
Membrane Association Modes of Natural Anticancer Peptides: Mechanistic Details on Helicity, Orientation, and Surface Coverage

 , , , , and
, , , , and
Abstract
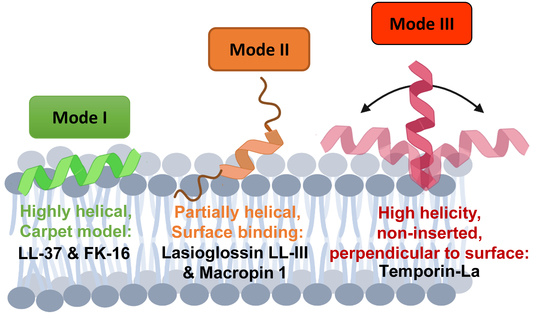
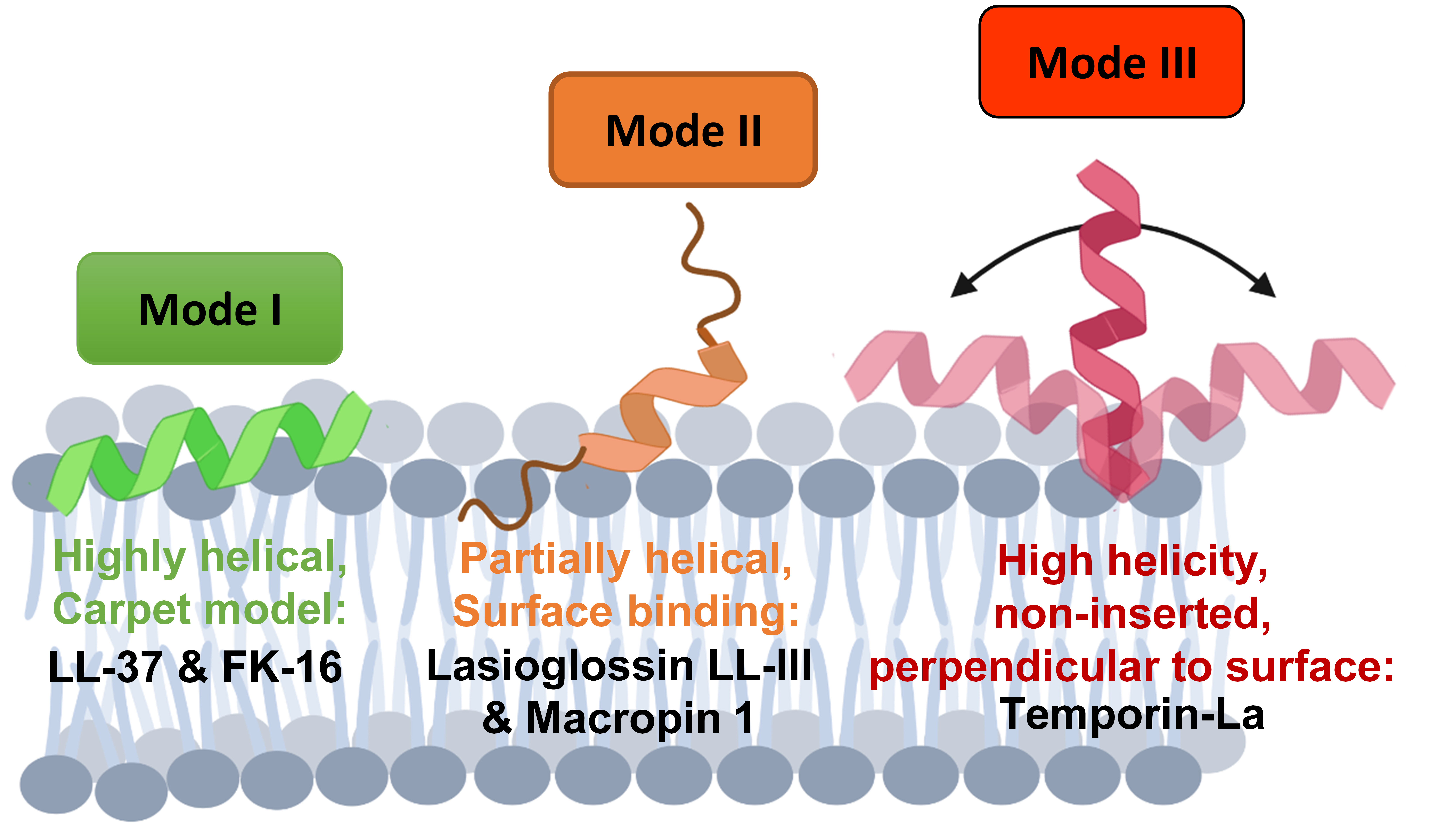
:
1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
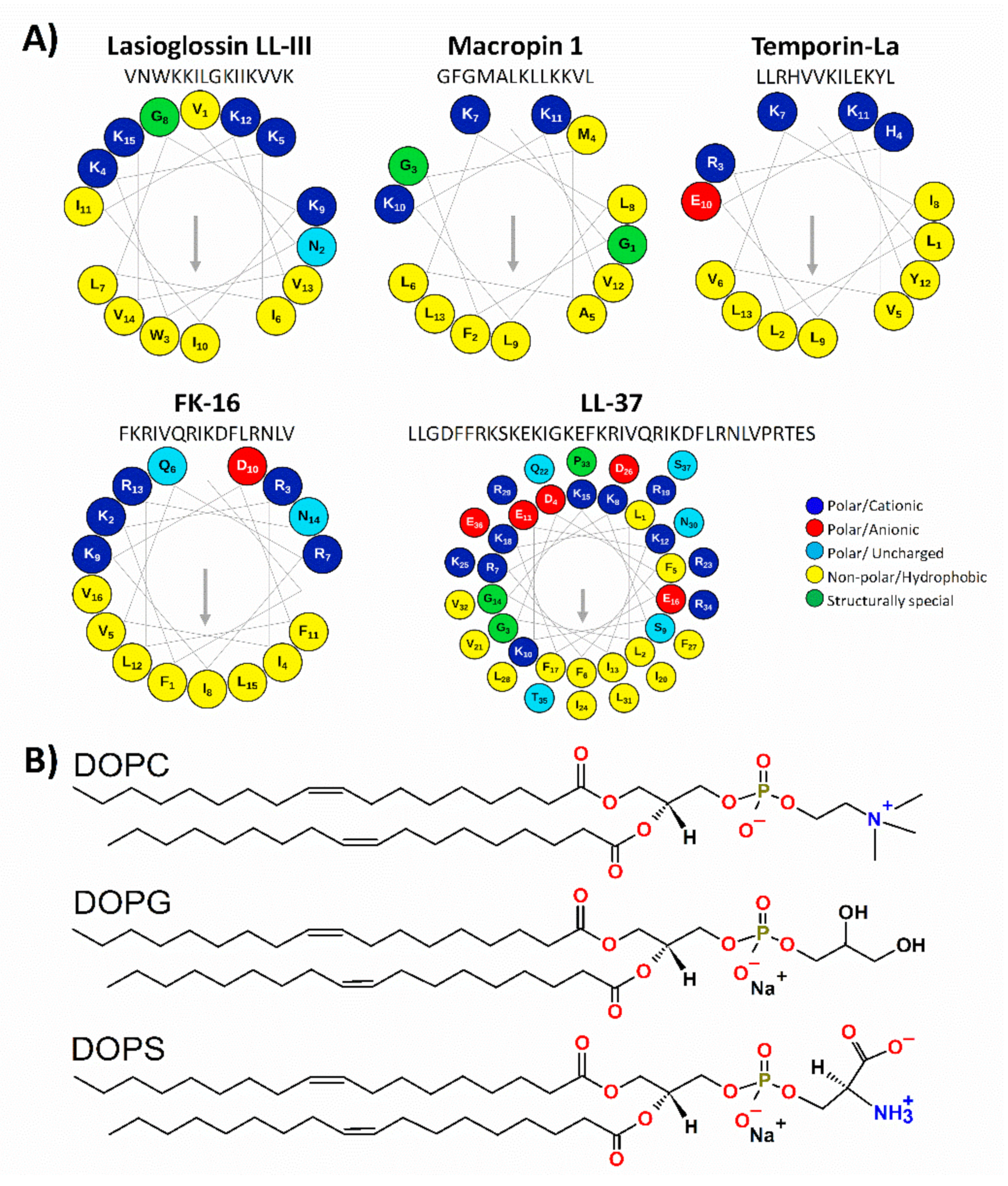
| Peptide | Family | Origin | Sequence a | Length (aa) | Net Charge b | <H> c | <μH> d |
|---|---|---|---|---|---|---|---|
| LASIO III | Lasioglossins | Lasioglossum laticeps (hymenopteran venom) | VNWKKILGKIIKVVK | 15 | +6 | 0.54 | 0.77 |
| MACRO1 | Macropins | Macropis fulvipes (hymenopteran venom) | GFGMALKLLKKVL | 13 | + 4 | 0.57 | 0.54 |
| TEMPO-LA | Temporins | Rana temporaria (anuran skin) | LLRHVVKILEKYL | 13 | +3 | 0.49 | 0.73 |
| FK-16 | Cathelicidins | Derived from the peptide LL-37 | FKRIVQRIKDFLRNLV | 16 | +5 | 0.32 | 0.78 |
| LL-37 | Cathelicidins | Homo sapiens (human) | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES e | 37 | +6 | 0.23 f | 0.60 f |
| 0.16 g | 0.73 g | ||||||
| 0.27 h | 0.49 h |
| Peptide | Antibacterial Mechanism | Proposed Anticancer Mechanism | Cancer Cell Line a | Haemolytic Activity b (LC50 [µM]) | Reference |
|---|---|---|---|---|---|
| LASIO III | Outer and inner membrane permeabilization. | Cell membrane penetration and enter cells. | PC12, L1210, CCRF-CEM T, HL-60, HeLa S3, SW480 | > 220 | [21,22,23,24] |
| MACRO1 | Membrane disruption and cell penetration. Permeabilization of the bacterial cell membrane. | - | CCRF-CEM, HeLa S3, SW480 | ~165 | [22,24,25,26] |
| TEMPO-LA | Insertion into the plasma membrane. | Electrostatic interactions. | HeLa S3, SM MC7721, BEL-7402, A549, SW1116, HepG-2, BGC-823, HL-7702, HEK-293T | > 250 | [2,27,28,29] |
| FK-16 | Increased permeabilization of the membrane. | Induces cell death by both caspase-independent apoptosis and autophagy. | LoVo, HCT116, HT-29 | ~125 c | [35,37,51,52,53] |
| LL-37 | Targets anionic bacterial membranes via the carpet or toroidal pore model. | Induces apoptosis and depends mostly on the ability to act as a ligand for different membrane receptors whose expression varies on different cancer cells. | HT-29, HCT116, SW1116, SW620, SW480, LoVo, AGS, TMK1, Jurkat | > 70 | [37,38,51,54,55,56,57,58,59,60,61] |

2. Results
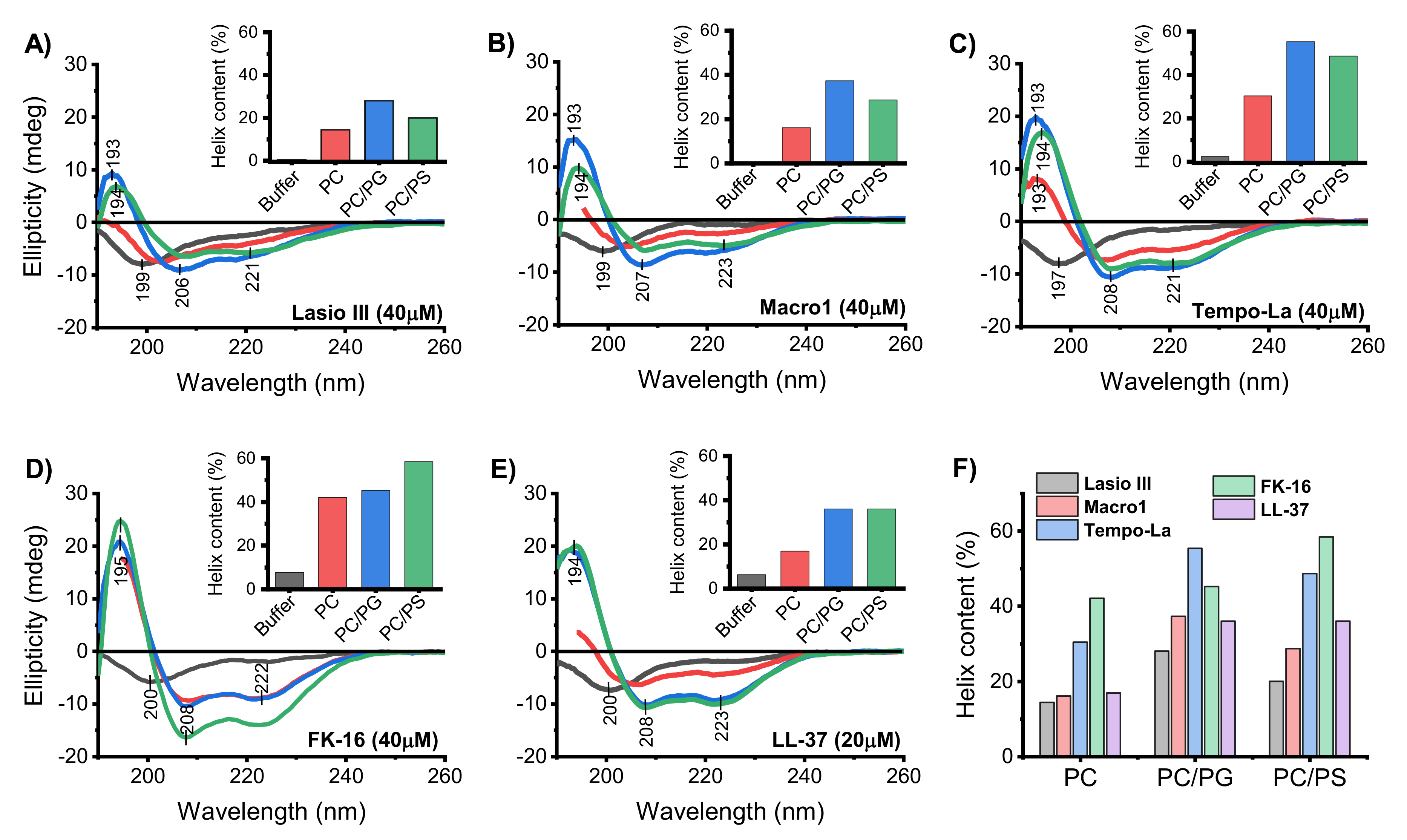
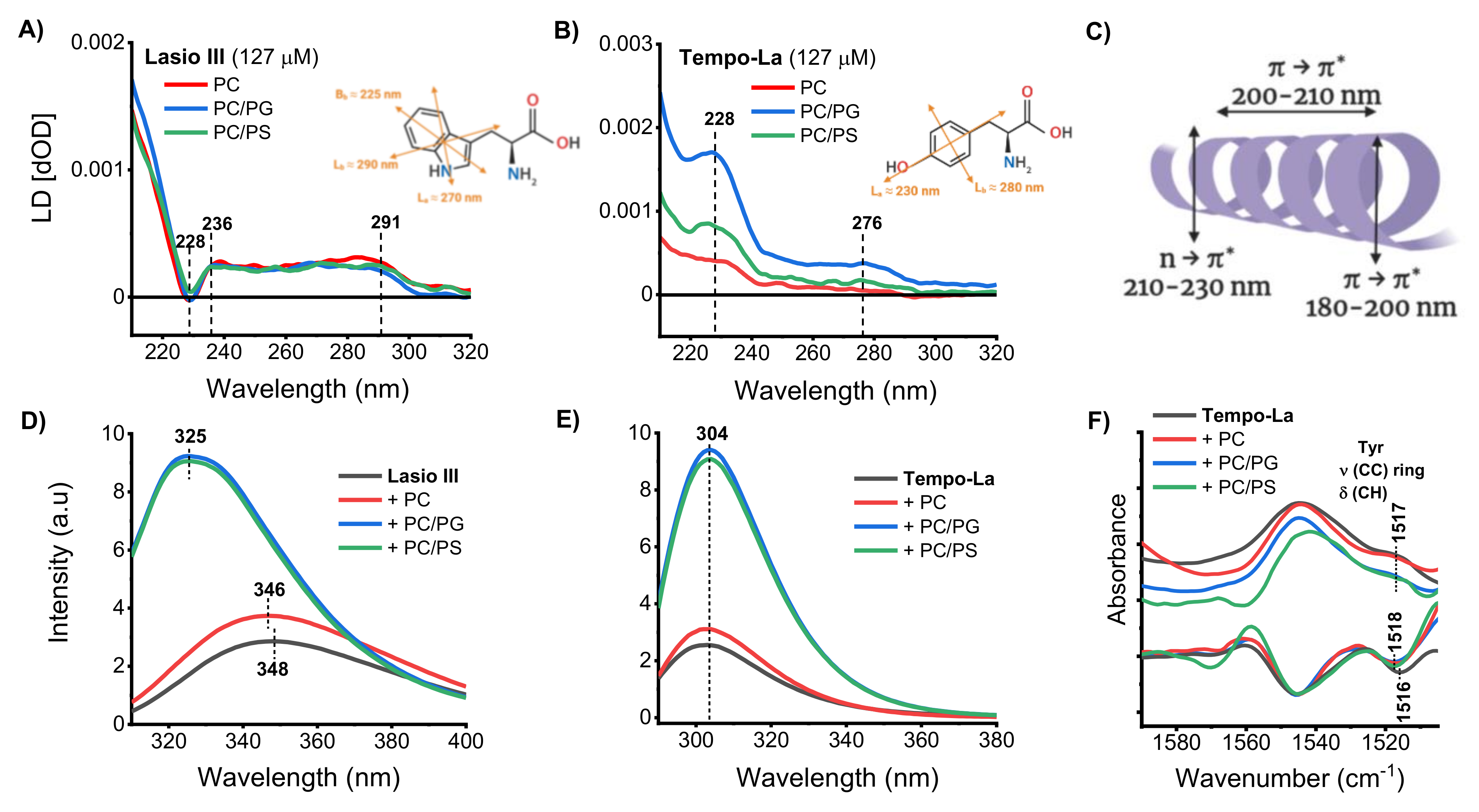
2.1. Peptide Structural Changes in the Presence of Lipid Membranes
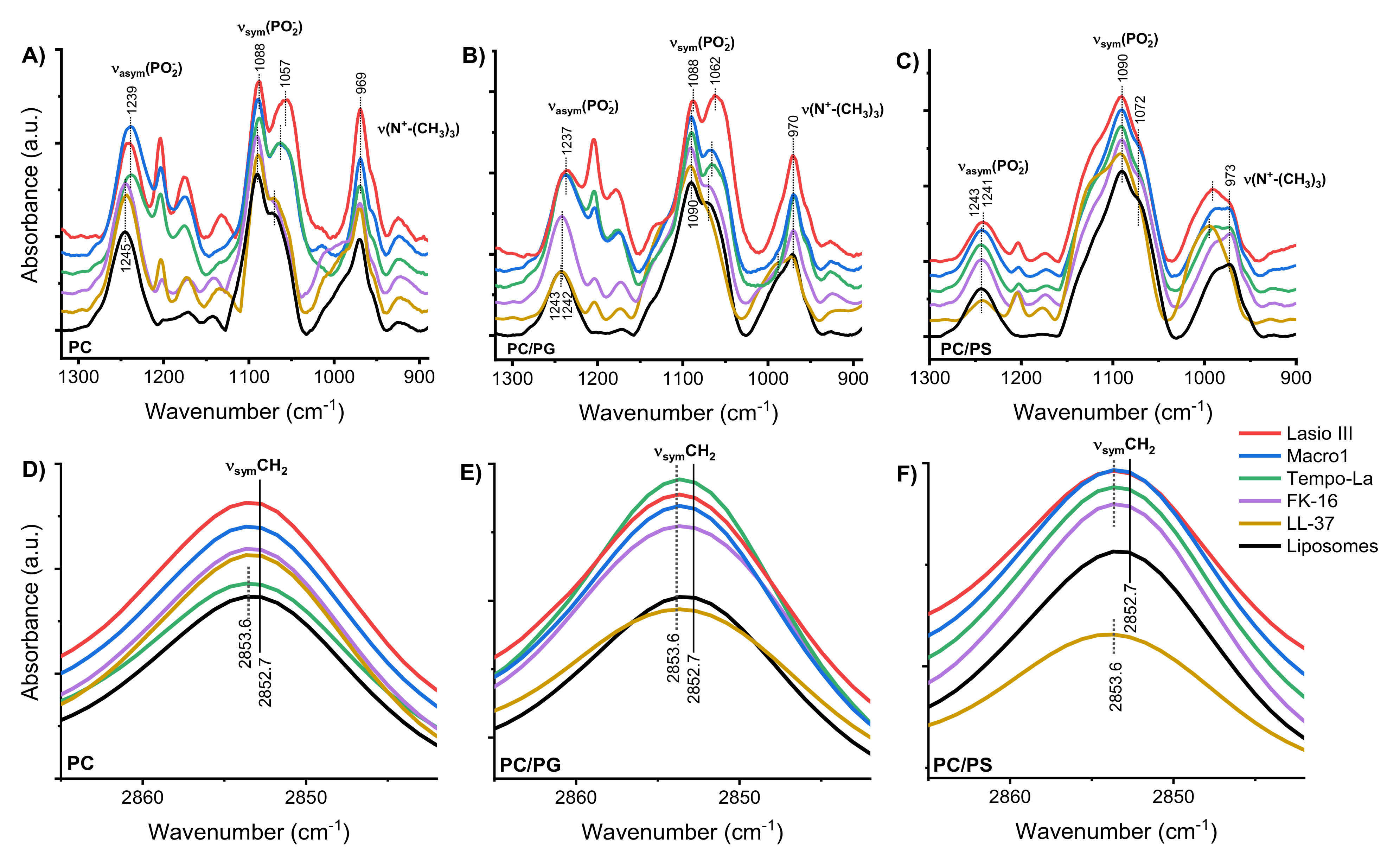
2.2. Position and Binding Depth of the Peptides in the Membrane/Interaction of the Peptides with Lipid Groups in the Membrane
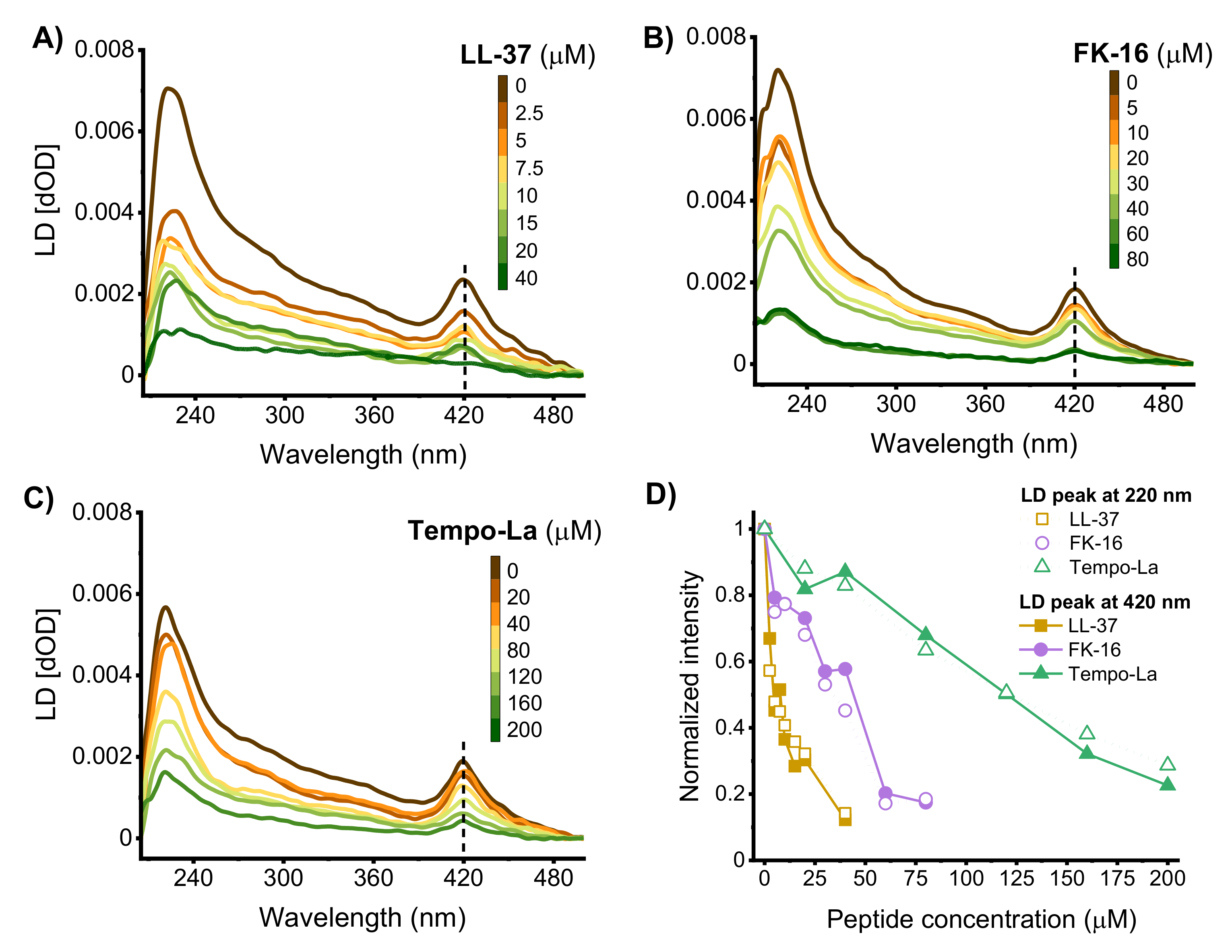
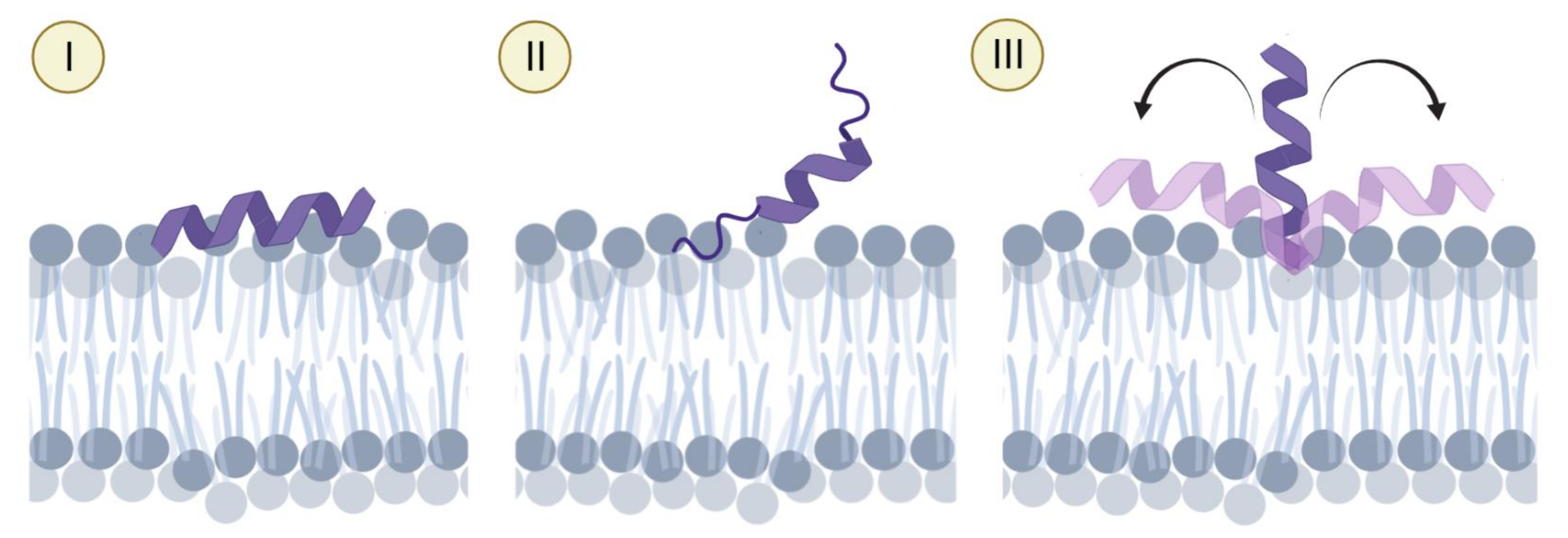
2.3. Orientation of the Peptides in the Membrane
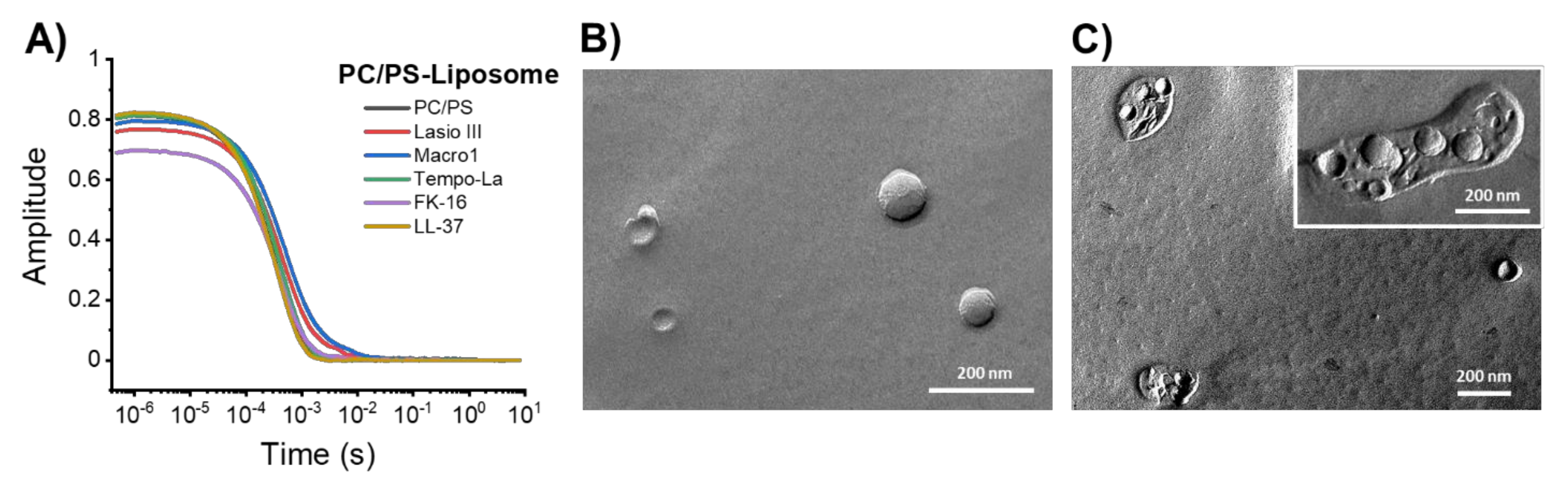
2.4. Effect of the Peptides on the Size and Morphology of Model Vesicles
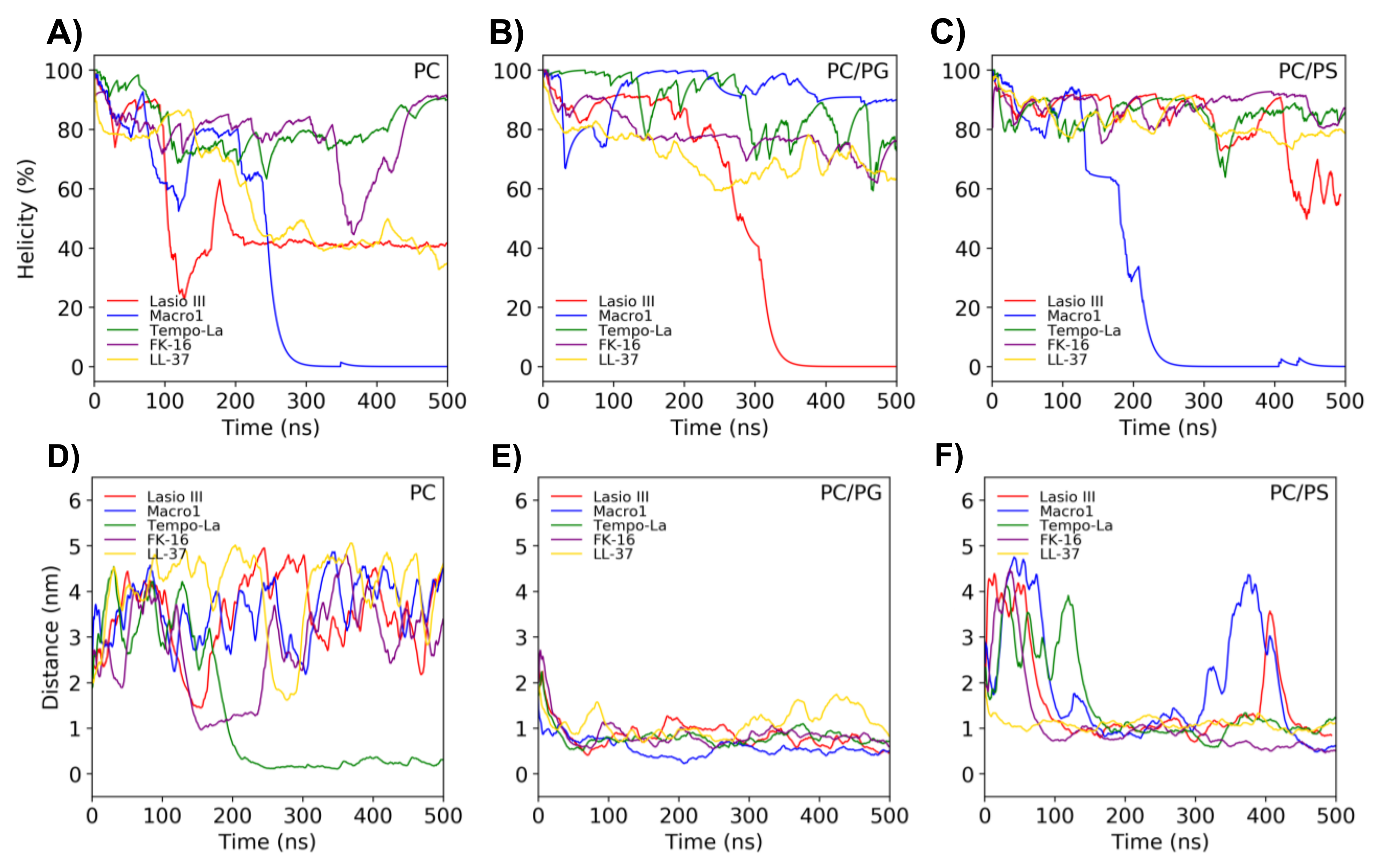
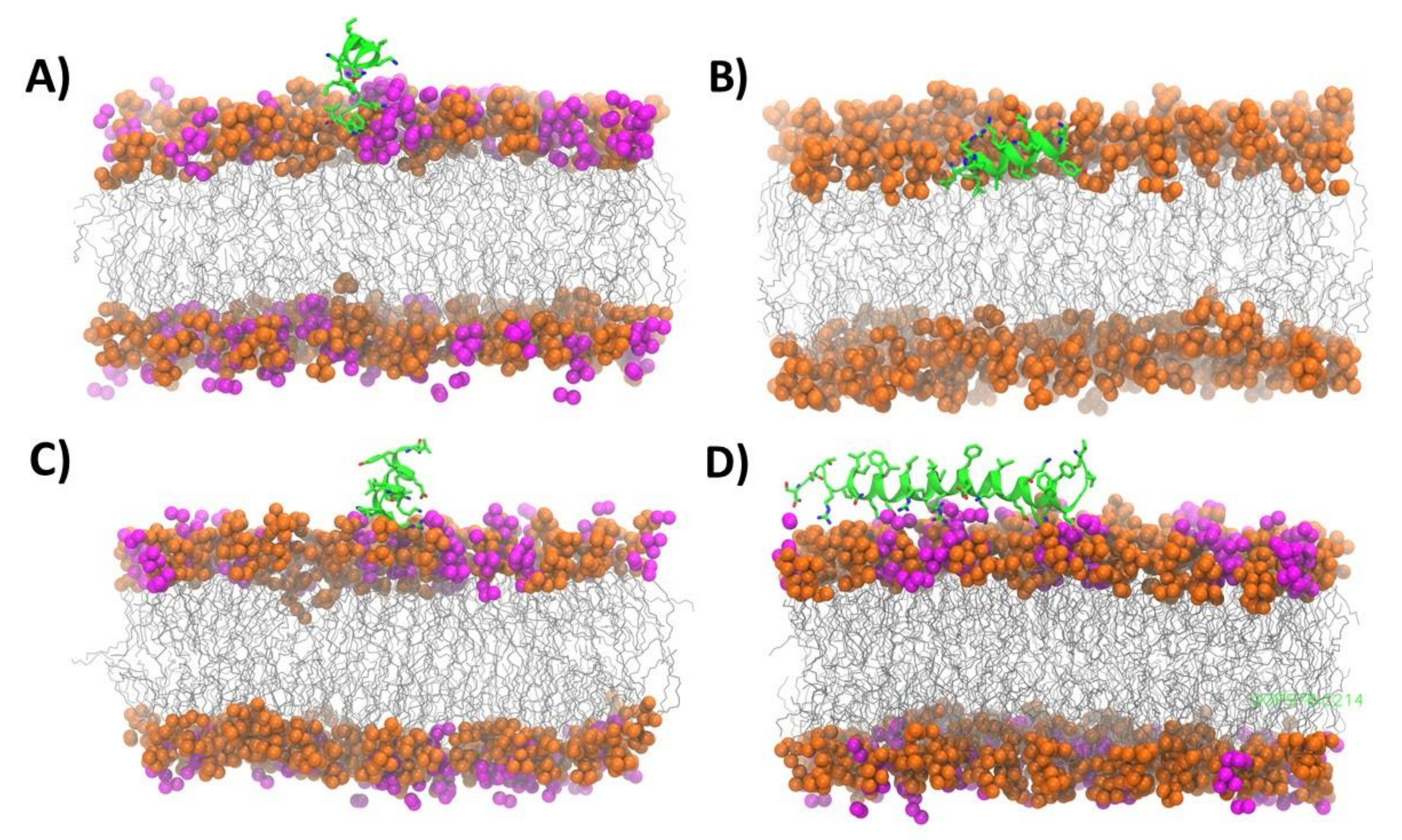
2.5. Molecular Dynamics Simulations on the Peptide-Lipid Interactions
2.6. Interaction of ACPs in a More Complex Membrane System
3. Discussion
3.1. Highly Helical—Carpet Model
