
Molecular Pathogenesis and Peripheral Monitoring of Adult Fragile X-Associated Syndromes


Abstract
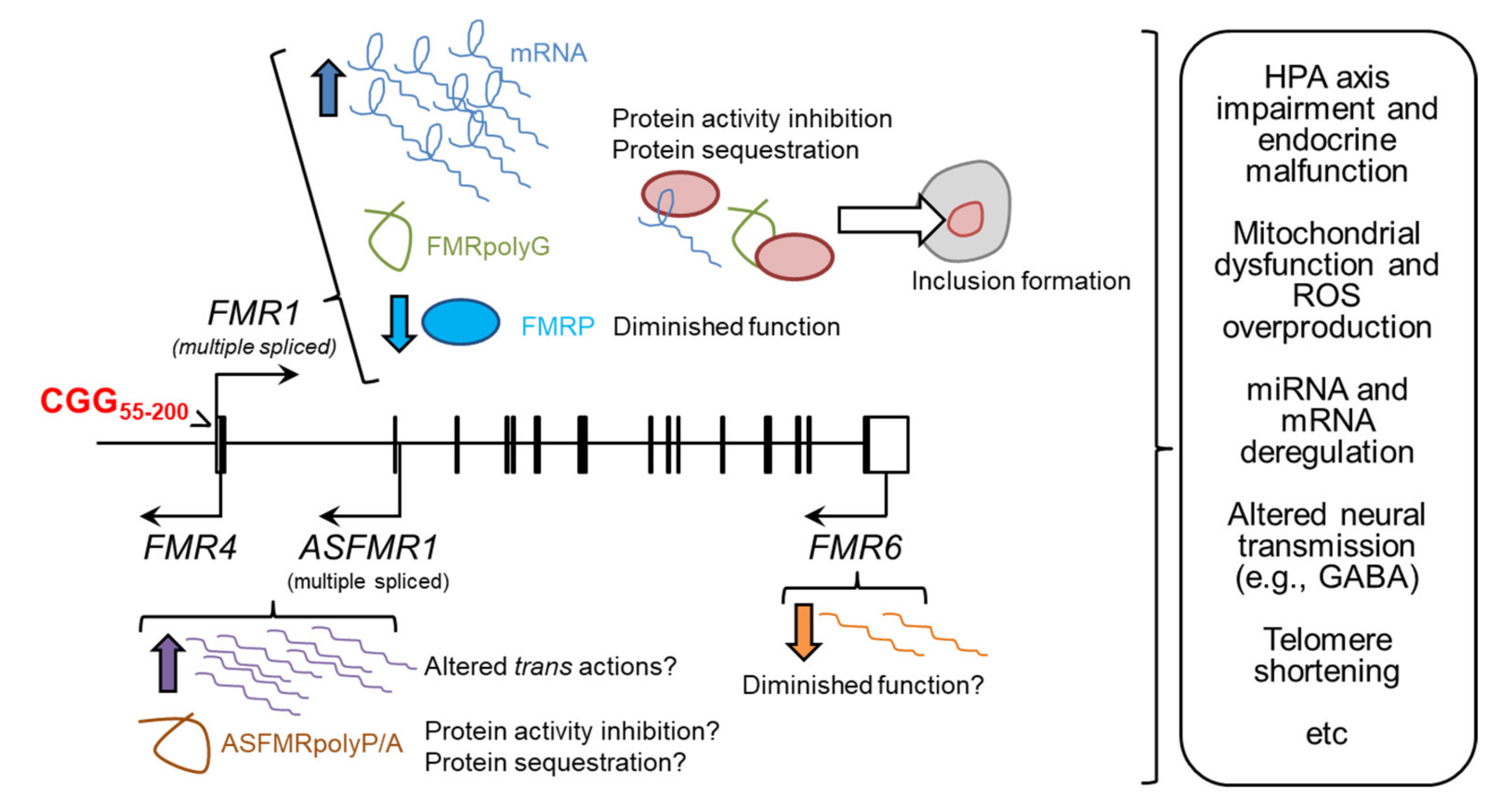
:1. The Multiple Faces of FMR1 in Pathology
1.1. Brief Description of the Fragile X-Associated Syndromes
1.2. The Molecular Pathogenesis of Adult Fragile X-Associated Syndromes
1.2.1. RNA Toxicity
1.2.2. Production of FMRpolyG Peptides
1.2.3. The Potential Role of Long Non-Coding FMR1 Isoforms
1.2.4. Reduction of FMRP Activity
1.3. Is There a Fragile X Spectrum Disorder?
2. Peripheral Blood as Source of Biomarkers for Adult Fragile X-Associated Syndromes
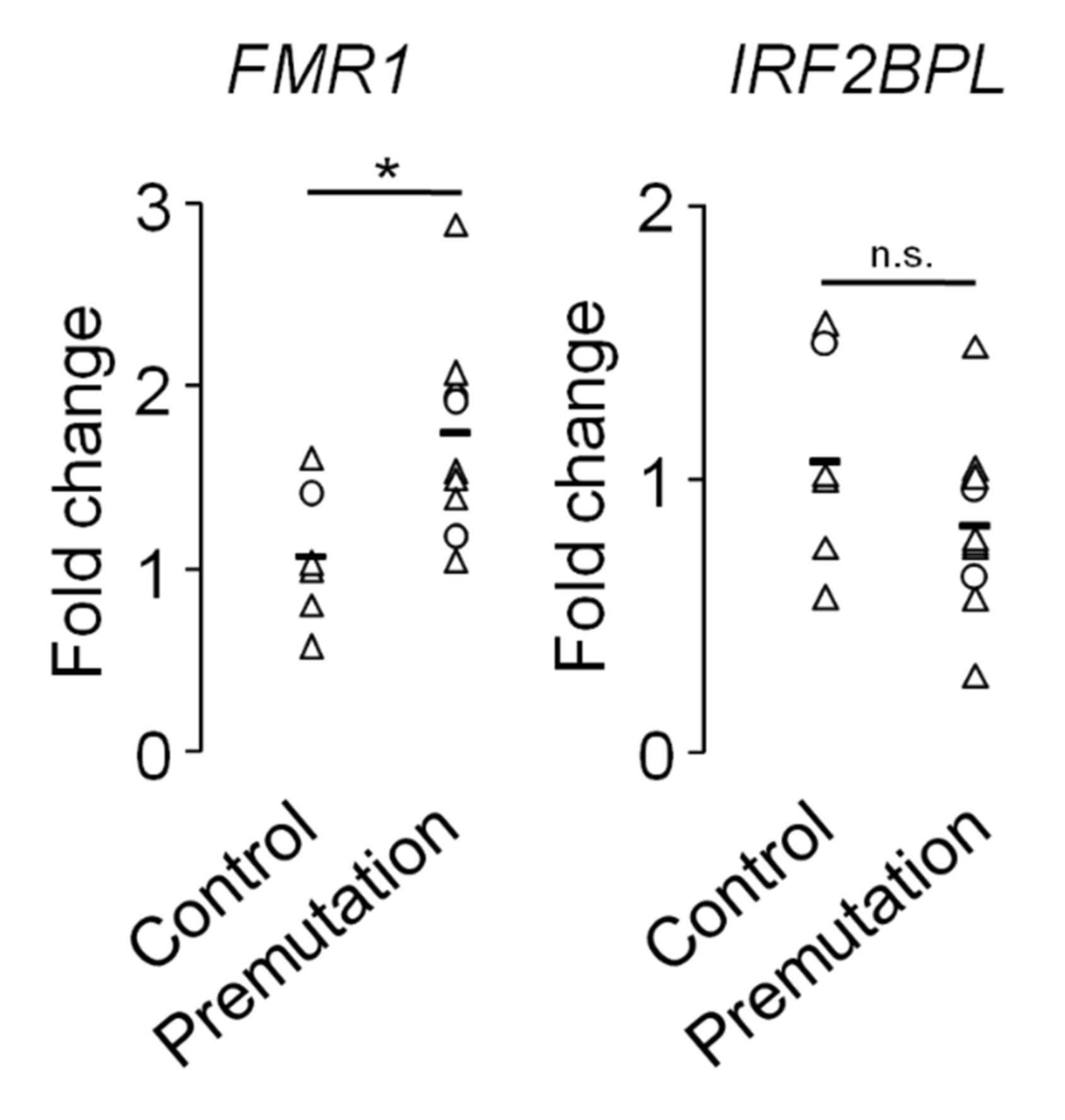
2.1. The Molecular Outputs of the FMR1 Locus as Biomarkers in Fragile X-Associated Pathology
2.2. Endocrine Biomarkers in Premutation Carriers
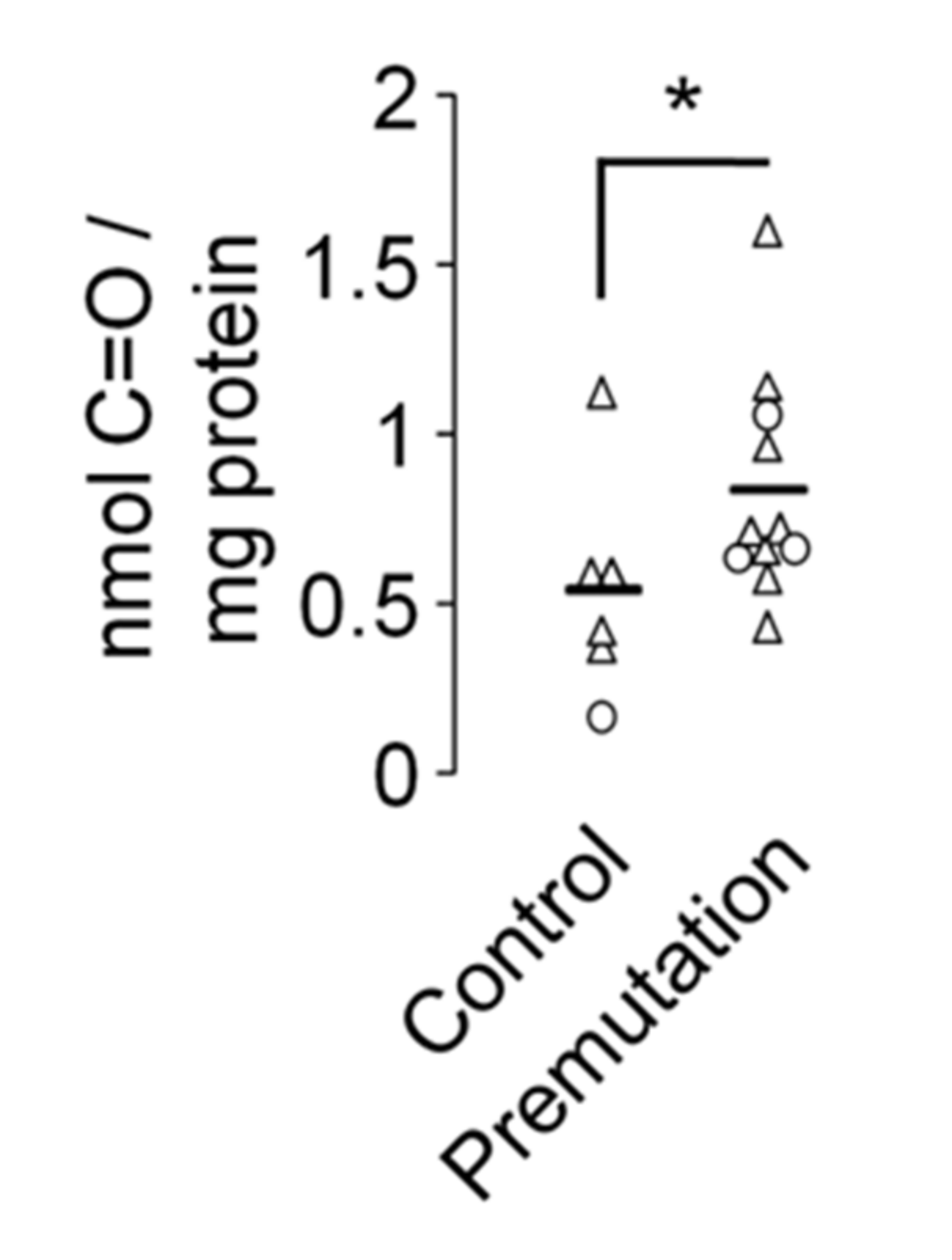
2.3. Mitochondrial Dysfunction and Overproduction of Reactive Oxygen Species in Premutation Carriers
2.4. Inflammation in the Premutation Condition
2.5. miRNAs as Potential Biomarkers in Fragile X-Associated Syndromes
2.6. Transcriptional Dysregulation in Fragile X-Associated Syndromes
2.7. The GABAergic Dysfuntion in Premutation Carriers
2.8. Telomere Shortening in Premutation Carriers
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A



{kind=link}
{kind=link}
{kind=link}
| miRNA | Ensembl ID | Fold Change (log2) | Adj p-Value |
|---|---|---|---|
| MIR375 | ENSG00000198973 | −2.58 | 5.04 × 10−5 |
| MIR483** | ENSG00000207805 | −2.72 | 1.06 × 10−2 |
| MIR4664 | ENSG00000265660 | −4.30 | 2.58 × 10−2 |
| MIR10B | ENSG00000207744 | −1.47 | 3.72 × 10−2 |
| MIR7159 | ENSG00000276824 | −2.95 | 3.72 × 10−2 |
| MIR125B2 | ENSG00000207863 | −1.67 | 4.82 × 10−2 |
| MIR6730 | ENSG00000276830 | −4.16 | 6.60 × 10−2 |
| ENSG00000280773 | −5.51 | 8.17 × 10−2 | |
| MIR501 | ENSG00000211538 | −2.10 | 9.14 × 10−2 |
| MIR885** | ENSG00000216135 | −2.63 | 9.99 × 10−2 |
| MIR27A* | ENSG00000207808 | 1.29 | 9.98 × 10−2 |
| MIR26A1 | ENSG00000199075 | 0.75 | 9.72 × 10−2 |
| MIR381 | ENSG00000199020 | 4.10 | 8.49 × 10−2 |
| MIR655 | ENSG00000207646 | 5.02 | 8.49 × 10−2 |
| MIR377 | ENSG00000199015 | 4.64 | 6.60 × 10−2 |
| MIR654 | ENSG00000207934 | 5.00 | 6.60 × 10−2 |
| MIR376C | ENSG00000283279 | 4.77 | 6.60 × 10−2 |
| MIR494 | ENSG00000194717 | 2.52 | 5.56 × 10−2 |
| MIR889 | ENSG00000216099 | 5.12 | 6.72 × 10−3 |
| MIR134 | ENSG00000207993 | 5.72 | 5.14 × 10−3 |
| MIR331 | ENSG00000199172 | 5.19 | 3.57 × 10−3 |
| MIR766 | ENSG00000211578 | 5.53 | 8.50 × 10−4 |
| MIR877 | ENSG00000216101 | 5.11 | 1.26 × 10−4 |
| MIR495 | ENSG00000207743 | 5.18 | 5.74 × 10−5 |
References
- Bagni, C.; Oostra, B.A. Fragile X syndrome: From protein function to therapy. Am. J. Med. Genet. A 2013, 161A, 2809–2821. [Google Scholar] [CrossRef]
- Madrigal, I.; Xuncla, M.; Tejada, M.I.; Martinez, F.; Fernandez-Carvajal, I.; Perez-Jurado, L.A.; Rodriguez-Revenga, L.; Mila, M. Intermediate FMR1 alleles and cognitive and/or behavioural phenotypes. Eur. J. Hum. Genet. 2011, 19, 921–923. [Google Scholar] [CrossRef] [Green Version]
- Hall, D.A.; Nag, S.; Ouyang, B.; Bennett, D.A.; Liu, Y.; Ali, A.; Zhou, L.; Berry-Kravis, E. Fragile X gray zone alleles are associated with signs of parkinsonism and earlier death. Mov. Disord. 2020, 35, 1448–1456. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Hagerman, P. (Eds.) Fragile-X Syndrome: Diagnosis, Treatment and Research, 3rd ed.; Johns Hopkins University Press: Baltimore, MD, USA, 2002. [Google Scholar]
- Hunter, J.; Rivero-Arias, O.; Angelov, A.; Kim, E.; Fotheringham, I.; Leal, J. Epidemiology of fragile X syndrome: A systematic review and meta-analysis. Am. J. Med. Genet. A 2014, 164A, 1648–1658. [Google Scholar] [CrossRef]
- Jacquemont, S.; Farzin, F.; Hall, D.; Leehey, M.; Tassone, F.; Gane, L.; Zhang, L.; Grigsby, J.; Jardini, T.; Lewin, F.; et al. Aging in individuals with the FMR1 mutation. Am. J. Ment. Retard. 2004, 109, 154–164. [Google Scholar] [CrossRef] [Green Version]
- Seltzer, M.M.; Baker, M.W.; Hong, J.; Maenner, M.; Greenberg, J.; Mandel, D. Prevalence of CGG expansions of the FMR1 gene in a US population-based sample. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159B, 589–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, A.; Summers, S.; Tassone, F.; Seritan, A.; Hessl, D.; Hagerman, P.; Hagerman, R. Women with fragile X-associated tremor/ataxia syndrome. Mov. Disord. Clin. Pract. 2020, 7, 910–919. [Google Scholar] [CrossRef]
- Mila, M.; Alvarez-Mora, M.I.; Madrigal, I.; Rodriguez-Revenga, L. Fragile X syndrome: An overview and update of the FMR1 gene. Clin. Genet. 2018, 93, 197–205. [Google Scholar] [CrossRef]
- Hagerman, P. Fragile X-associated tremor/ataxia syndrome (FXTAS): Pathology and mechanisms. Acta Neuropathol. 2013, 126, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Brunberg, J.A.; Jacquemont, S.; Hagerman, R.J.; Berry-Kravis, E.M.; Grigsby, J.; Leehey, M.A.; Tassone, F.; Brown, W.T.; Greco, C.M.; Hagerman, P.J. Fragile X premutation carriers: Characteristic MR imaging findings of adult male patients with progressive cerebellar and cognitive dysfunction. AJNR Am. J. Neuroradiol. 2002, 23, 1757–1766. [Google Scholar]
- Morales, H.; Tomsick, T. Middle cerebellar peduncles: Magnetic resonance imaging and pathophysiologic correlate. World J. Radiol. 2015, 7, 438–447. [Google Scholar] [CrossRef]
- Wang, J.Y.; Hessl, D.; Hagerman, R.J.; Simon, T.J.; Tassone, F.; Ferrer, E.; Rivera, S.M. Abnormal trajectories in cerebellum and brainstem volumes in carriers of the fragile X premutation. Neurobiol. Aging 2017, 55, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greco, C.M.; Hagerman, R.J.; Tassone, F.; Chudley, A.E.; Del Bigio, M.R.; Jacquemont, S.; Leehey, M.; Hagerman, P.J. Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain 2002, 125, 1760–1771. [Google Scholar] [CrossRef]
- Gokden, M.; Al-Hinti, J.T.; Harik, S.I. Peripheral nervous system pathology in fragile X tremor/ataxia syndrome (FXTAS). Neuropathology 2009, 29, 280–284. [Google Scholar] [CrossRef]
- Hunsaker, M.R.; Greco, C.M.; Spath, M.A.; Smits, A.P.; Navarro, C.S.; Tassone, F.; Kros, J.M.; Severijnen, L.A.; Berry-Kravis, E.M.; Berman, R.F.; et al. Widespread non-central nervous system organ pathology in fragile X premutation carriers with fragile X-associated tremor/ataxia syndrome and CGG knock-in mice. Acta Neuropathol. 2011, 122, 467–479. [Google Scholar] [CrossRef] [Green Version]
- Wittenberger, M.D.; Hagerman, R.J.; Sherman, S.L.; McConkie-Rosell, A.; Welt, C.K.; Rebar, R.W.; Corrigan, E.C.; Simpson, J.L.; Nelson, L.M. The FMR1 premutation and reproduction. Fertil. Steril. 2007, 87, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Fink, D.A.; Nelson, L.M.; Pyeritz, R.; Johnson, J.; Sherman, S.L.; Cohen, Y.; Elizur, S.E. Fragile X associated primary ovarian insufficiency (FXPOI): Case report and literature review. Front. Genet. 2018, 9, 529. [Google Scholar] [CrossRef] [Green Version]
- Hunter, J.E.; Rohr, J.K.; Sherman, S.L. Co-occurring diagnoses among FMR1 premutation allele carriers. Clin. Genet. 2010, 77, 374–381. [Google Scholar] [CrossRef] [Green Version]
