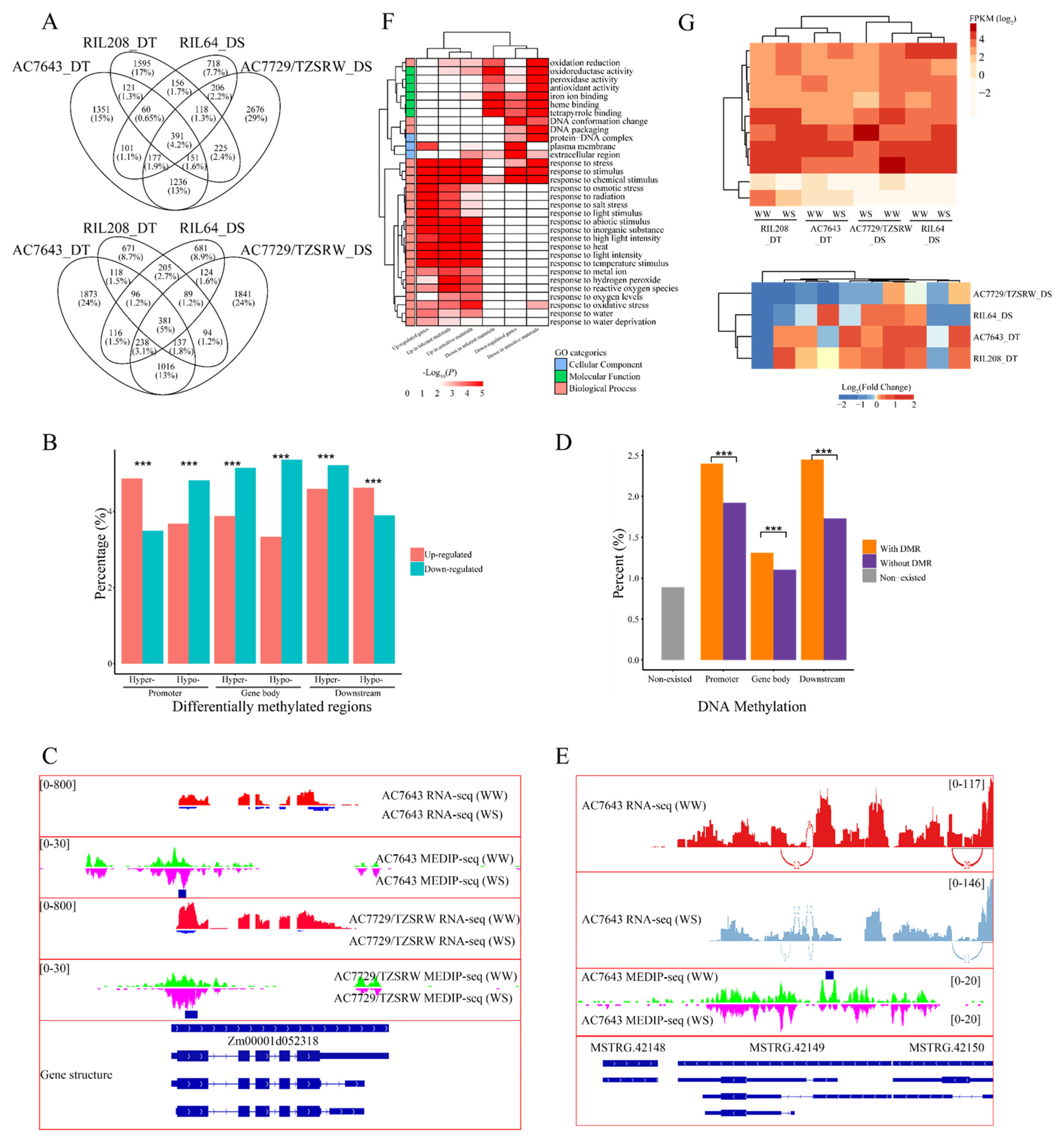
2.1. Genome-Wide Distribution Patterns of DNA Methylation in Maize Roots
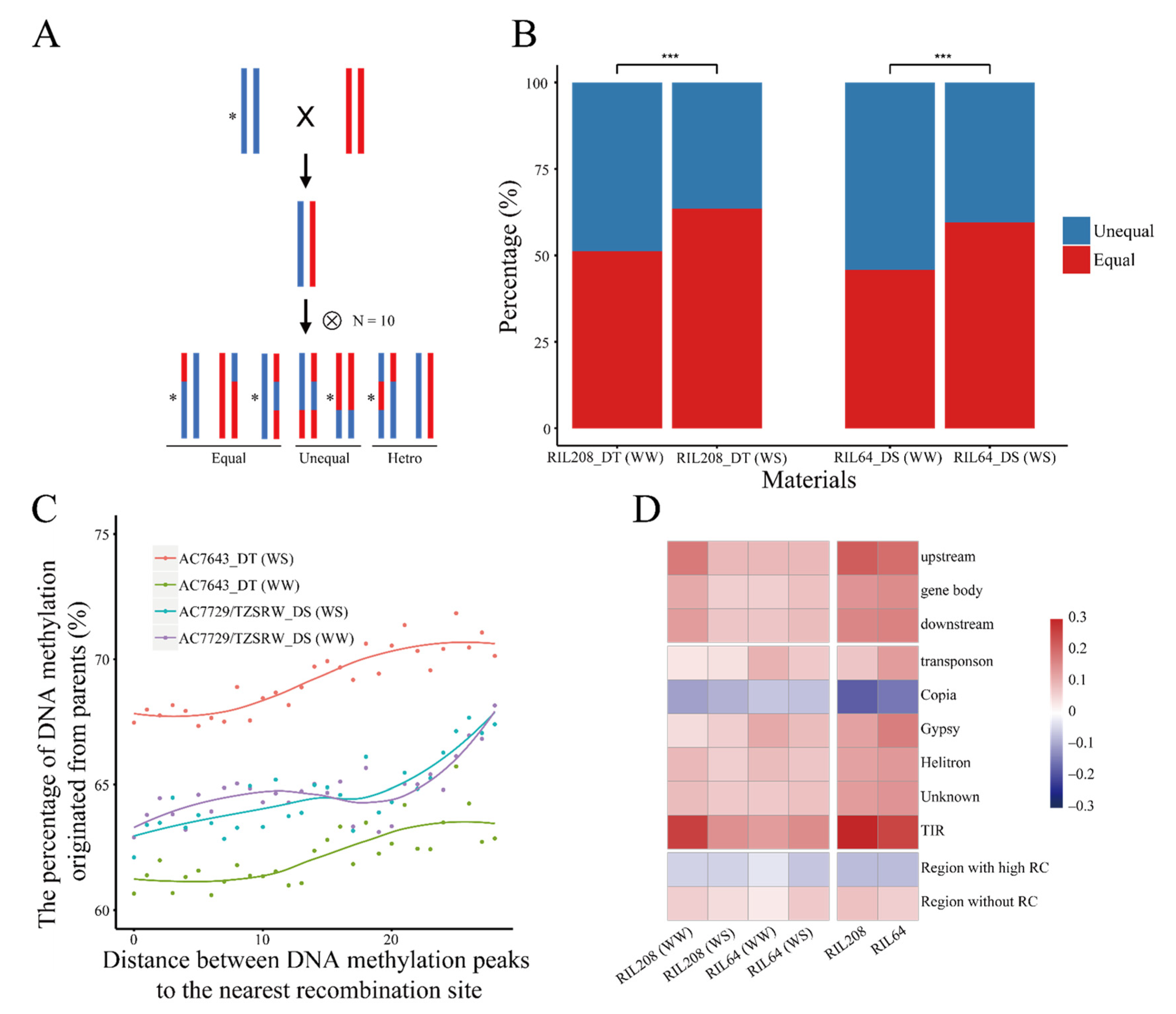
2.2. Inheritance of DNA Methylation Modifications Increased under Drought Stress
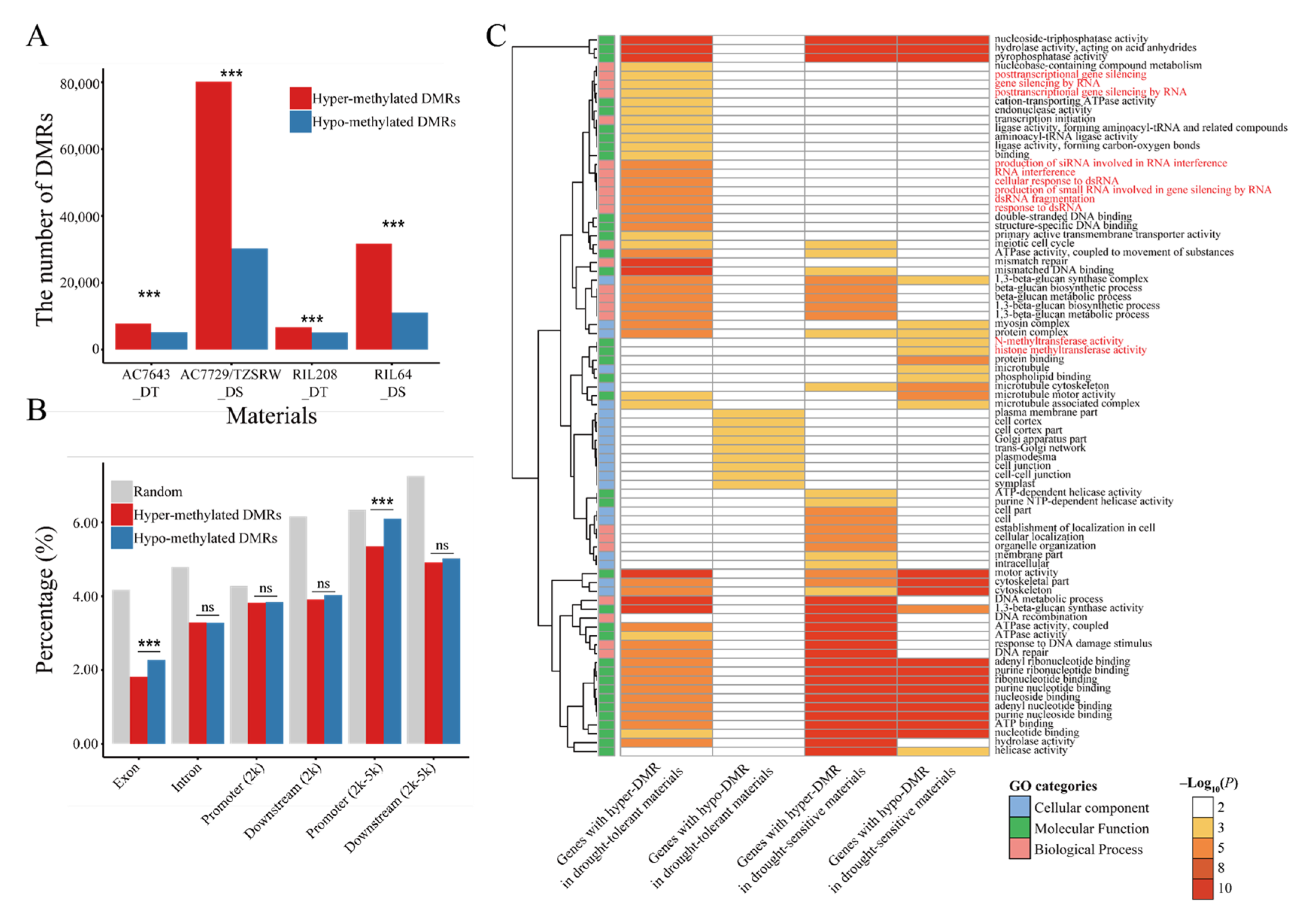
2.3. DNA Methylation Varied in Different Genotypes and Genomic Regions under Drought Stress
2.4. Potential Role of DNA Methylation in Drought Stress Response Regulation
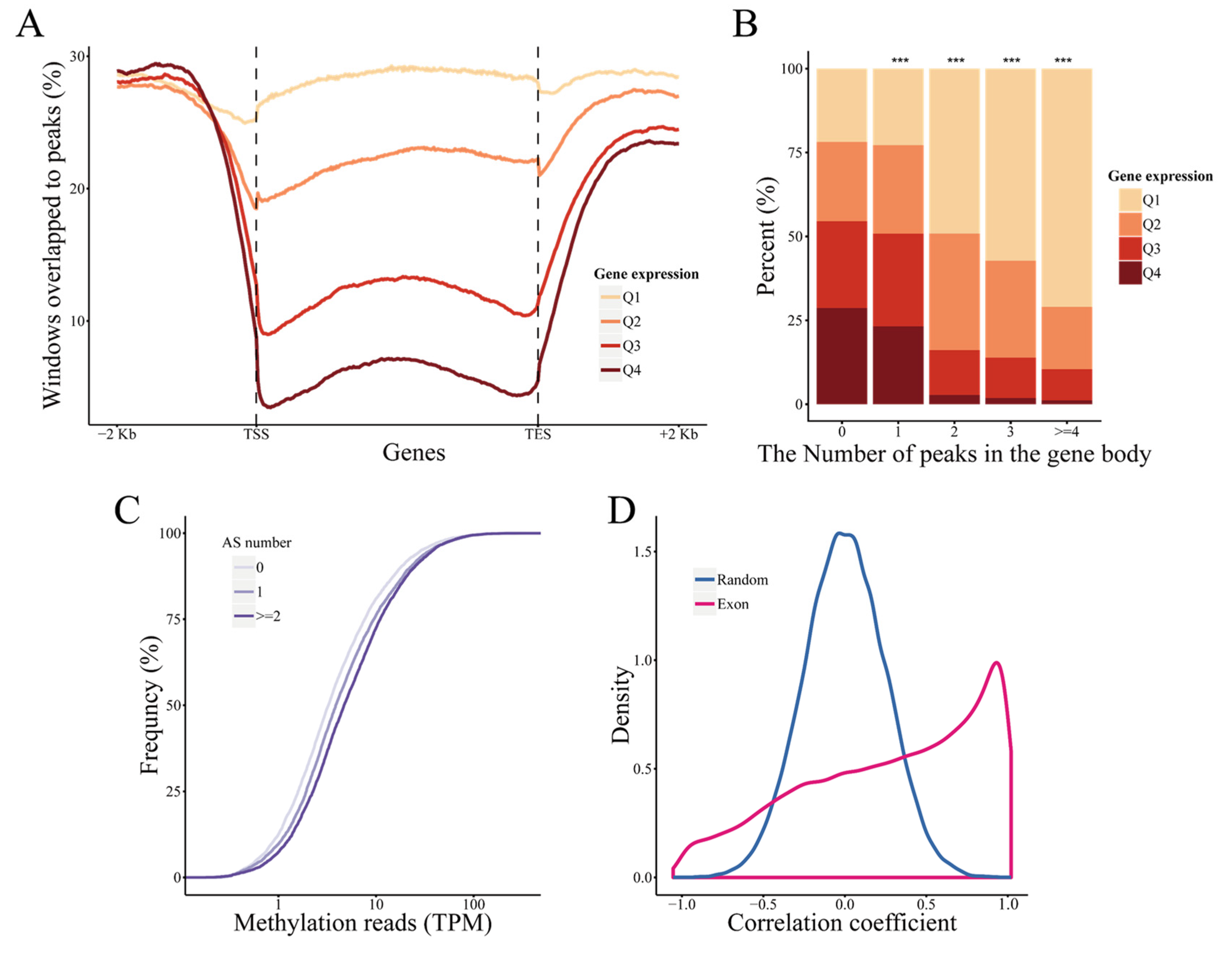
2.5. DNA Methylation Regulates Gene Expression and Alternative Splicing in Response to Drought Stress
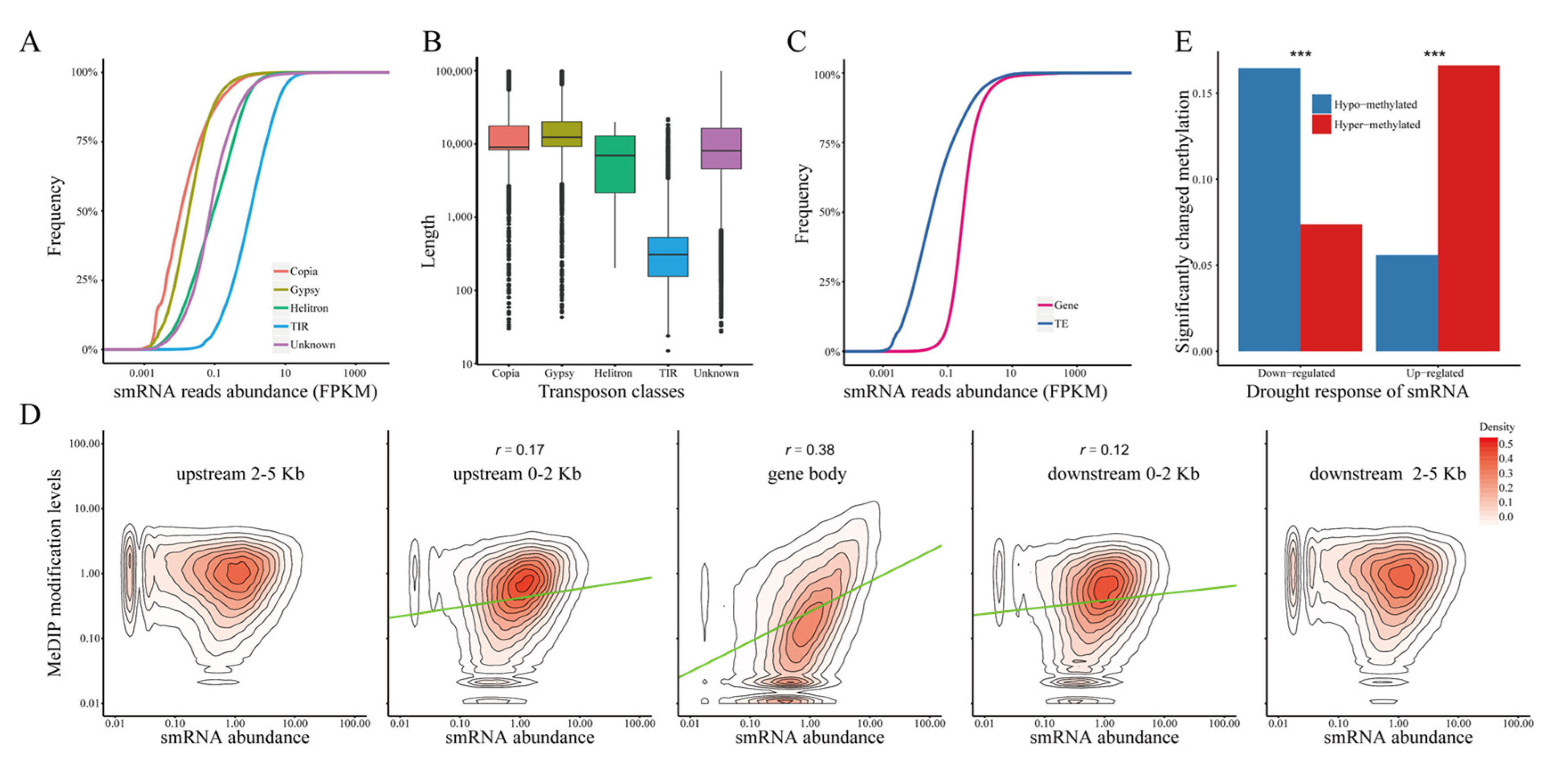
2.6. The Stress Response of DNA Methylation Is Significantly Correlated with siRNA Expression
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. DNA Methylation Peak Calling
5.2. Construction and Validation of Bin Map
5.3. Differentially Methylated Region Calling
5.4. Identification of Differentially Expressed Genes
5.5. Identification of Alternative Splicing Events
5.6. siRNA Abundance and Their Fold Change
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- FAO. World Food and Agriculture—Statistical Yearbook 2020; FAO: Rome, Italy, 2020; p. 11. [Google Scholar]
- Fahad, S.; Bajwa, A.; Nazir, U.; Anjum, S.A.; Farooq, A.; Zohaib, A.; Sadia, S.; Nasim, W.; Adkins, S.; Saud, S.; et al. Crop Production under Drought and Heat Stress: Plant Responses and Management Options. Front. Plant Sci. 2017, 8, 1147. [Google Scholar] [CrossRef] [Green Version]
- Lesk, C.; Rowhani, P.; Ramankutty, N. Influence of extreme weather disasters on global crop production. Nature 2016, 529, 84–87. [Google Scholar] [CrossRef]
- Bartels, A.; Han, Q.; Nair, P.; Stacey, L.; Gaynier, H.; Mosley, M.; Huang, Q.Q.; Pearson, J.K.; Hsieh, T.-F.; An, Y.-Q.C.; et al. Dynamic DNA Methylation in Plant Growth and Development. Int. J. Mol. Sci. 2018, 19, 2144. [Google Scholar] [CrossRef] [Green Version]
- Tirnaz, S.; Batley, J. DNA Methylation: Toward Crop Disease Resistance Improvement. Trends Plant Sci. 2019, 24, 1137–1150. [Google Scholar] [CrossRef]
- Liu, J.; He, Z. Small DNA Methylation, Big Player in Plant Abiotic Stress Responses and Memory. Front. Plant Sci. 2020, 11, 1977. [Google Scholar] [CrossRef]