3.2. Partially Helical, Surface Binding
3.3. High Helicity, Non-Inserted, Perpendicular to Surface
4. Materials and Methods
4.1. Peptide Solutions
4.2. Lipid Solutions
4.3. Assay Conditions
4.4. Red Blood-Cell Derived Extracellular Vesicles (REVs) Isolation
4.5. Circular Dichroism (CD) Spectroscopy
4.6. Linear Dichroism (LD) Spectroscopy
4.7. Dynamic Light Scattering (DLS)
4.8. Attenuated Total Reflection-Fourier Transform Infrared (ATR-FTIR) Spectroscopy
4.9. Transmission Electron Microscopy Combined with Freeze Fracture (FF-TEM)
4.10. Fluorescence Spectroscopy
4.11. Molecular Dynamics Simulations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bandyopadhyay, S.; Lee, M.; Sivaraman, J.; Chatterjee, C. Model membrane interaction and DNA-binding of antimicrobial peptide Lasioglossin II derived from bee venom. Biochem. Biophys. Res. Commun. 2013, 430, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Gabernet, G.; Müller, A.T.; Hiss, J.A.; Schneider, G. Membranolytic anticancer peptides. MedChemComm 2016, 7, 2232–2245. [Google Scholar] [CrossRef]
- Gaspar, D.; Veiga, A.S.; Castanho, M.A. From antimicrobial to anticancer peptides. A review. Front. Microbiol. 2013, 4, 294. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Riedl, S.; Zweytick, D.; Lohner, K. Membrane-active host defense peptides–challenges and perspectives for the development of novel anticancer drugs. Chem. Phys. Lipids 2011, 164, 766–781. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Liu, D.; Yang, Y. Anti-cancer peptides: Classification, mechanism of action, reconstruction and modification. Open Biol. 2020, 10, 200004. [Google Scholar] [CrossRef]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging biological principles of metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [Green Version]
- Hilchie, A.; Hoskin, D.; Coombs, M.P. Anticancer activities of natural and synthetic peptides. Antimicrob. Pept. 2019, 1117, 131–147. [Google Scholar]
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L. The value of antimicrobial peptides in the age of resistance. Lancet Infect. Dis. 2020, 20, e216–e230. [Google Scholar] [CrossRef]
- Pan, X.; Xu, J.; Jia, X. Research progress evaluating the function and mechanism of anti-tumor peptides. Cancer Manag. Res. 2020, 12, 397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirtskhalava, M.; Amstrong, A.A.; Grigolava, M.; Chubinidze, M.; Alimbarashvili, E.; Vishnepolsky, B.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, M. DBAASP v3: Database of antimicrobial/cytotoxic activity and structure of peptides as a resource for development of new therapeutics. Nucleic Acids Res. 2021, 49, D288–D297. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Sahl, H.-G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B.; Lohner, K. Detergent-like actions of linear amphipathic cationic antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1529–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweizer, F. Cationic amphiphilic peptides with cancer-selective toxicity. Eur. J. Pharmacol. 2009, 625, 190–194. [Google Scholar] [CrossRef]