- Van der Stege, J.G.; Groen, H.; van Zadelhoff, S.J.; Lambalk, C.B.; Braat, D.D.; van Kasteren, Y.M.; van Santbrink, E.J.; Apperloo, M.J.; Weijmar Schultz, W.C.; Hoek, A. Decreased androgen concentrations and diminished general and sexual well-being in women with premature ovarian failure. Menopause 2008, 15, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, J.C. Effect of early menopause on bone mineral density and fractures. Menopause 2007, 14, 567–571. [Google Scholar] [CrossRef]
- Atsma, F.; Bartelink, M.L.; Grobbee, D.E.; van der Schouw, Y.T. Postmenopausal status and early menopause as independent risk factors for cardiovascular disease: A meta-analysis. Menopause 2006, 13, 265–279. [Google Scholar] [CrossRef]
- Mondul, A.M.; Rodriguez, C.; Jacobs, E.J.; Calle, E.E. Age at natural menopause and cause-specific mortality. Am. J. Epidemiol. 2005, 162, 1089–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalantaridou, S.N.; Naka, K.K.; Papanikolaou, E.; Kazakos, N.; Kravariti, M.; Calis, K.A.; Paraskevaidis, E.A.; Sideris, D.A.; Tsatsoulis, A.; Chrousos, G.P.; et al. Impaired endothelial function in young women with premature ovarian failure: Normalization with hormone therapy. J. Clin. Endocrinol. Metab. 2004, 89, 3907–3913. [Google Scholar] [CrossRef] [PubMed]
- Nobile, V.; Pucci, C.; Chiurazzi, P.; Neri, G.; Tabolacci, E. DNA methylation, mechanisms of FMR1 inactivation and therapeutic perspectives for fragile X syndrome. Biomolecules 2021, 11, 296. [Google Scholar] [CrossRef]
- Swinnen, B.; Robberecht, W.; Van Den Bosch, L. RNA toxicity in non-coding repeat expansion disorders. EMBO J. 2020, 39, e101112. [Google Scholar] [CrossRef]
- Tassone, F.; Iwahashi, C.; Hagerman, P.J. FMR1 RNA within the intranuclear inclusions of fragile X-associated tremor/ataxia syndrome (FXTAS). RNA Biol. 2004, 1, 103–105. [Google Scholar] [CrossRef] [Green Version]
- Elizur, S.E.; Lebovitz, O.; Derech-Haim, S.; Dratviman-Storobinsky, O.; Feldman, B.; Dor, J.; Orvieto, R.; Cohen, Y. Elevated levels of FMR1 mRNA in granulosa cells are associated with low ovarian reserve in FMR1 premutation carriers. PLoS ONE 2014, 9, e105121. [Google Scholar] [CrossRef]
- Eslami, H.; Eslami, A.; Favaedi, R.; Asadpour, U.; Zari Moradi, S.; Eftekhari-Yazdi, P.; Madani, T.; Shahhoseini, M.; Mohseni Meybodi, A. Epigenetic aberration of FMR1 gene in infertile women with diminished ovarian reserve. Cell J. 2018, 20, 78–83. [Google Scholar] [CrossRef]
- Solvsten, C.; Nielsen, A.L. FMR1 CGG repeat lengths mediate different regulation of reporter gene expression in comparative transient and locus specific integration assays. Gene 2011, 486, 15–22. [Google Scholar] [CrossRef]
- Tassone, F.; De Rubeis, S.; Carosi, C.; La Fata, G.; Serpa, G.; Raske, C.; Willemsen, R.; Hagerman, P.J.; Bagni, C. Differential usage of transcriptional start sites and polyadenylation sites in FMR1 premutation alleles. Nucleic Acids Res. 2011, 39, 6172–6185. [Google Scholar] [CrossRef]
- Zongaro, S.; Hukema, R.; D’Antoni, S.; Davidovic, L.; Barbry, P.; Catania, M.V.; Willemsen, R.; Mari, B.; Bardoni, B. The 3′ UTR of FMR1 mRNA is a target of miR-101, miR-129-5p and miR-221: Implications for the molecular pathology of FXTAS at the synapse. Hum. Mol. Genet. 2013, 22, 1971–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolskiy, A.A.; Yarushkin, A.A.; Grishchenko, I.V.; Lemskaya, N.A.; Pindyurin, A.V.; Boldyreva, L.V.; Pustylnyak, V.O.; Yudkin, D.V. miRNA expression and interaction with the 3’UTR of FMR1 in FRAXopathy pathogenesis. Noncoding RNA Res. 2021, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Li, Y.; Allen, E.G.; Jin, P. Therapeutic development for CGG repeat expansion-associated neurodegeneration. Front. Cell. Neurosci. 2021, 15, 655568. [Google Scholar] [CrossRef]
- Glineburg, M.R.; Todd, P.K.; Charlet-Berguerand, N.; Sellier, C. Repeat-associated non-AUG (RAN) translation and other molecular mechanisms in Fragile X tremor ataxia syndrome. Brain Res. 2018, 1693, 43–54. [Google Scholar] [CrossRef]
- Iwahashi, C.K.; Yasui, D.H.; An, H.J.; Greco, C.M.; Tassone, F.; Nannen, K.; Babineau, B.; Lebrilla, C.B.; Hagerman, R.J.; Hagerman, P.J. Protein composition of the intranuclear inclusions of FXTAS. Brain 2006, 129, 256–271. [Google Scholar] [CrossRef]
- Jin, P.; Duan, R.; Qurashi, A.; Qin, Y.; Tian, D.; Rosser, T.C.; Liu, H.; Feng, Y.; Warren, S.T. Pur alpha binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron 2007, 55, 556–564. [Google Scholar] [CrossRef] [Green Version]
- Sofola, O.A.; Jin, P.; Qin, Y.; Duan, R.; Liu, H.; de Haro, M.; Nelson, D.L.; Botas, J. RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron 2007, 55, 565–571. [Google Scholar] [CrossRef] [Green Version]
- Sellier, C.; Freyermuth, F.; Tabet, R.; Tran, T.; He, F.; Ruffenach, F.; Alunni, V.; Moine, H.; Thibault, C.; Page, A.; et al. Sequestration of DROSHA and DGCR8 by expanded CGG RNA repeats alters microRNA processing in fragile X-associated tremor/ataxia syndrome. Cell Rep. 2013, 3, 869–880. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Herren, A.W.; Espinal, G.; Randol, J.; McLaughlin, B.; Martinez-Cerdeno, V.; Pessah, I.N.; Hagerman, R.J.; Hagerman, P.J. Composition of the intranuclear inclusions of fragile X-associated tremor/ataxia syndrome. Acta Neuropathol. Commun. 2019, 7, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cid-Samper, F.; Gelabert-Baldrich, M.; Lang, B.; Lorenzo-Gotor, N.; Ponti, R.D.; Severijnen, L.; Bolognesi, B.; Gelpi, E.; Hukema, R.K.; Botta-Orfila, T.; et al. An integrative study of protein-RNA condensates identifies scaffolding RNAs and reveals players in fragile X-associated tremor/ataxia syndrome. Cell Rep. 2018, 25, 3422–3434. [Google Scholar] [CrossRef] [Green Version]
- Todd, P.K.; Oh, S.Y.; Krans, A.; He, F.; Sellier, C.; Frazer, M.; Renoux, A.J.; Chen, K.C.; Scaglione, K.M.; Basrur, V.; et al. CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron 2013, 78, 440–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonapace, G.; Gullace, R.; Concolino, D.; Iannello, G.; Procopio, R.; Gagliardi, M.; Arabia, G.; Barbagallo, G.; Lupo, A.; Manfredini, L.I.; et al. Intracellular FMRpolyG-Hsp70 complex in fibroblast cells from a patient affected by fragile X tremor ataxia syndrome. Heliyon 2019, 5, e01954. [Google Scholar] [CrossRef] [Green Version]
- Castelli, L.M.; Huang, W.P.; Lin, Y.H.; Chang, K.Y.; Hautbergue, G.M. Mechanisms of repeat-associated non-AUG translation in neurological microsatellite expansion disorders. Biochem. Soc. Trans. 2021, 49, 775–792. [Google Scholar] [CrossRef] [PubMed]
- Buijsen, R.A.; Sellier, C.; Severijnen, L.A.; Oulad-Abdelghani, M.; Verhagen, R.F.; Berman, R.F.; Charlet-Berguerand, N.; Willemsen, R.; Hukema, R.K. FMRpolyG-positive inclusions in CNS and non-CNS organs of a fragile X premutation carrier with fragile X-associated tremor/ataxia syndrome. Acta Neuropathol. Commun. 2014, 2, 162. [Google Scholar] [CrossRef] [Green Version]
- Friedman-Gohas, M.; Elizur, S.E.; Dratviman-Storobinsky, O.; Aizer, A.; Haas, J.; Raanani, H.; Orvieto, R.; Cohen, Y. FMRpolyG accumulates in FMR1 premutation granulosa cells. J. Ovarian Res. 2020, 13, 22. [Google Scholar] [CrossRef] [Green Version]
- Dijkstra, A.A.; Haify, S.N.; Verwey, N.A.; Prins, N.D.; van der Toorn, E.C.; Rozemuller, A.J.M.; Bugiani, M.; den Dunnen, W.F.A.; Todd, P.K.; Charlet-Berguerand, N.; et al. Neuropathology of FMR1-premutation carriers presenting with dementia and neuropsychiatric symptoms. Brain Commun. 2021, 3, fcab007. [Google Scholar] [CrossRef]