- Shen, X.; De Jonge, J.; Forsberg, S.; Pettersson, M.; Sheng, Z.; Hennig, L.; Carlborg, Ö. Natural CMT2 Variation Is Associated with Genome-Wide Methylation Changes and Temperature Seasonality. PLoS Genet. 2014, 10, e1004842. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Sun, Y.; Cheng, B.; Xue, S.; Cheng, D.; Liu, L.; Meng, L.; Qiang, S. Variation inICE1Methylation Primarily Determines Phenotypic Variation in Freezing Tolerance in Arabidopsis thaliana. Plant Cell Physiol. 2018, 60, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Conde, D.; Le Gac, A.; Perales, M.; Dervinis, C.; Kirst, M.; Maury, S.; González-Melendi, P.; Allona, I. Chilling-responsive DEMETER-LIKE DNA demethylase mediates in poplar bud break. Plant Cell Environ. 2017, 40, 2236–2249. [Google Scholar] [CrossRef] [PubMed]
- Bharti, P.; Mahajan, M.; Vishwakarma, A.K.; Bhardwaj, J.; Yadav, S.K. AtROS1 overexpression provides evidence for epigenetic regulation of genes encoding enzymes of flavonoid biosynthesis and antioxidant pathways during salt stress in transgenic tobacco. J. Exp. Bot. 2015, 66, 5959–5969. [Google Scholar] [CrossRef] [Green Version]
- Gahlaut, V.; Samtani, H.; Khurana, P. Genome-wide identification and expression profiling of cytosine-5 DNA methyl-transferases during drought and heat stress in wheat (Triticum aestivum). Genomics 2020, 112, 4796–4807. [Google Scholar] [CrossRef]
- Lu, X.; Wang, X.; Chen, X.; Shu, N.; Wang, J.; Wang, D.; Wang, S.; Fan, W.; Guo, L.; Guo, X.; et al. Single-base resolution methylomes of upland cotton (Gossypium hirsutum L.) reveal epigenome modifications in response to drought stress. BMC Genom. 2017, 18, 297. [Google Scholar] [CrossRef] [Green Version]
- Garg, R.; Chevala, V.N.; Shankar, R.; Jain, M. Divergent DNA methylation patterns associated with gene expression in rice cultivars with contrasting drought and salinity stress response. Sci. Rep. 2015, 5, 14922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Qin, Q.; Sun, F.; Wang, Y.; Xu, D.; Li, Z.; Fu, B. Genome-Wide Differences in DNA Methylation Changes in Two Contrasting Rice Genotypes in Response to Drought Conditions. Front. Plant Sci. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Chen, L.; Li, M.; Lou, Q.; Xia, H.; Wang, P.; Li, T.; Liu, H.; Luo, L. Transgenerational Variations in DNA Methylation Induced by Drought Stress in Two Rice Varieties with Distinguished Difference to Drought Resistance. PLoS ONE 2013, 8, e80253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, H.; Wang, H.; Liu, S.; Li, Z.; Yang, X.; Yan, J.; Li, J.; Tran, L.-S.; Qin, F. A transposable element in a NAC gene is associated with drought tolerance in maize seedlings. Nat. Commun. 2015, 6, 8326. [Google Scholar] [CrossRef] [Green Version]
- Sallam, N.; Moussa, M. DNA methylation changes stimulated by drought stress in ABA-deficient maize mutant vp10. Plant Physiol. Biochem. 2021, 160, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lang, Z.; Zhu, J.-K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506. [Google Scholar] [CrossRef]
- Zilberman, D. An evolutionary case for functional gene body methylation in plants and animals. Genome Biol. 2017, 18, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bewick, A.J.; Schmitz, R.J. Gene body DNA methylation in plants. Curr. Opin. Plant Biol. 2017, 36, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Bewick, A.J.; Ji, L.; Niederhuth, C.; Willing, E.-M.; Hofmeister, B.T.; Shi, X.; Wang, L.; Lu, Z.; Rohr, N.A.; Hartwig, B.; et al. On the origin and evolutionary consequences of gene body DNA methylation. Proc. Natl. Acad. Sci. USA 2016, 113, 9111–9116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neri, F.; Rapelli, S.; Krepelova, A.; Incarnato, D.; Parlato, C.; Basile, G.; Maldotti, M.; Anselmi, F.; Oliviero, S. Intragenic DNA methylation prevents spurious transcription initiation. Nature 2017, 543, 72–77. [Google Scholar] [CrossRef]
- Maunakea, A.K.; Chepelev, I.; Cui, K.; Zhao, K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013, 23, 1256–1269. [Google Scholar] [CrossRef] [Green Version]