- Matsuzaki, K. Membrane permeabilization mechanisms. Antimicrob. Pept. 2019, 1117, 9–16. [Google Scholar]
- Epand, R.M.; Vogel, H.J. Diversity of antimicrobial peptides and their mechanisms of action. Biochim. Biophys. Acta Biomembr. 1999, 1462, 11–28. [Google Scholar] [CrossRef] [Green Version]
- Hallock, K.J.; Lee, D.-K.; Omnaas, J.; Mosberg, H.I.; Ramamoorthy, A. Membrane composition determines pardaxin’s mechanism of lipid bilayer disruption. Biophys. J. 2002, 83, 1004–1013. [Google Scholar] [CrossRef] [Green Version]
- Zelezetsky, I.; Tossi, A. Alpha-helical antimicrobial peptides—Using a sequence template to guide structure–activity relationship studies. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1436–1449. [Google Scholar] [CrossRef] [Green Version]
- Almeida, P.F.; Pokorny, A. Mechanisms of antimicrobial, cytolytic, and cell-penetrating peptides: From kinetics to thermodynamics. Biochemistry 2009, 48, 8083–8093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevcsik, E.; Pabst, G.; Richter, W.; Danner, S.; Amenitsch, H.; Lohner, K. Interaction of LL-37 with model membrane systems of different complexity: Influence of the lipid matrix. Biophys. J. 2008, 94, 4688–4699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Čeřovský, V.; Buděšínský, M.; Hovorka, O.; Cvačka, J.; Voburka, Z.; Slaninová, J.; Borovičková, L.; Fučík, V.; Bednárová, L.; Votruba, I. Lasioglossins: Three novel antimicrobial peptides from the venom of the eusocial bee Lasioglossum laticeps (Hymenoptera: Halictidae). ChemBioChem 2009, 10, 2089–2099. [Google Scholar] [CrossRef]
- Tonk, M.; Vilcinskas, A.; Rahnamaeian, M. Insect antimicrobial peptides: Potential tools for the prevention of skin cancer. Appl. Microbiol. Biotechnol. 2016, 100, 7397–7405. [Google Scholar] [CrossRef] [Green Version]
- Mishra, B.; Basu, A.; Saravanan, R.; Xiang, L.; Yang, L.K.; Leong, S.S.J. Lasioglossin-III: Antimicrobial characterization and feasibility study for immobilization applications. RSC Adv. 2013, 3, 9534–9543. [Google Scholar] [CrossRef]
- Slaninová, J.; Mlsová, V.; Kroupová, H.; Alán, L.; Tůmová, T.; Monincová, L.; Borovičková, L.; Fučík, V.; Čeřovský, V. Toxicity study of antimicrobial peptides from wild bee venom and their analogs toward mammalian normal and cancer cells. Peptides 2012, 33, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.J.; Kim, M.K.; Bang, J.K.; Seo, C.H.; Luchian, T.; Park, Y. Macropis fulvipes venom component Macropin exerts its antibacterial and anti-biofilm properties by damaging the plasma membranes of drug resistant bacteria. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monincová, L.; Veverka, V.; Slaninová, J.; Buděšínský, M.; Fučík, V.; Bednárová, L.; Straka, J.; Čeřovský, V. Structure–activity study of macropin, a novel antimicrobial peptide from the venom of solitary bee Macropis fulvipes (Hymenoptera: Melittidae). J. Pept. Sci. 2014, 20, 375–384. [Google Scholar] [CrossRef]