- Sellier, C.; Buijsen, R.A.M.; He, F.; Natla, S.; Jung, L.; Tropel, P.; Gaucherot, A.; Jacobs, H.; Meziane, H.; Vincent, A.; et al. Translation of expanded CGG repeats into FMRpolyG is pathogenic and may contribute to fragile X tremor ataxia syndrome. Neuron 2017, 93, 331–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haify, S.N.; Mankoe, R.S.D.; Boumeester, V.; van der Toorn, E.C.; Verhagen, R.F.M.; Willemsen, R.; Hukema, R.K.; Bosman, L.W.J. Lack of a clear behavioral phenotype in an inducible FXTAS mouse model despite the presence of neuronal FMRpolyG-positive aggregates. Front. Mol. Biosci. 2020, 7, 599101. [Google Scholar] [CrossRef]
- Hoem, G.; Bowitz Larsen, K.; Overvatn, A.; Brech, A.; Lamark, T.; Sjottem, E.; Johansen, T. The FMRpolyGlycine protein mediates aggregate formation and toxicity independent of the CGG mRNA hairpin in a cellular model for FXTAS. Front. Genet. 2019, 10, 249. [Google Scholar] [CrossRef]
- Oh, S.Y.; He, F.; Krans, A.; Frazer, M.; Taylor, J.P.; Paulson, H.L.; Todd, P.K. RAN translation at CGG repeats induces ubiquitin proteasome system impairment in models of fragile X-associated tremor ataxia syndrome. Hum. Mol. Genet. 2015, 24, 4317–4326. [Google Scholar] [CrossRef] [Green Version]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef] [PubMed]
- Ladd, P.D.; Smith, L.E.; Rabaia, N.A.; Moore, J.M.; Georges, S.A.; Hansen, R.S.; Hagerman, R.J.; Tassone, F.; Tapscott, S.J.; Filippova, G.N. An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum. Mol. Genet. 2007, 16, 3174–3187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, A.M.; Faghihi, M.A.; Modarresi, F.; Brothers, S.P.; Wahlestedt, C. A novel RNA transcript with antiapoptotic function is silenced in fragile X syndrome. PLoS ONE 2008, 3, e1486. [Google Scholar] [CrossRef] [PubMed]
- Pastori, C.; Peschansky, V.J.; Barbouth, D.; Mehta, A.; Silva, J.P.; Wahlestedt, C. Comprehensive analysis of the transcriptional landscape of the human FMR1 gene reveals two new long noncoding RNAs differentially expressed in Fragile X syndrome and Fragile X-associated tremor/ataxia syndrome. Hum. Genet. 2014, 133, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Elizur, S.E.; Dratviman-Storobinsky, O.; Derech-Haim, S.; Lebovitz, O.; Dor, J.; Orvieto, R.; Cohen, Y. FMR6 may play a role in the pathogenesis of fragile X-associated premature ovarian insufficiency. Gynecol. Endocrinol. 2016, 32, 334–337. [Google Scholar] [CrossRef]
- Peschansky, V.J.; Pastori, C.; Zeier, Z.; Wentzel, K.; Velmeshev, D.; Magistri, M.; Silva, J.P.; Wahlestedt, C. The long non-coding RNA FMR4 promotes proliferation of human neural precursor cells and epigenetic regulation of gene expression in trans. Mol. Cell. Neurosci. 2016, 74, 49–57. [Google Scholar] [CrossRef]
- Peschansky, V.J.; Pastori, C.; Zeier, Z.; Motti, D.; Wentzel, K.; Velmeshev, D.; Magistri, M.; Bixby, J.L.; Lemmon, V.P.; Silva, J.P.; et al. Changes in expression of the long non-coding RNA FMR4 associate with altered gene expression during differentiation of human neural precursor cells. Front. Genet. 2015, 6, 263. [Google Scholar] [CrossRef] [Green Version]
- Krans, A.; Kearse, M.G.; Todd, P.K. Repeat-associated non-AUG translation from antisense CCG repeats in fragile X tremor/ataxia syndrome. Ann. Neurol. 2016, 80, 871–881. [Google Scholar] [CrossRef]
- Tassone, F.; Hagerman, R.J.; Taylor, A.K.; Gane, L.W.; Godfrey, T.E.; Hagerman, P.J. Elevated levels of FMR1 mRNA in carrier males: A new mechanism of involvement in the fragile-X syndrome. Am. J. Hum. Genet. 2000, 66, 6–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenneson, A.; Zhang, F.; Hagedorn, C.H.; Warren, S.T. Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers. Hum. Mol. Genet. 2001, 10, 1449–1454. [Google Scholar] [CrossRef] [Green Version]
- Oostra, B.A.; Willemsen, R. A fragile balance: FMR1 expression levels. Hum. Mol. Genet. 2003, 12, R249–R257. [Google Scholar] [CrossRef] [Green Version]
- Bagni, C.; Zukin, R.S. A synaptic perspective of fragile X syndrome and autism spectrum disorders. Neuron 2019, 101, 1070–1088. [Google Scholar] [CrossRef] [Green Version]
- Schneider, A.; Winarni, T.I.; Cabal-Herrera, A.M.; Bacalman, S.; Gane, L.; Hagerman, P.; Tassone, F.; Hagerman, R. Elevated FMR1-mRNA and lowered FMRP—A double-hit mechanism for psychiatric features in men with FMR1 premutations. Transl. Psychiatry 2020, 10, 205. [Google Scholar] [CrossRef]
- Storey, E.; Bui, M.Q.; Stimpson, P.; Tassone, F.; Atkinson, A.; Loesch, D.Z. Relationships between motor scores and cognitive functioning in FMR1 female premutation X carriers indicate early involvement of cerebello-cerebral pathways. Cerebellum Ataxias 2021, 8, 15. [Google Scholar] [CrossRef]
- Greco, C.M.; Soontrapornchai, K.; Wirojanan, J.; Gould, J.E.; Hagerman, P.J.; Hagerman, R.J. Testicular and pituitary inclusion formation in fragile X associated tremor/ataxia syndrome. J. Urol. 2007, 177, 1434–1437. [Google Scholar] [CrossRef]
- Lozano, R.; Summers, S.; Lozano, C.; Mu, Y.; Hessl, D.; Nguyen, D.; Tassone, F.; Hagerman, R. Association between macroorchidism and intelligence in FMR1 premutation carriers. Am. J. Med. Genet. A 2014, 164A, 2206–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, Y.T.; Aliaga, S.M.; Arpone, M.; Francis, D.; Li, X.; Chong, B.; Slater, H.R.; Rogers, C.; Bretherton, L.; Hunter, M.; et al. Partially methylated alleles, microdeletion, and tissue mosaicism in a fragile X male with tremor and ataxia at 30 years of age: A case report. Am. J. Med. Genet. A 2016, 170, 3327–3332. [Google Scholar] [CrossRef] [Green Version]
- Bailey, D.B., Jr.; Raspa, M.; Olmsted, M.; Holiday, D.B. Co-occurring conditions associated with FMR1 gene variations: Findings from a national parent survey. Am. J. Med. Genet. A 2008, 146A, 2060–2069. [Google Scholar] [CrossRef]
- Hagerman, R.; Hoem, G.; Hagerman, P. Fragile X and autism: Intertwined at the molecular level leading to targeted treatments. Mol. Autism 2010, 1, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagerman, R.; Au, J.; Hagerman, P. FMR1 premutation and full mutation molecular mechanisms related to autism. J. Neurodev. Disord. 2011, 3, 211–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagerman, R.J.; Protic, D.; Rajaratnam, A.; Salcedo-Arellano, M.J.; Aydin, E.Y.; Schneider, A. Fragile X-Associated Neuropsychiatric Disorders (FXAND). Front. Psychiatry 2018, 9, 564. [Google Scholar] [CrossRef] [Green Version]
- Schneider, A.; Johnston, C.; Tassone, F.; Sansone, S.; Hagerman, R.J.; Ferrer, E.; Rivera, S.M.; Hessl, D. Broad autism spectrum and obsessive-compulsive symptoms in adults with the fragile X premutation. Clin. Neuropsychol. 2016, 30, 929–943. [Google Scholar] [CrossRef] [Green Version]
- Strimbu, K.; Tavel, J.A. What are biomarkers? Curr. Opin. HIV AIDS 2010, 5, 463–466. [Google Scholar] [CrossRef]
- Alvarez-Mora, M.I.; Rodriguez-Revenga, L.; Madrigal, I.; Torres-Silva, F.; Mateu-Huertas, E.; Lizano, E.; Friedlander, M.R.; Marti, E.; Estivill, X.; Mila, M. MicroRNA expression profiling in blood from fragile X-associated tremor/ataxia syndrome patients. Genes Brain Behav. 2013, 12, 595–603. [Google Scholar] [CrossRef] [Green Version]