- Shayevitch, R.; Askayo, D.; Keydar, I.; Ast, G. The importance of DNA methylation of exons on alternative splicing. RNA 2018, 24, 1351–1362. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Hu, L.; Wang, X.; Li, N.; Xu, C.; Gong, L.; Liu, B. DNA methylation affects gene alternative splicing in plants: An example from rice. Mol. Plant 2016, 9, 305–307. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Chang, F.; You, C.; Cui, J.; Zhu, G.; Wang, L.; Zheng, Y.; Qi, J.; Ma, H. Whole-genome DNA methylation patterns and complex associations with gene structure and expression dur-ing flower development in Arabidopsis. Plant J. 2015, 81, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Liu, J.; Xia, A.; Springer, N.M.; He, Y. Maize decrease in DNA methylation 1 targets RNA-directed DNA methylation on active chromatin. Plant Cell 2021. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Liu, R.; Niu, Q.; Tang, K.; Zhang, B.; Zhang, H.; Chen, K.; Zhu, J.-K.; Lang, Z. Global increase in DNA methylation during orange fruit development and ripening. Proc. Natl. Acad. Sci. USA 2019, 116, 1430–1436. [Google Scholar] [CrossRef] [Green Version]
- Lang, Z.; Wang, Y.; Tang, K.; Tang, D.; Datsenka, T.; Cheng, J.; Zhang, Y.; Handa, A.K.; Zhu, J.-K. Critical roles of DNA demethylation in the activation of ripening-induced genes and inhibition of ripen-ing-repressed genes in tomato fruit. Proc. Natl. Acad. Sci. USA 2017, 114, E4511–E4519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Secco, D.; Wang, C.; Shou, H.; Schultz, M.D.; Chiarenza, S.; Nussaume, L.; Ecker, J.; Whelan, J.; Lister, R. Stress induced gene expression drives transient DNA methylation changes at adjacent repetitive elements. eLife 2015, 4, e09343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.-Y.; Zhao, H.-X.; Wu, S.-H.; Huang, F.; Wu, K.-T.; Zeng, X.-F.; Chen, X.-Q.; Xu, P.-Z.; Wu, X.-J. Global Methylation Patterns and Their Relationship with Gene Expression and Small RNA in Rice Lines with Different Ploidy. Front. Plant Sci. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-M.; Sasaki, T.; Ueda, M.; Sako, K.; Seki, M. Chromatin changes in response to drought, salinity, heat, and cold stresses in plants. Front. Plant Sci. 2015, 6, 114. [Google Scholar] [CrossRef] [Green Version]
- Eichten, S.R.; Springer, N.M. Minimal evidence for consistent changes in maize DNA methylation patterns following en-vironmental stress. Front. Plant Sci. 2015, 6, 308. [Google Scholar] [CrossRef] [Green Version]