- Zhao, R.-L.; Han, J.-Y.; Han, W.-Y.; He, H.-X.; Ma, J.-F. Effects of Two Novel Peptides from Skin of Lithobates catesbeianus on Tumor cell Morphology and Proliferation. In Molecular Cloning–Selected Applications in Medicine and Biology; IntechOpen: London, UK, 2011; p. 73. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.-L.; Han, J.-Y.; Han, W.-Y.; Lei, L.-C.; Sun, C.-J.; Feng, X.; Jiang, L.-N.; Qiao, H.-W.; Cai, L.-J. Molecular cloning of two novel temporins from Lithobates catesbeianus and studying of their antimicrobial mechanisms. Prog. Biochem. Biophys. 2009, 36, 1064–1070. [Google Scholar] [CrossRef]
- Diao, Y.; Han, W.; Zhao, H.; Zhu, S.; Liu, X.; Feng, X.; Gu, J.; Yao, C.; Liu, S.; Sun, C.; et al. Designed synthetic analogs of the α-helical peptide temporin-La with improved antitumor efficacies via charge modification and incorporation of the integrin αvβ3 homing domain. J. Pept. Sci. 2012, 18, 476–486. [Google Scholar] [CrossRef]
- Zsila, F.; Kohut, G.; Beke-Somfai, T. Disorder-to-helix conformational conversion of the human immunomodulatory peptide LL-37 induced by antiinflammatory drugs, food dyes and some metabolites. Int. J. Biol. Macromol. 2019, 129, 50–60. [Google Scholar] [CrossRef] [Green Version]
- Burton, M.F.; Steel, P.G. The chemistry and biology of LL-37. Nat. Prod. Rep. 2009, 26, 1572–1584. [Google Scholar] [CrossRef]
- Kuroda, K.; Okumura, K.; Isogai, H.; Isogai, E. The human cathelicidin antimicrobial peptide LL-37 and mimics are potential anticancer drugs. Front. Oncol. 2015, 5, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verjans, E.-T.; Zels, S.; Luyten, W.; Landuyt, B.; Schoofs, L. Molecular mechanisms of LL-37-induced receptor activation: An. overview. Peptides 2016, 85, 16–26. [Google Scholar] [CrossRef]
- Sørensen, O.E.; Follin, P.; Johnsen, A.H.; Calafat, J.; Tjabringa, G.S.; Hiemstra, P.S.; Borregaard, N. Human cathelicidin, hCAP-18, is processed to the antimicrobial peptide LL-37 by extracellular cleavage with proteinase 3. Blood 2001, 97, 3951–3959. [Google Scholar] [CrossRef] [Green Version]
- Ren, S.X.; Shen, J.; Cheng, A.S.; Lu, L.; Chan, R.L.; Li, Z.J.; Wang, X.J.; Wong, C.C.; Zhang, L.; Ng, S.S. FK-16 derived from the anticancer peptide LL-37 induces caspase-independent apoptosis and autophagic cell death in colon cancer cells. PLoS ONE 2013, 8, e63641. [Google Scholar] [CrossRef] [Green Version]
- Mishra, B.; Wang, G. Titanium surfaces immobilized with the major antimicrobial fragment FK-16 of human cathelicidin LL-37 are potent against multiple antibiotic-resistant bacteria. Biofouling 2017, 33, 544–555. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, Y.; Han, H.; Miller, D.W.; Wang, G. Solution structures of human LL-37 fragments and NMR-based identification of a minimal membrane-targeting antimicrobial and anticancer region. J. Am. Chem. Soc. 2006, 128, 5776–5785. [Google Scholar] [CrossRef]
- Wang, G. Structures of human host defense cathelicidin LL-37 and its smallest antimicrobial peptide KR-12 in lipid micelles. J. Biol. Chem. 2008, 283, 32637–32643. [Google Scholar] [CrossRef] [Green Version]
- Sok, M.; Šentjurc, M.; Schara, M. Membrane fluidity characteristics of human lung cancer. Cancer Lett. 1999, 139, 215–220. [Google Scholar] [CrossRef]