- Mateu-Huertas, E.; Rodriguez-Revenga, L.; Alvarez-Mora, M.I.; Madrigal, I.; Willemsen, R.; Mila, M.; Marti, E.; Estivill, X. Blood expression profiles of fragile X premutation carriers identify candidate genes involved in neurodegenerative and infertility phenotypes. Neurobiol. Dis. 2014, 65, 43–54. [Google Scholar] [CrossRef]
- Cao, Y.; Peng, Y.; Kong, H.E.; Allen, E.G.; Jin, P. Metabolic alterations in FMR1 premutation carriers. Front. Mol. Biosci. 2020, 7, 571092. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Napoli, E.; Wong, S.; Hagerman, R.; Liu, S.; Tassone, F.; Giulivi, C. Altered redox mitochondrial biology in the neurodegenerative disorder fragile X-tremor/ataxia syndrome: Use of antioxidants in precision medicine. Mol. Med. 2016, 22, 548–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napoli, E.; McLennan, Y.A.; Schneider, A.; Tassone, F.; Hagerman, R.J.; Giulivi, C. Characterization of the metabolic, clinical and neuropsychological phenotype of female carriers of the premutation in the X-Linked FMR1 gene. Front. Mol. Biosci. 2020, 7, 578640. [Google Scholar] [CrossRef]
- Allen, E.G.; Sullivan, A.K.; Marcus, M.; Small, C.; Dominguez, C.; Epstein, M.P.; Charen, K.; He, W.; Taylor, K.C.; Sherman, S.L. Examination of reproductive aging milestones among women who carry the FMR1 premutation. Hum. Reprod. 2007, 22, 2142–2152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouwer, J.R.; Huizer, K.; Severijnen, L.A.; Hukema, R.K.; Berman, R.F.; Oostra, B.A.; Willemsen, R. CGG-repeat length and neuropathological and molecular correlates in a mouse model for fragile X-associated tremor/ataxia syndrome. J. Neurochem. 2008, 107, 1671–1682. [Google Scholar] [CrossRef] [Green Version]
- White, P.J.; Borts, R.H.; Hirst, M.C. Stability of the human fragile X (CGG)(n) triplet repeat array in Saccharomyces cerevisiae deficient in aspects of DNA metabolism. Mol. Cell. Biol. 1999, 19, 5675–5684. [Google Scholar] [CrossRef] [Green Version]
- Kononenko, A.V.; Ebersole, T.; Vasquez, K.M.; Mirkin, S.M. Mechanisms of genetic instability caused by (CGG)n repeats in an experimental mammalian system. Nat. Struct. Mol. Biol. 2018, 25, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.; Hughes, A. Pathogenic mechanisms in Huntington’s disease. Int. Rev. Neurobiol. 2011, 98, 373–418. [Google Scholar] [CrossRef]
- Savic Pavicevic, D.; Miladinovic, J.; Brkusanin, M.; Svikovic, S.; Djurica, S.; Brajuskovic, G.; Romac, S. Molecular genetics and genetic testing in myotonic dystrophy type 1. Biomed. Res. Int. 2013, 2013, 391821. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.H.M.; Pearson, C.E. Disease-associated repeat instability and mismatch repair. DNA Repair 2016, 38, 117–126. [Google Scholar] [CrossRef]
- Lokanga, R.A.; Entezam, A.; Kumari, D.; Yudkin, D.; Qin, M.; Smith, C.B.; Usdin, K. Somatic expansion in mouse and human carriers of fragile X premutation alleles. Hum. Mutat. 2013, 34, 157–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tassone, F.; Hagerman, R.J.; Taylor, A.K.; Mills, J.B.; Harris, S.W.; Gane, L.W.; Hagerman, P.J. Clinical involvement and protein expression in individuals with the FMR1 premutation. Am. J. Med. Genet. 2000, 91, 144–152. [Google Scholar] [CrossRef]
- Allen, E.G.; Sherman, S.; Abramowitz, A.; Leslie, M.; Novak, G.; Rusin, M.; Scott, E.; Letz, R. Examination of the effect of the polymorphic CGG repeat in the FMR1 gene on cognitive performance. Behav. Genet. 2005, 35, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Hessl, D.; Tassone, F.; Loesch, D.Z.; Berry-Kravis, E.; Leehey, M.A.; Gane, L.W.; Barbato, I.; Rice, C.; Gould, E.; Hall, D.A.; et al. Abnormal elevation of FMR1 mRNA is associated with psychological symptoms in individuals with the fragile X premutation. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2005, 139B, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.; Todorova-Koteva, K.; Pandya, S.; Bernard, B.; Ouyang, B.; Walsh, M.; Pounardjian, T.; Deburghraeve, C.; Zhou, L.; Losh, M.; et al. Neurological and endocrine phenotypes of fragile X carrier women. Clin. Genet. 2016, 89, 60–67. [Google Scholar] [CrossRef] [Green Version]
- Moore, C.J.; Daly, E.M.; Tassone, F.; Tysoe, C.; Schmitz, N.; Ng, V.; Chitnis, X.; McGuire, P.; Suckling, J.; Davies, K.E.; et al. The effect of pre-mutation of X chromosome CGG trinucleotide repeats on brain anatomy. Brain 2004, 127, 2672–2681. [Google Scholar] [CrossRef] [PubMed]
- Koldewyn, K.; Hessl, D.; Adams, J.; Tassone, F.; Hagerman, P.J.; Hagerman, R.J.; Rivera, S.M. Reduced hippocampal activation during recall is associated with elevated FMR1 mRNA and psychiatric symptoms in men with the fragile X premutation. Brain Imaging Behav. 2008, 2, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, R.; Backer, K.C.; Tassone, F.; Hagerman, R.J.; Rivera, S.M. An fMRI study of the prefrontal activity during the performance of a working memory task in premutation carriers of the fragile X mental retardation 1 gene with and without fragile X-associated tremor/ataxia syndrome (FXTAS). J. Psychiatr. Res. 2011, 45, 36–43. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.M.; Koldewyn, K.; Hashimoto, R.; Schneider, A.; Le, L.; Tassone, F.; Cheung, K.; Hagerman, P.; Hessl, D.; Rivera, S.M. Male carriers of the FMR1 premutation show altered hippocampal-prefrontal function during memory encoding. Front. Hum. Neurosci. 2012, 6, 297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.Y.; Hashimoto, R.; Tassone, F.; Simon, T.J.; Rivera, S.M. Altered neural activity of magnitude estimation processing in adults with the fragile X premutation. J. Psychiatr. Res. 2013, 47, 1909–1916. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.Y.; Hessl, D.; Schneider, A.; Tassone, F.; Hagerman, R.J.; Rivera, S.M. Fragile X-associated tremor/ataxia syndrome: Influence of the FMR1 gene on motor fiber tracts in males with normal and premutation alleles. JAMA Neurol. 2013, 70, 1022–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hocking, D.R.; Birch, R.C.; Bui, Q.M.; Menant, J.C.; Lord, S.R.; Georgiou-Karistianis, N.; Godler, D.E.; Wen, W.; Hackett, A.; Rogers, C.; et al. Cerebellar volume mediates the relationship between FMR1 mRNA levels and voluntary step initiation in males with the premutation. Neurobiol. Aging 2017, 50, 5–12. [Google Scholar] [CrossRef]
- Brown, S.S.G.; Basu, S.; Whalley, H.C.; Kind, P.C.; Stanfield, A.C. Age-related functional brain changes in FMR1 premutation carriers. Neuroimage Clin. 2018, 17, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Loesch, D.Z.; Godler, D.E.; Evans, A.; Bui, Q.M.; Gehling, F.; Kotschet, K.E.; Trost, N.; Storey, E.; Stimpson, P.; Kinsella, G.; et al. Evidence for the toxicity of bidirectional transcripts and mitochondrial dysfunction in blood associated with small CGG expansions in the FMR1 gene in patients with parkinsonism. Genet. Med. 2011, 13, 392–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vittal, P.; Pandya, S.; Sharp, K.; Berry-Kravis, E.; Zhou, L.; Ouyang, B.; Jackson, J.; Hall, D.A. ASFMR1 splice variant: A predictor of fragile X-associated tremor/ataxia syndrome. Neurol. Genet. 2018, 4, e246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Olaby, R.R.; Tang, H.T.; Durbin-Johnson, B.; Schneider, A.; Hessl, D.; Rivera, S.M.; Tassone, F. Assessment of molecular measures in non-FXTAS male premutation carriers. Front. Genet. 2018, 9, 302. [Google Scholar] [CrossRef] [Green Version]