- Matzke, M.A.; Mosher, R. RNA-directed DNA methylation: An epigenetic pathway of increasing complexity. Nat. Rev. Genet. 2014, 15, 394–408. [Google Scholar] [CrossRef]
- Du, J.; Zhong, X.; Bernatavichute, Y.V.; Stroud, H.; Feng, S.; Caro, E.; Vashisht, A.A.; Terragni, J.; Chin, H.G.; Tu, A.; et al. Dual Binding of Chromomethylase Domains to H3K9me2-Containing Nucleosomes Directs DNA Methylation in Plants. Cell 2012, 151, 167–180. [Google Scholar] [CrossRef] [Green Version]
- Stelpflug, S.C.; Eichten, S.R.; Hermanson, P.J.; Springer, N.M.; Kaeppler, S.M. Consistent and Heritable Alterations of DNA Methylation Are Induced by Tissue Culture in Maize. Genetics 2014, 198, 209–218. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Eichten, S.R.; Hermanson, P.J.; Springer, N.M. Inheritance Patterns and Stability of DNA Methylation Variation in Maize Near-Isogenic Lines. Genetics 2014, 196, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, M.; Paszkowski, J. Identification of genes preventing transgenerational transmission of stress-induced epigenetic states. Proc. Natl. Acad. Sci. USA 2014, 111, 8547–8552. [Google Scholar] [CrossRef] [Green Version]
- Dubin, M.J.; Zhang, P.; Meng, D.; Remigereau, M.-S.; Osborne, E.J.; Casale, F.P.; Drewe, P.; Kahles, A.; Jean, G.; Vilhjalmsson, B.; et al. DNA methylation in Arabidopsis has a genetic basis and shows evidence of local adaptation. eLife 2015, 4, e05255. [Google Scholar] [CrossRef]
- Xu, J.; Chen, G.; Hermanson, P.J.; Xu, Q.; Sun, C.; Chen, W.; Kan, Q.; Li, M.; Crisp, P.; Yan, J.; et al. Population-level analysis reveals the widespread occurrence and phenotypic consequence of DNA methylation variation not tagged by genetic variation in maize. Genome Biol. 2019, 20, 243. [Google Scholar] [CrossRef]
- Kawakatsu, T.; Huang, S.-S.C.; Jupe, F.; Sasaki, E.; Schmitz, R.; Urich, M.A.; Castanon, R.; Nery, J.R.; Barragan, C.; He, Y.; et al. Epigenomic Diversity in a Global Collection of Arabidopsis thaliana Accessions. Cell 2016, 166, 492–505. [Google Scholar] [CrossRef] [Green Version]