- Hoskin, D.W.; Ramamoorthy, A. Studies on anticancer activities of antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 2008, 1778, 357–375. [Google Scholar] [CrossRef] [Green Version]
- Riedl, S.; Rinner, B.; Asslaber, M.; Schaider, H.; Walzer, S.; Novak, A.; Lohner, K.; Zweytick, D. In search of a novel target—phosphatidylserine exposed by non-apoptotic tumor cells and metastases of malignancies with poor treatment efficacy. Biochim. Biophys. Acta Biomembr. 2011, 1808, 2638–2645. [Google Scholar] [CrossRef] [Green Version]
- Papo, N.; Shai, Y. Host defense peptides as new weapons in cancer treatment. Cell. Mol. Life Sci. 2005, 62, 784–790. [Google Scholar] [CrossRef]
- Tripisciano, C.; Weiss, R.; Karuthedom George, S.; Fischer, M.B.; Weber, V. Extracellular Vesicles Derived From Platelets, Red Blood Cells, and Monocyte-Like Cells Differ. Regarding Their Ability to Induce Factor XII-Dependent Thrombin Generation. Front. Cell Dev. Biol. 2020, 8, 298. [Google Scholar] [CrossRef]
- Jørgensen, M.M.; Bæk, R.; Varming, K. Potentials and capabilities of the Extracellular Vesicle (EV) Array. J. Extracell. Vesicles 2015, 4, 26048. [Google Scholar] [CrossRef]
- Thangaraju, K.; Neerukonda, S.N.; Katneni, U.; Buehler, P.W. Extracellular Vesicles from Red Blood Cells and Their Evolving Roles in Health, Coagulopathy and Therapy. Int. J. Mol. Sci. 2021, 22, 153. [Google Scholar] [CrossRef]
- Usman, W.M.; Pham, T.C.; Kwok, Y.Y.; Vu, L.T.; Ma, V.; Peng, B.; San Chan, Y.; Wei, L.; Chin, S.M.; Azad, A. Efficient RNA drug delivery using red blood cell extracellular vesicles. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Fauchere, J. Hydrophobic Parameters Pi of Amino-Acid Side Chains from The Partitioning Of N-Acetyl-Amino Amides. Eur. J. Med. Chem. 1983, 18, 369–375. [Google Scholar]
- Gautier, R.; Douguet, D.; Antonny, B.; Drin, G. Heliquest: A web server to screen sequences with specific α-helical properties. Bioinformatics 2008, 24, 2101–2102. [Google Scholar] [CrossRef]
- Eisenberg, D.; Schwarz, E.; Komaromy, M.; Wall, R. Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J. Mol. Biol. 1984, 179, 125–142. [Google Scholar] [CrossRef]
- Eisenberg, D.; Weiss, R.M.; Terwilliger, T.C. The helical hydrophobic moment: A measure of the amphiphilicity of a helix. Nature 1982, 299, 371–374. [Google Scholar] [CrossRef]
- Piktel, E.; Niemirowicz, K.; Wnorowska, U.; Wątek, M.; Wollny, T.; Głuszek, K.; Góźdź, S.; Levental, I.; Bucki, R. The role of cathelicidin LL-37 in cancer development. Arch. Immunol. Ther. Exp. 2016, 64, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammed, I.; Said, D.G.; Nubile, M.; Mastropasqua, L.; Dua, H.S. Cathelicidin-derived synthetic peptide improves therapeutic potential of vancomycin against Pseudomonas aeruginosa. Front. Microbiol. 2019, 10, 2190. [Google Scholar] [CrossRef] [Green Version]
- Quemé-Peña, M.; Ricci, M.; Juhász, T.; Horváti, K.; Bősze, S.; Biri-Kovács, B.; Szeder, B.; Zsila, F.; Beke-Somfai, T. Old Polyanionic Drug Suramin Suppresses Detrimental Cytotoxicity of the Host Defense Peptide LL-37. ACS Pharmacol. Transl. Sci. 2020, 4, 155–167. [Google Scholar] [CrossRef]