- Zafarullah, M.; Tang, H.T.; Durbin-Johnson, B.; Fourie, E.; Hessl, D.; Rivera, S.M.; Tassone, F. FMR1 locus isoforms: Potential biomarker candidates in fragile X-associated tremor/ataxia syndrome (FXTAS). Sci. Rep. 2020, 10, 11099. [Google Scholar] [CrossRef]
- Pretto, D.I.; Eid, J.S.; Yrigollen, C.M.; Tang, H.T.; Loomis, E.W.; Raske, C.; Durbin-Johnson, B.; Hagerman, P.J.; Tassone, F. Differential increases of specific FMR1 mRNA isoforms in premutation carriers. J. Med. Genet. 2015, 52, 42–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, E.; Tang, H.T.; AlOlaby, R.R.; Hickey, L.; Tassone, F. Altered expression of the FMR1 splicing variants landscape in premutation carriers. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 1117–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasilyev, N.; Polonskaia, A.; Darnell, J.C.; Darnell, R.B.; Patel, D.J.; Serganov, A. Crystal structure reveals specific recognition of a G-quadruplex RNA by a beta-turn in the RGG motif of FMRP. Proc. Natl. Acad. Sci. USA 2015, 112, E5391–E5400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yrigollen, C.M.; Durbin-Johnson, B.; Gane, L.; Nelson, D.L.; Hagerman, R.; Hagerman, P.J.; Tassone, F. AGG interruptions within the maternal FMR1 gene reduce the risk of offspring with fragile X syndrome. Genet. Med. 2012, 14, 729–736. [Google Scholar] [CrossRef] [Green Version]
- Cornish, K.M.; Kraan, C.M.; Bui, Q.M.; Bellgrove, M.A.; Metcalfe, S.A.; Trollor, J.N.; Hocking, D.R.; Slater, H.R.; Inaba, Y.; Li, X.; et al. Novel methylation markers of the dysexecutive-psychiatric phenotype in FMR1 premutation women. Neurology 2015, 84, 1631–1638. [Google Scholar] [CrossRef] [Green Version]
- Shelton, A.L.; Cornish, K.M.; Kolbe, S.; Clough, M.; Slater, H.R.; Li, X.; Kraan, C.M.; Bui, Q.M.; Godler, D.E.; Fielding, J. Brain structure and intragenic DNA methylation are correlated, and predict executive dysfunction in fragile X premutation females. Transl. Psychiatry 2016, 6, e984. [Google Scholar] [CrossRef] [Green Version]
- Amer Abed, F.; Ezzat Maroof, R.; Al-Nakkash, U.M.A. Comparing the diagnostic accuracy of anti-mullerian hormone and follicle stimulating hormone in detecting premature ovarian failure in Iraqi women by ROC analysis. Rep. Biochem. Mol. Biol. 2019, 8, 126–131. [Google Scholar]
- Lee, D.H.; Pei, C.Z.; Song, J.Y.; Lee, K.J.; Yun, B.S.; Kwack, K.B.; Lee, E.I.; Baek, K.H. Identification of serum biomarkers for premature ovarian failure. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 219–226. [Google Scholar] [CrossRef]
- Welt, C.K.; Smith, P.C.; Taylor, A.E. Evidence of early ovarian aging in fragile X premutation carriers. J. Clin. Endocrinol. Metab. 2004, 89, 4569–4574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, A.; Webb, J.; MacSwiney, F.; Shipley, E.L.; Morton, N.E.; Conway, G.S. Serum concentrations of follicle stimulating hormone may predict premature ovarian failure in FRAXA premutation women. Hum. Reprod. 1999, 14, 1217–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hundscheid, R.D.; Braat, D.D.; Kiemeney, L.A.; Smits, A.P.; Thomas, C.M. Increased serum FSH in female fragile X premutation carriers with either regular menstrual cycles or on oral contraceptives. Hum. Reprod. 2001, 16, 457–462. [Google Scholar] [CrossRef] [Green Version]
- Barasoain, M.; Barrenetxea, G.; Huerta, I.; Telez, M.; Carrillo, A.; Perez, C.; Criado, B.; Arrieta, I. Study of FMR1 gene association with ovarian dysfunction in a sample from the Basque Country. Gene 2013, 521, 145–149. [Google Scholar] [CrossRef]
- Hoffman, G.E.; Le, W.W.; Entezam, A.; Otsuka, N.; Tong, Z.B.; Nelson, L.; Flaws, J.A.; McDonald, J.H.; Jafar, S.; Usdin, K. Ovarian abnormalities in a mouse model of fragile X primary ovarian insufficiency. J. Histochem. Cytochem. 2012, 60, 439–456. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Lin, L.; Tan, H.; Wu, H.; Sherman, S.L.; Gao, F.; Jin, P.; Chen, D. Fragile X premutation RNA is sufficient to cause primary ovarian insufficiency in mice. Hum. Mol. Genet. 2012, 21, 5039–5047. [Google Scholar] [CrossRef] [Green Version]
- Rohr, J.; Allen, E.G.; Charen, K.; Giles, J.; He, W.; Dominguez, C.; Sherman, S.L. Anti-Mullerian hormone indicates early ovarian decline in fragile X mental retardation (FMR1) premutation carriers: A preliminary study. Hum. Reprod. 2008, 23, 1220–1225. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.Y.; Liu, Z.; Cahill, N.; Richards, J.S. Targeted disruption of Pten in ovarian granulosa cells enhances ovulation and extends the life span of luteal cells. Mol. Endocrinol. 2008, 22, 2128–2140. [Google Scholar] [CrossRef] [Green Version]
- Makker, A.; Goel, M.M.; Mahdi, A.A. PI3K/PTEN/Akt and TSC/mTOR signaling pathways, ovarian dysfunction, and infertility: An update. J. Mol. Endocrinol. 2014, 53, R103–R118. [Google Scholar] [CrossRef] [Green Version]
- Rose, B.I.; Brown, S.E. An explanation of the mechanisms underlying fragile X-associated premature ovarian insufficiency. J. Assist. Reprod. Genet. 2020, 37, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Louis, E.; Moskowitz, C.; Friez, M.; Amaya, M.; Vonsattel, J.P. Parkinsonism, dysautonomia, and intranuclear inclusions in a fragile X carrier: A clinical-pathological study. Mov. Disord. 2006, 21, 420–425. [Google Scholar] [CrossRef]
- Brouwer, J.R.; Severijnen, E.; de Jong, F.H.; Hessl, D.; Hagerman, R.J.; Oostra, B.A.; Willemsen, R. Altered hypothalamus-pituitary-adrenal gland axis regulation in the expanded CGG-repeat mouse model for fragile X-associated tremor/ataxia syndrome. Psychoneuroendocrinology 2008, 33, 863–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spath, M.A.; Feuth, T.B.; Allen, E.G.; Smits, A.P.; Yntema, H.G.; van Kessel, A.G.; Braat, D.D.; Sherman, S.L.; Thomas, C.M. Intra-individual stability over time of standardized anti-Mullerian hormone in FMR1 premutation carriers. Hum. Reprod. 2011, 26, 2185–2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creus, M.; Penarrubia, J.; Fabregues, F.; Vidal, E.; Carmona, F.; Casamitjana, R.; Vanrell, J.A.; Balasch, J. Day 3 serum inhibin B and FSH and age as predictors of assisted reproduction treatment outcome. Hum. Reprod. 2000, 15, 2341–2346. [Google Scholar] [CrossRef]
- Maslow, B.S.; Davis, S.; Engmann, L.; Nulsen, J.C.; Benadiva, C.A. Correlation of normal-range FMR1 repeat length or genotypes and reproductive parameters. J. Assist. Reprod. Genet. 2016, 33, 1149–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Bazhin, A.V.; Werner, J.; Karakhanova, S. Reactive oxygen species in the immune system. Int. Rev. Immunol. 2013, 32, 249–270. [Google Scholar] [CrossRef] [PubMed]
- Terzi, A.; Suter, D.M. The role of NADPH oxidases in neuronal development. Free Radic. Biol. Med. 2020, 154, 33–47. [Google Scholar] [CrossRef]
- Yun, H.R.; Jo, Y.H.; Kim, J.; Shin, Y.; Kim, S.S.; Choi, T.G. Roles of autophagy in oxidative stress. Int. J. Mol. Sci. 2020, 21, 3289. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative stress: A key modulator in neurodegenerative diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [Green Version]