- Lei, M.; Zhang, H.; Julian, R.; Tang, K.; Xie, S.; Zhu, J.-K. Regulatory link between DNA methylation and active demethylation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2015, 112, 3553–3557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, C.J.; Scheibe, M.; Wongpalee, S.P.; Liu, W.; Cornett, E.M.; Vaughan, R.M.; Li, X.; Chen, W.; Xue, Y.; Zhong, Z.; et al. A DNA methylation reader complex that enhances gene transcription. Science 2018, 362, 1182–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichten, S.R.; Briskine, R.; Song, J.; Li, Q.; Swanson-Wagner, R.; Hermanson, P.J.; Waters, A.J.; Starr, E.; West, P.T.; Tiffin, P.; et al. Epigenetic and Genetic Influences on DNA Methylation Variation in Maize Populations. Plant Cell 2013, 25, 2783–2797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichten, S.; Swanson-Wagner, R.A.; Schnable, J.; Waters, A.J.; Hermanson, P.J.; Liu, S.; Yeh, C.-T.; Jia, Y.; Gendler, K.; Freeling, M.; et al. Heritable Epigenetic Variation among Maize Inbreds. PLoS Genet. 2011, 7, e1002372. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Guan, X.; Chen, Z.J. Dynamic Roles for Small RNAs and DNA Methylation during Ovule and Fiber Development in Allotetraploid Cotton. PLoS Genet. 2015, 11, e1005724. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Wu, W.; Zinta, G.; Yang, L.; Wang, D.; Liu, R.; Zhang, H.; Zheng, Z.; Huang, H.; Zhang, Q.; et al. A naturally occurring epiallele associates with leaf senescence and local climate adaptation in Arabidopsis accessions. Nat. Commun. 2018, 9, 460. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yu, Y.; Tao, F.; Zhang, J.; Copetti, D.; Kudrna, D.; Talag, J.; Lee, S.; Wing, R.A.; Fan, C. DNA methylation changes facilitated evolution of genes derived from Mutator-like transposable elements. Genome Biol. 2016, 17, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, M.; Hong, C.; Zhang, B.; Lowdon, R.F.; Xing, X.; Li, D.; Zhou, X.; Lee, H.J.; Maire, C.L.; Ligon, K.L.; et al. DNA hypomethylation within specific transposable element families associates with tissue-specific enhancer landscape. Nat. Genet. 2013, 45, 836–841. [Google Scholar] [CrossRef]