- Nan, Y.H.; Bang, J.-K.; Jacob, B.; Park, I.-S.; Shin, S.Y. Prokaryotic selectivity and LPS-neutralizing activity of short antimicrobial peptides designed from the human antimicrobial peptide LL-37. Peptides 2012, 35, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Oren, Z.; Lerman, J.C.; Gudmundsson, G.H.; Agerberth, B.; Shai, Y. Structure and organization of the human antimicrobial peptide LL-37 in phospholipid membranes: Relevance to the molecular basis for its non-cell-selective activity. Biochem. J. 1999, 341, 501–513. [Google Scholar] [CrossRef]
- Ciornei, C.D.; Sigurdardóttir, T.; Schmidtchen, A.; Bodelsson, M. Antimicrobial and chemoattractant activity, lipopolysaccharide neutralization, cytotoxicity, and inhibition by serum of analogs of human cathelicidin LL-37. Antimicrob. Agents Chemother. 2005, 49, 2845–2850. [Google Scholar] [CrossRef] [Green Version]
- Ren, S.X.; Cheng, A.S.; To, K.F.; Tong, J.H.; Li, M.S.; Shen, J.; Wong, C.C.; Zhang, L.; Chan, R.L.; Wang, X.J. Host immune defense peptide LL-37 activates caspase-independent apoptosis and suppresses colon cancer. Cancer Res. 2012, 72, 6512–6523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.K.K.; Sung, J.J.Y.; To, K.F.; Yu, L.; Li, H.T.; Li, Z.J.; Chu, K.M.; Yu, J.; Cho, C.H. The host defense peptide LL-37 activates the tumor-suppressing bone morphogenetic protein signaling via inhibition of proteasome in gastric cancer cells. J. Cell. Physiol. 2010, 223, 178–186. [Google Scholar] [CrossRef]
- Mader, J.S.; Mookherjee, N.; Hancock, R.E.; Bleackley, R.C. The Human Host Defense Peptide LL-37 Induces Apoptosis in a Calpain-and Apoptosis-Inducing Factor–Dependent Manner Involving Bax Activity. Mol. Cancer Res. 2009, 7, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Narayana, J.L.; Mishra, B.; Zhang, Y.; Wang, F.; Wang, C.; Zarena, D.; Lushnikova, T.; Wang, X. Design of antimicrobial peptides: Progress made with human cathelicidin LL-37. Antimicrob. Pept. 2019, 1117, 215–240. [Google Scholar] [CrossRef]
- Henzler Wildman, K.A.; Lee, D.-K.; Ramamoorthy, A. Mechanism of lipid bilayer disruption by the human antimicrobial peptide, LL-37. Biochemistry 2003, 42, 6545–6558. [Google Scholar] [CrossRef]
- Reißer, S.; Prock, S.; Heinzmann, H.; Ulrich, A.S. Protein ORIGAMI: A program for the creation of 3D paper models of folded peptides. Biochem. Mol. Biol. Educ. 2018, 46, 403–409. [Google Scholar] [CrossRef]
- Xhindoli, D.; Pacor, S.; Benincasa, M.; Scocchi, M.; Gennaro, R.; Tossi, A. The human cathelicidin LL-37—A pore-forming antibacterial peptide and host-cell modulator. Biochim. Biophys. Acta Biomembr. 2016, 1858, 546–566. [Google Scholar] [CrossRef]
- Johansson, J.; Gudmundsson, G.H.; Rottenberg, M.n.E.; Berndt, K.D.; Agerberth, B. Conformation-dependent antibacterial activity of the naturally occurring human peptide LL-37. J. Biol. Chem. 1998, 273, 3718–3724. [Google Scholar] [CrossRef] [Green Version]
- Woody, R.W. Circular dichroism of intrinsically disordered proteins. In Instrumental Analysis of Intrinsically Disordered Proteins: Assessing Structure and Conformation; John Wiley & Sons: Hoboken, NJ, USA, 2010; pp. 303–321. [Google Scholar]