- Panth, N.; Paudel, K.R.; Parajuli, K. Reactive oxygen species: A key hallmark of cardiovascular disease. Adv. Med. 2016, 2016, 9152732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Bekay, R.; Romero-Zerbo, Y.; Decara, J.; Sanchez-Salido, L.; Del Arco-Herrera, I.; Rodriguez-de Fonseca, F.; de Diego-Otero, Y. Enhanced markers of oxidative stress, altered antioxidants and NADPH-oxidase activation in brains from Fragile X mental retardation 1-deficient mice, a pathological model for Fragile X syndrome. Eur. J. Neurosci. 2007, 26, 3169–3180. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, E.S.; Cao, Z.; Hulsizer, S.; Tassone, F.; Berman, R.F.; Hagerman, P.J.; Pessah, I.N. Early mitochondrial abnormalities in hippocampal neurons cultured from Fmr1 pre-mutation mouse model. J. Neurochem. 2012, 123, 613–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napoli, E.; Ross-Inta, C.; Wong, S.; Omanska-Klusek, A.; Barrow, C.; Iwahashi, C.; Garcia-Arocena, D.; Sakaguchi, D.; Berry-Kravis, E.; Hagerman, R.; et al. Altered zinc transport disrupts mitochondrial protein processing/import in fragile X-associated tremor/ataxia syndrome. Hum. Mol. Genet. 2011, 20, 3079–3092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napoli, E.; Song, G.; Wong, S.; Hagerman, R.; Giulivi, C. Altered bioenergetics in primary dermal fibroblasts from adult carriers of the FMR1 premutation before the onset of the neurodegenerative disease fragile X-associated tremor/ataxia syndrome. Cerebellum 2016, 15, 552–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross-Inta, C.; Omanska-Klusek, A.; Wong, S.; Barrow, C.; Garcia-Arocena, D.; Iwahashi, C.; Berry-Kravis, E.; Hagerman, R.J.; Hagerman, P.J.; Giulivi, C. Evidence of mitochondrial dysfunction in fragile X-associated tremor/ataxia syndrome. Biochem. J. 2010, 429, 545–552. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Mora, M.I.; Rodriguez-Revenga, L.; Madrigal, I.; Guitart-Mampel, M.; Garrabou, G.; Mila, M. Impaired mitochondrial function and dynamics in the pathogenesis of FXTAS. Mol. Neurobiol. 2017, 54, 6896–6902. [Google Scholar] [CrossRef]
- Nobile, V.; Palumbo, F.; Lanni, S.; Ghisio, V.; Vitali, A.; Castagnola, M.; Marzano, V.; Maulucci, G.; De Angelis, C.; De Spirito, M.; et al. Altered mitochondrial function in cells carrying a premutation or unmethylated full mutation of the FMR1 gene. Hum. Genet. 2020, 139, 227–245. [Google Scholar] [CrossRef] [PubMed]
- Loesch, D.Z.; Annesley, S.J.; Trost, N.; Bui, M.Q.; Lay, S.T.; Storey, E.; De Piazza, S.W.; Sanislav, O.; Francione, L.M.; Hammersley, E.M.; et al. Novel blood biomarkers are associated with white matter lesions in fragile X- associated tremor/ataxia syndrome. Neurodegener. Dis. 2017, 17, 22–30. [Google Scholar] [CrossRef]
- Napoli, E.; Schneider, A.; Hagerman, R.; Song, G.; Wong, S.; Tassone, F.; Giulivi, C. Impact of FMR1 premutation on neurobehavior and bioenergetics in young monozygotic twins. Front. Genet. 2018, 9, 338. [Google Scholar] [CrossRef] [PubMed]
- Alfatni, A.; Riou, M.; Charles, A.L.; Meyer, A.; Barnig, C.; Andres, E.; Lejay, A.; Talha, S.; Geny, B. Peripheral blood mononuclear cells and platelets mitochondrial dysfunction, oxidative stress, and circulating mtDNA in cardiovascular diseases. J. Clin. Med. 2020, 9, 311. [Google Scholar] [CrossRef] [Green Version]
- Afrifa, J.; Zhao, T.; Yu, J. Circulating mitochondria DNA, a non-invasive cancer diagnostic biomarker candidate. Mitochondrion 2019, 47, 238–243. [Google Scholar] [CrossRef]
- Gambardella, S.; Limanaqi, F.; Ferese, R.; Biagioni, F.; Campopiano, R.; Centonze, D.; Fornai, F. ccf-mtDNA as a potential link between the brain and immune system in neuro-immunological disorders. Front. Immunol. 2019, 10, 1064. [Google Scholar] [CrossRef] [Green Version]
- Lebedeva, M.A.; Eaton, J.S.; Shadel, G.S. Loss of p53 causes mitochondrial DNA depletion and altered mitochondrial reactive oxygen species homeostasis. Biochim. Biophys. Acta 2009, 1787, 328–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Zhang, L.; Yu, X.; Zhou, H.; Luo, Y.; Wang, W.; Wang, L. Clinical application of plasma mitochondrial DNA content in patients with lung cancer. Oncol. Lett. 2018, 16, 7074–7081. [Google Scholar] [CrossRef]
- Alvarez-Mora, M.I.; Podlesniy, P.; Gelpi, E.; Hukema, R.; Madrigal, I.; Pagonabarraga, J.; Trullas, R.; Mila, M.; Rodriguez-Revenga, L. Fragile X-associated tremor/ataxia syndrome: Regional decrease of mitochondrial DNA copy number relates to clinical manifestations. Genes Brain Behav. 2019, 18, e12565. [Google Scholar] [CrossRef] [PubMed]
- Kala, M.; Shaikh, M.V.; Nivsarkar, M. Equilibrium between anti-oxidants and reactive oxygen species: A requisite for oocyte development and maturation. Reprod. Med. Biol. 2017, 16, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Conca Dioguardi, C.; Uslu, B.; Haynes, M.; Kurus, M.; Gul, M.; Miao, D.Q.; De Santis, L.; Ferrari, M.; Bellone, S.; Santin, A.; et al. Granulosa cell and oocyte mitochondrial abnormalities in a mouse model of fragile X primary ovarian insufficiency. Mol. Hum. Reprod. 2016, 22, 384–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gohel, D.; Sripada, L.; Prajapati, P.; Singh, K.; Roy, M.; Kotadia, D.; Tassone, F.; Charlet-Berguerand, N.; Singh, R. FMRpolyG alters mitochondrial transcripts level and respiratory chain complex assembly in Fragile X associated tremor/ataxia syndrome [FXTAS]. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- Gohel, D.; Berguerand, N.C.; Tassone, F.; Singh, R. The emerging molecular mechanisms for mitochondrial dysfunctions in FXTAS. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165918. [Google Scholar] [CrossRef]
- Gohel, D.; Sripada, L.; Prajapati, P.; Currim, F.; Roy, M.; Singh, K.; Shinde, A.; Mane, M.; Kotadia, D.; Tassone, F.; et al. Expression of expanded FMR1-CGG repeats alters mitochondrial miRNAs and modulates mitochondrial functions and cell death in cellular model of FXTAS. Free Radic. Biol. Med. 2021, 165, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Hukema, R.K.; Buijsen, R.A.; Raske, C.; Severijnen, L.A.; Nieuwenhuizen-Bakker, I.; Minneboo, M.; Maas, A.; de Crom, R.; Kros, J.M.; Hagerman, P.J.; et al. Induced expression of expanded CGG RNA causes mitochondrial dysfunction in vivo. Cell Cycle 2014, 13, 2600–2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giulivi, C.; Napoli, E.; Tassone, F.; Halmai, J.; Hagerman, R. Plasma biomarkers for monitoring brain pathophysiology in FMR1 premutation carriers. Front. Mol. Neurosci. 2016, 9, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giulivi, C.; Napoli, E.; Tassone, F.; Halmai, J.; Hagerman, R. Plasma metabolic profile delineates roles for neurodegeneration, pro-inflammatory damage and mitochondrial dysfunction in the FMR1 premutation. Biochem. J. 2016, 473, 3871–3888. [Google Scholar] [CrossRef]
- Suzuki, Y.J.; Carini, M.; Butterfield, D.A. Protein carbonylation. Antioxid. Redox Signal. 2010, 12, 323–325. [Google Scholar] [CrossRef]
- Marrocco, I.; Altieri, F.; Peluso, I. Measurement and clinical significance of biomarkers of oxidative stress in humans. Oxid. Med. Cell. Longev. 2017, 2017, 6501046. [Google Scholar] [CrossRef]