- Xu, J.; Wang, Q.; Freeling, M.; Zhang, X.; Xu, Y.; Mao, Y.; Tang, X.; Wu, F.; Lan, H.; Cao, M.; et al. Natural antisense transcripts are significantly involved in regulation of drought stress in maize. Nucleic Acids Res. 2017, 45, 5126–5141. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [Green Version]
- Gonda, I.; Ashrafi, H.; Lyon, D.A.; Strickler, S.R.; Hulse-Kemp, A.M.; Ma, Q.; Sun, H.; Stoffel, K.; Powell, A.F.; Futrell, S.; et al. Sequencing-Based Bin Map Construction of a Tomato Mapping Population, Facilitating High-Resolution Quantitative Trait Loci Detection. Plant Genome 2018, 12, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Dell’Acqua, M.; Gatti, D.M.; Pea, G.; Cattonaro, F.; Coppens, F.; Magris, G.; Hlaing, A.L.; Aung, H.H.; Nelissen, H.; Baute, J.; et al. Genetic properties of the MAGIC maize population: A new platform for high definition QTL mapping in Zea mays. Genome Biol. 2015, 16, 167. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. agriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; I Love, M.; Irizarry, M.I.L.R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trincado, J.L.; Entizne, J.C.; Hysenaj, G.; Singh, B.; Skalic, M.; Elliott, D.J.; Eyras, E. SUPPA2: Fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol. 2018, 19, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hyper-Methylated DMRs | Hypo-Methylated DMRs | |||||
|---|---|---|---|---|---|---|
| TE-related genes | Non-TE genes | P value (χ2 test) | TE-related genes | Non-TE genes | P value (χ2 test) | |
| Genes | 2963 (8.94%) | 501 (3.97%) | 5.44E72 | 1708 (5.15%) | 353 (2.80%) | 2.32E27 |
| Exon | 1563 (4.72%) | 313 (2.48%) | 5.74E27 | 923 (2.78%) | 245 (1.94%) | 3.78E07 |
| Intron | 1877 (5.66%) | 291 (2.31%) | 2.06E51 | 1202 (3.63%) | 211 (1.67%) | 4.64E27 |
| Promoter 2k | 3050 (9.20%) | 986 (7.81%) | 3.09E06 | 1478 (4.46%) | 500 (3.96%) | 2.08E02 |
| Promoter 2k–5k | 4469 (13.48%) | 1363 (10.80%) | 1.63E14 | 2349 (7.09%) | 843 (6.68%) | 1.32E01 |
| Downstream 2k | 2874 (8.67%) | 696 (5.51%) | 2.86E29 | 1636 (4.94%) | 397 (3.15%) | 1.22E16 |
| Downstream 2k–5k | 3682 (11.11%) | 936 (7.42%) | 1.25E31 | 2025 (6.11%) | 589 (4.67%) | 3.23E09 |
| AC7643_DT | AC7729/TZSRW_DS | RIL208_DT | RIL64_DS | |||||