- Nordén, B.; Rodger, A.; Dafforn, T. Linear Dichroism and Circular Dichroism; The Royal Society of Chemistry: London, UK, 2010. [Google Scholar]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.-H.; Goto, Y.; Réfrégiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E3095–E3103. [Google Scholar] [CrossRef] [Green Version]
- Mihály, J.; Deák, R.; Szigyártó, I.C.; Bóta, A.; Beke-Somfai, T.; Varga, Z. Characterization of extracellular vesicles by IR spectroscopy: Fast and simple classification based on amide and CH stretching vibrations. Biochim. Biophys. Acta Biomembr. 2017, 1859, 459–466. [Google Scholar] [CrossRef]
- Deák, R.; Mihály, J.; Szigyártó, I.C.; Wacha, A.; Lelkes, G.; Bóta, A. Physicochemical characterization of artificial nanoerythrosomes derived from erythrocyte ghost membranes. Colloids Surf. B Biointerfaces 2015, 135, 225–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigano, C.; Manciu, L.; Buyse, F.; Goormaghtigh, E.; Ruysschaert, J.M. Attenuated total reflection IR spectroscopy as a tool to investigate the structure, orientation and tertiary structure changes in peptides and membrane proteins. Pept. Sci. 2000, 55, 373–380. [Google Scholar] [CrossRef]
- Dluhy, R.A.; Stephens, S.M.; Widayati, S.; Williams, A.D. Vibrational spectroscopy of biophysical monolayers. Applications of IR and Raman spectroscopy to biomembrane model systems at interfaces. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 1995, 51, 1413–1447. [Google Scholar] [CrossRef]
- Schibli, D.J.; Epand, R.F.; Vogel, H.J.; Epand, R.M. Tryptophan-rich antimicrobial peptides: Comparative properties and membrane interactions. Biochem. Cell Biol. 2002, 80, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Bóta, A.; Wacha, A.; Varga, Z.; Csilla Szigyártó, I.; Kristyán, S.; Lőrincz, A.; Szabó, P.; Kálmán, M.; Naszályi-Nagy , L.; Mihály, J. Role of oligo (malic acid) on the formation of unilamellar vesicles. J. Colloid Interface Sci. 2018, 532, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.N.; McElhaney, R.N. Membrane lipid phase transitions and phase organization studied by Fourier transform infrared spectroscopy. Biochim. Biophys. Acta Biomembr. 2013, 1828, 2347–2358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolini, C.; Kraineva, J.; Khurana, M.; Periasamy, N.; Funari, S.S.; Winter, R. Temperature and pressure effects on structural and conformational properties of POPC/SM/cholesterol model raft mixtures—A FT-IR, SAXS, DSC, PPC and Laurdan fluorescence spectroscopy study. Biochim. Biophys. Acta Biomembr. 2006, 1758, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.J.; Sills, R.H.; Patel, N.; Mendelsohn, R. Conformational order of phospholipids incorporated into human erythrocytes: An FTIR spectroscopy study. Biochemistry 1996, 35, 229–235. [Google Scholar] [CrossRef]
- Rocha, S.; Kogan, M.; Beke-Somfai, T.; Nordén, B. Probing Microscopic Orientation in Membranes by Linear Dichroism. Langmuir 2016, 32, 2841–2846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esbjörner, E.K.; Oglecka, K.; Lincoln, P.; Gräslund, A.; Nordén, B. Membrane binding of pH-sensitive influenza fusion peptides. Positioning, configuration, and induced leakage in a lipid vesicle model. Biochemistry 2007, 46, 13490–13504. [Google Scholar] [CrossRef] [PubMed]