- Moselhy, H.F.; Reid, R.G.; Yousef, S.; Boyle, S.P. A specific, accurate, and sensitive measure of total plasma malondialdehyde by HPLC. J. Lipid Res. 2013, 54, 852–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez Cerdeno, V.; Hong, T.; Amina, S.; Lechpammer, M.; Ariza, J.; Tassone, F.; Noctor, S.C.; Hagerman, P.; Hagerman, R. Microglial cell activation and senescence are characteristic of the pathology FXTAS. Mov. Disord. 2018, 33, 1887–1894. [Google Scholar] [CrossRef]
- Dufour, B.D.; Amina, S.; Martinez-Cerdeno, V. FXTAS presents with upregulation of the cytokines IL12 and TNFalpha. Parkinsonism Relat. Disord. 2021, 82, 117–120. [Google Scholar] [CrossRef]
- Porro, C.; Cianciulli, A.; Panaro, M.A. The regulatory role of IL-10 in neurodegenerative diseases. Biomolecules 2020, 10, 1017. [Google Scholar] [CrossRef]
- Marek, D.; Papin, S.; Ellefsen, K.; Niederhauser, J.; Isidor, N.; Ransijn, A.; Poupon, L.; Spertini, F.; Pantaleo, G.; Bergmann, S.; et al. Carriers of the fragile X mental retardation 1 (FMR1) premutation allele present with increased levels of cytokine IL-10. J. Neuroinflammation 2012, 9, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michlewski, G.; Caceres, J.F. Post-transcriptional control of miRNA biogenesis. RNA 2019, 25, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salloum-Asfar, S.; Satheesh, N.J.; Abdulla, S.A. Circulating miRNAs, small but promising biomarkers for autism spectrum disorder. Front. Mol. Neurosci. 2019, 12, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, G.L.; Platania, C.B.M.; Drago, F.; Salomone, S.; Ragusa, M.; Barbagallo, C.; Di Pietro, C.; Purrello, M.; Reibaldi, M.; Avitabile, T.; et al. Retinal and circulating miRNAs in age-related macular degeneration: An in vivo animal and human study. Front. Pharmacol. 2017, 8, 168. [Google Scholar] [CrossRef] [Green Version]
- Low, Y.H.; Asi, Y.; Foti, S.C.; Lashley, T. Heterogeneous nuclear ribonucleoproteins: Implications in neurological diseases. Mol. Neurobiol. 2021, 58, 631–646. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Poidevin, M.; Li, H.; Chen, D.; Jin, P. MicroRNA-277 modulates the neurodegeneration caused by Fragile X premutation rCGG repeats. PLoS Genet. 2012, 8, e1002681. [Google Scholar] [CrossRef] [PubMed]
- Schermelleh, L.; Carlton, P.M.; Haase, S.; Shao, L.; Winoto, L.; Kner, P.; Burke, B.; Cardoso, M.C.; Agard, D.A.; Gustafsson, M.G.; et al. Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science 2008, 320, 1332–1336. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.L.; Poleshko, A.; Epstein, J.A. The nuclear periphery is a scaffold for tissue-specific enhancers. Nucleic Acids Res. 2021, 49, 6181–6195. [Google Scholar] [CrossRef]
- Garcia-Arocena, D.; Yang, J.E.; Brouwer, J.R.; Tassone, F.; Iwahashi, C.; Berry-Kravis, E.M.; Goetz, C.G.; Sumis, A.M.; Zhou, L.; Nguyen, D.V.; et al. Fibroblast phenotype in male carriers of FMR1 premutation alleles. Hum. Mol. Genet. 2010, 19, 299–312. [Google Scholar] [CrossRef] [Green Version]
- Hoem, G.; Raske, C.R.; Garcia-Arocena, D.; Tassone, F.; Sanchez, E.; Ludwig, A.L.; Iwahashi, C.K.; Kumar, M.; Yang, J.E.; Hagerman, P.J. CGG-repeat length threshold for FMR1 RNA pathogenesis in a cellular model for FXTAS. Hum. Mol. Genet. 2011, 20, 2161–2170. [Google Scholar] [CrossRef]
- Arocena, D.G.; Iwahashi, C.K.; Won, N.; Beilina, A.; Ludwig, A.L.; Tassone, F.; Schwartz, P.H.; Hagerman, P.J. Induction of inclusion formation and disruption of lamin A/C structure by premutation CGG-repeat RNA in human cultured neural cells. Hum. Mol. Genet. 2005, 14, 3661–3671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachman, M.; Uribe-Lewis, S.; Yang, X.; Williams, M.; Murrell, A.; Balasubramanian, S. 5-Hydroxymethylcytosine is a predominantly stable DNA modification. Nat. Chem. 2014, 6, 1049–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, B.; Lin, L.; Street, R.C.; Zalewski, Z.A.; Galloway, J.N.; Wu, H.; Nelson, D.L.; Jin, P. Genome-wide alteration of 5-hydroxymethylcytosine in a mouse model of fragile X-associated tremor/ataxia syndrome. Hum. Mol. Genet. 2014, 23, 1095–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrade-Navarro, M.A.; Muhlenberg, K.; Spruth, E.J.; Mah, N.; Gonzalez-Lopez, A.; Andreani, T.; Russ, J.; Huska, M.R.; Muro, E.M.; Fontaine, J.F.; et al. RNA Sequencing of human peripheral blood cells indicates upregulation of immune-related genes in Huntington’s disease. Front. Neurol. 2020, 11, 573560. [Google Scholar] [CrossRef] [PubMed]
- Heger, S.; Mastronardi, C.; Dissen, G.A.; Lomniczi, A.; Cabrera, R.; Roth, C.L.; Jung, H.; Galimi, F.; Sippell, W.; Ojeda, S.R. Enhanced at puberty 1 (EAP1) is a new transcriptional regulator of the female neuroendocrine reproductive axis. J. Clin. Investig. 2007, 117, 2145–2154. [Google Scholar] [CrossRef]
- Marcogliese, P.C.; Shashi, V.; Spillmann, R.C.; Stong, N.; Rosenfeld, J.A.; Koenig, M.K.; Martinez-Agosto, J.A.; Herzog, M.; Chen, A.H.; Dickson, P.I.; et al. IRF2BPL is associated with neurological phenotypes. Am. J. Hum. Genet. 2018, 103, 245–260. [Google Scholar] [CrossRef] [Green Version]
- Tran Mau-Them, F.; Guibaud, L.; Duplomb, L.; Keren, B.; Lindstrom, K.; Marey, I.; Mochel, F.; van den Boogaard, M.J.; Oegema, R.; Nava, C.; et al. De novo truncating variants in the intronless IRF2BPL are responsible for developmental epileptic encephalopathy. Genet. Med. 2019, 21, 1008–1014. [Google Scholar] [CrossRef]
- Alvarez-Mora, M.I.; Rodriguez-Revenga, L.; Madrigal, I.; Garcia-Garcia, F.; Duran, M.; Dopazo, J.; Estivill, X.; Mila, M. Deregulation of key signaling pathways involved in oocyte maturation in FMR1 premutation carriers with Fragile X-associated primary ovarian insufficiency. Gene 2015, 571, 52–57. [Google Scholar] [CrossRef]
- Schur, R.R.; Draisma, L.W.; Wijnen, J.P.; Boks, M.P.; Koevoets, M.G.; Joels, M.; Klomp, D.W.; Kahn, R.S.; Vinkers, C.H. Brain GABA levels across psychiatric disorders: A systematic literature review and meta-analysis of (1) H-MRS studies. Hum. Brain Mapp. 2016, 37, 3337–3352. [Google Scholar] [CrossRef] [Green Version]
- Chiapponi, C.; Piras, F.; Caltagirone, C.; Spalletta, G. GABA system in schizophrenia and mood disorders: A mini review on third-generation imaging studies. Front. Psychiatry 2016, 7, 61. [Google Scholar] [CrossRef] [Green Version]
- Prevot, T.; Sibille, E. Altered GABA-mediated information processing and cognitive dysfunctions in depression and other brain disorders. Mol. Psychiatry 2021, 26, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Conde, V.; Palomar, F.J.; Lama, M.J.; Martinez, R.; Carrillo, F.; Pintado, E.; Mir, P. Abnormal GABA-mediated and cerebellar inhibition in women with the fragile X premutation. J. Neurophysiol. 2013, 109, 1315–1322. [Google Scholar] [CrossRef] [Green Version]
- Gantois, I.; Vandesompele, J.; Speleman, F.; Reyniers, E.; D’Hooge, R.; Severijnen, L.A.; Willemsen, R.; Tassone, F.; Kooy, R.F. Expression profiling suggests underexpression of the GABA(A) receptor subunit delta in the fragile X knockout mouse model. Neurobiol. Dis. 2006, 21, 346–357. [Google Scholar] [CrossRef] [PubMed]
- D’Hulst, C.; De Geest, N.; Reeve, S.P.; Van Dam, D.; De Deyn, P.P.; Hassan, B.A.; Kooy, R.F. Decreased expression of the GABAA receptor in fragile X syndrome. Brain Res. 2006, 1121, 238–245. [Google Scholar] [CrossRef]