|---|---|---|---|---|---|---|---|---|
| Up-regulated | Down-regulated | Up-regulated | Down-regulated | Up-regulated | Down-regulated | Up-regulated | Down-regulated | |
| Total genes | 3588 (7.84%) | 3975 (8.69%) | 5180 (11.32%) | 3920 (8.56%) | 2817 (6.15%) | 1791 (3.91%) | 1927 (4.21%) | 1930 (4.22%) |
| TE-related genes | 2586 (7.80%) | 2755 (8.31%) | 3641 (10.98%) | 2720 (8.21%) | 2215 (6.68%) | 1242 (3.75%) | 1439 (4.34%) | 1347 (4.06%) |
| p value | 0.855 | 0.065 | 0.145 | 0.075 | 0.003 | 0.238 | 0.379 | 0.295 |
| Non-TE genes | 1002 (7.94%) | 1220 (9.67%) | 1539 (12.19%) | 1200 (9.51%) | 602 (4.77%) | 549 (4.35%) | 488 (3.87%) | 583 (4.62%) |
| p value | 0.726 | 0.001 | 0.007 | 0.001 | 0.000 | 0.029 | 0.091 | 0.051 |
| p value (TE-related/Non-TE) | 0.638 | 0.000 | 0.000 | 0.000 | 0.000 | 0.003 | 0.026 | 0.009 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Xu, J.; Pu, X.; Lv, H.; Liu, Y.; Ma, H.; Wu, F.; Wang, Q.; Feng, X.; Liu, T.; et al. Maize DNA Methylation in Response to Drought Stress Is Involved in Target Gene Expression and Alternative Splicing. Int. J. Mol. Sci. 2021, 22, 8285. https://doi.org/10.3390/ijms22158285
Wang Q, Xu J, Pu X, Lv H, Liu Y, Ma H, Wu F, Wang Q, Feng X, Liu T, et al. Maize DNA Methylation in Response to Drought Stress Is Involved in Target Gene Expression and Alternative Splicing. International Journal of Molecular Sciences. 2021; 22(15):8285. https://doi.org/10.3390/ijms22158285
Chicago/Turabian StyleWang, Qi, Jie Xu, Xuemei Pu, Haozhe Lv, Yanjun Liu, Huili Ma, Fengkai Wu, Qingjun Wang, Xuanjun Feng, Tianhong Liu, and et al. 2021. "Maize DNA Methylation in Response to Drought Stress Is Involved in Target Gene Expression and Alternative Splicing" International Journal of Molecular Sciences 22, no. 15: 8285. https://doi.org/10.3390/ijms22158285
APA StyleWang, Q., Xu, J., Pu, X., Lv, H., Liu, Y., Ma, H., Wu, F., Wang, Q., Feng, X., Liu, T., Tang, Q., Liu, Y., & Lu, Y. (2021). Maize DNA Methylation in Response to Drought Stress Is Involved in Target Gene Expression and Alternative Splicing. International Journal of Molecular Sciences, 22(15), 8285. https://doi.org/10.3390/ijms22158285