- D’Hulst, C.; Heulens, I.; Brouwer, J.R.; Willemsen, R.; De Geest, N.; Reeve, S.P.; De Deyn, P.P.; Hassan, B.A.; Kooy, R.F. Expression of the GABAergic system in animal models for fragile X syndrome and fragile X associated tremor/ataxia syndrome (FXTAS). Brain Res. 2009, 1253, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Hulsizer, S.; Tassone, F.; Tang, H.T.; Hagerman, R.J.; Rogawski, M.A.; Hagerman, P.J.; Pessah, I.N. Clustered burst firing in FMR1 premutation hippocampal neurons: Amelioration with allopregnanolone. Hum. Mol. Genet. 2012, 21, 2923–2935. [Google Scholar] [CrossRef] [Green Version]
- Coyne, L.; Lees, G.; Nicholson, R.A.; Zheng, J.; Neufield, K.D. The sleep hormone oleamide modulates inhibitory ionotropic receptors in mammalian CNS in vitro. Br. J. Pharmacol. 2002, 135, 1977–1987. [Google Scholar] [CrossRef] [Green Version]
- Akanmu, M.A.; Adeosun, S.O.; Ilesanmi, O.R. Neuropharmacological effects of oleamide in male and female mice. Behav. Brain Res. 2007, 182, 88–94. [Google Scholar] [CrossRef]
- Wang, J.Y.; Trivedi, A.M.; Carrillo, N.R.; Yang, J.; Schneider, A.; Giulivi, C.; Adams, P.; Tassone, F.; Kim, K.; Rivera, S.M.; et al. Open-label allopregnanolone treatment of men with fragile X-associated tremor/ataxia syndrome. Neurotherapeutics 2017, 14, 1073–1083. [Google Scholar] [CrossRef] [PubMed]
- Napoli, E.; Schneider, A.; Wang, J.Y.; Trivedi, A.; Carrillo, N.R.; Tassone, F.; Rogawski, M.; Hagerman, R.J.; Giulivi, C. Allopregnanolone treatment improves plasma metabolomic profile associated with GABA metabolism in fragile X-associated tremor/ataxia syndrome: A pilot study. Mol. Neurobiol. 2019, 56, 3702–3713. [Google Scholar] [CrossRef]
- Blackburn, E.H.; Epel, E.S.; Lin, J. Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science 2015, 350, 1193–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, M.T. Telomere biology in aging and cancer: Early history and perspectives. Genes Genet. Syst. 2018, 92, 107–118. [Google Scholar] [CrossRef] [Green Version]
- Martinez, P.; Blasco, M.A. Heart-breaking telomeres. Circ. Res. 2018, 123, 787–802. [Google Scholar] [CrossRef]
- Chen, M.S.; Lee, R.T.; Garbern, J.C. Senescence mechanisms and targets in the heart. Cardiovasc. Res. 2021, in press. [Google Scholar] [CrossRef]
- Vodicka, P.; Andera, L.; Opattova, A.; Vodickova, L. The interactions of DNA repair, telomere homeostasis, and p53 mutational status in solid cancers: Risk, prognosis, and prediction. Cancers 2021, 13, 479. [Google Scholar] [CrossRef] [PubMed]
- Vaiserman, A.; Krasnienkov, D. Telomere length as a marker of biological age: State-of-the-art, open issues, and future perspectives. Front. Genet. 2020, 11, 630186. [Google Scholar] [CrossRef]
- Cai, Z.; Yan, L.J.; Ratka, A. Telomere shortening and Alzheimer’s disease. Neuromol. Med. 2013, 15, 25–48. [Google Scholar] [CrossRef] [PubMed]
- Stock, C.J.W.; Renzoni, E.A. Telomeres in interstitial lung disease. J. Clin. Med. 2021, 10, 1384. [Google Scholar] [CrossRef]
- Jenkins, E.C.; Tassone, F.; Ye, L.; Gu, H.; Xi, M.; Velinov, M.; Brown, W.T.; Hagerman, R.J.; Hagerman, P.J. Reduced telomere length in older men with premutation alleles of the fragile X mental retardation 1 gene. Am. J. Med. Genet. A 2008, 146A, 1543–1546. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, E.C.; Tassone, F.; Ye, L.; Hoogeveen, A.T.; Brown, W.T.; Hagerman, R.J.; Hagerman, P.J. Reduced telomere length in individuals with FMR1 premutations and full mutations. Am. J. Med. Genet. A 2012, 158A, 1060–1065. [Google Scholar] [CrossRef] [Green Version]
- Albizua, I.; Chopra, P.; Allen, E.G.; He, W.; Amin, A.S.; Sherman, S.L. Study of telomere length in men who carry a fragile X premutation or full mutation allele. Hum. Genet. 2020, 139, 1531–1539. [Google Scholar] [CrossRef]
- Albizua, I.; Rambo-Martin, B.L.; Allen, E.G.; He, W.; Amin, A.S.; Sherman, S.L. Women who carry a fragile X premutation are biologically older than noncarriers as measured by telomere length. Am. J. Med. Genet. A 2017, 173, 2985–2994. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Furtado, C.L.; Luchiari, H.R.; Chielli Pedroso, D.C.; Kogure, G.S.; Caetano, L.C.; Santana, B.A.; Santana, V.P.; Benetti-Pinto, C.L.; Reis, F.M.; Maciel, M.A.; et al. Skewed X-chromosome inactivation and shorter telomeres associate with idiopathic premature ovarian insufficiency. Fertil. Steril. 2018, 110, 476–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.C.; Im, J.A.; Kim, J.H.; Lee, H.R.; Shim, J.Y. Effect of long-term hormone therapy on telomere length in postmenopausal women. Yonsei Med. J. 2005, 46, 471–479. [Google Scholar] [CrossRef] [Green Version]
- Levine, R.L.; Garland, D.; Oliver, C.N.; Amici, A.; Climent, I.; Lenz, A.G.; Ahn, B.W.; Shaltiel, S.; Stadtman, E.R. Determination of carbonyl content in oxidatively modified proteins. Methods Enzym. 1990, 186, 464–478. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]

| PM Carriers | Correlated Signs | FMR1 Output | Reference |
|---|---|---|---|
| Males | Less voxel density in grey and white matter of brain areas, including cerebellum, brainstem and others | CGG repeats (no correlation with mRNA) | [92] |
| Diminished grey matter of amygdala and hippocampal complex, left thalamus and brainstem | % FMRP+-lymphocytes (no correlation with mRNA) | ||
| Males | Reduced left hippocampal activation during recall task, correlated with psychiatric assessment in the absence of hippocampal volume change | mRNA (no correlation with CGG repeats) | [93] |
| Males without FXTAS | Reduced activity of right ventral inferior frontal cortex in verbal working memory in both carriers | mRNA | [94] |
| Males | Decreased parahippocampal activation in working memory task | Blood FMRP levels | [95] |
| Females without FXTAS | Activity of right dorsolateral prefrontal cortex in correct encoded trials | mRNA | [96] |
| Decreased fronto-parietal activity in a magnitude estimation task | CGG repeats (no correlation with mRNA) | ||
| Males with/without FXTAS | White matter structural connectivity of thesuperior cerebellar peduncles in both carriers | CGG repeats and mRNA | [97] |
| Males | Cerebellar volume | CGG repeats | [98] |
| Defective anticipatory postural responses during stepping | mRNA and CGG repeats | ||
| Males without FXTAS | Motor dysfunction: tremor, balance and brain activation during random finger tapping | (no correlation with mRNA) | [99] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valor, L.M.; Morales, J.C.; Hervás-Corpión, I.; Marín, R. Molecular Pathogenesis and Peripheral Monitoring of Adult Fragile X-Associated Syndromes. Int. J. Mol. Sci. 2021, 22, 8368. https://doi.org/10.3390/ijms22168368
Valor LM, Morales JC, Hervás-Corpión I, Marín R. Molecular Pathogenesis and Peripheral Monitoring of Adult Fragile X-Associated Syndromes. International Journal of Molecular Sciences. 2021; 22(16):8368. https://doi.org/10.3390/ijms22168368
Chicago/Turabian StyleValor, Luis M., Jorge C. Morales, Irati Hervás-Corpión, and Rosario Marín. 2021. "Molecular Pathogenesis and Peripheral Monitoring of Adult Fragile X-Associated Syndromes" International Journal of Molecular Sciences 22, no. 16: 8368. https://doi.org/10.3390/ijms22168368
APA StyleValor, L. M., Morales, J. C., Hervás-Corpión, I., & Marín, R. (2021). Molecular Pathogenesis and Peripheral Monitoring of Adult Fragile X-Associated Syndromes. International Journal of Molecular Sciences, 22(16), 8368. https://doi.org/10.3390/ijms22168368