- Svensson, F.R.; Lincoln, P.; Nordén, B.; Esbjörner, E.K. Tryptophan orientations in membrane-bound gramicidin and melittin—a comparative linear dichroism study on transmembrane and surface-bound peptides. Biochim. Biophys. Acta Biomembr. 2011, 1808, 219–228. [Google Scholar] [CrossRef] [Green Version]
- Fornander, L.H.; Feng, B.; Beke-Somfai, T.S.; Nordén, B. UV transition moments of tyrosine. J. Phys. Chem. B 2014, 118, 9247–9257. [Google Scholar] [CrossRef]
- Caesar, C.E.; Esbjörner, E.K.; Lincoln, P.; Nordén, B. Membrane interactions of cell-penetrating peptides probed by tryptophan fluorescence and dichroism techniques: Correlations of structure to cellular uptake. Biochemistry 2006, 45, 7682–7692. [Google Scholar] [CrossRef]
- Rodger, A.; Dorrington, G.; Ang, D.L. Linear dichroism as a probe of molecular structure and interactions. Analyst 2016, 141, 6490–6498. [Google Scholar] [CrossRef]
- Hicks, M.R.; Kowałski, J.; Rodger, A. LD spectroscopy of natural and synthetic biomaterials. Chem. Soc. Rev. 2010, 39, 3380–3393. [Google Scholar] [CrossRef]
- Caesar, C.E.; Esbjörner, E.K.; Lincoln, P.; Nordén, B. Assigning membrane binding geometry of cytochrome C by polarized light spectroscopy. Biophys. J. 2009, 96, 3399–3411. [Google Scholar] [CrossRef] [Green Version]
- Brattwall, C.E.; Lincoln, P.; Nordén, B. Orientation and conformation of cell-penetrating peptide penetratin in phospholipid vesicle membranes determined by polarized-light spectroscopy. J. Am. Chem. Soc. 2003, 125, 14214–14215. [Google Scholar] [CrossRef]
- Arias, M.; Nguyen, L.T.; Kuczynski, A.M.; Lejon, T.; Vogel, H.J. Position-dependent influence of the three trp residues on the membrane activity of the antimicrobial peptide, tritrpticin. Antibiotics 2014, 3, 595–616. [Google Scholar] [CrossRef] [Green Version]
- Kohn, E.M.; Shirley, D.J.; Arotsky, L.; Picciano, A.M.; Ridgway, Z.; Urban, M.W.; Carone, B.R.; Caputo, G.A. Role of cationic side chains in the antimicrobial activity of C18G. Molecules 2018, 23, 329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.H.; Starr, C.G.; Guha, S.; Wimley, W.C.; Ulmschneider, M.B.; Ulmschneider, J.P. Tuning of a membrane-perforating antimicrobial peptide to selectively target membranes of different lipid composition. J. Membr. Biol. 2021, 254, 75–96. [Google Scholar] [CrossRef]
- Ricci, M.; Horváti, K.; Juhász, T.; Szigyártó, I.; Török, G.; Sebák, F.; Bodor, A.; Homolya, L.; Henczkó, J.; Pályi, B.; et al. Anionic food color tartrazine enhances antibacterial efficacy of histatin-derived peptide DHVAR4 by fine-tuning its membrane activity. Q. Rev. Biophys. 2020, 53, E5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.X.; Kiomourtzis, T.; Lam, C.K.; Le, M.T. The biology and therapeutic applications of red blood cell extracellular vesicles. In Erythrocyte; IntechOpen: London, UK, 2019. [Google Scholar]
- Deák, R.; Mihály, J.; Szigyártó, I.C.; Beke-Somfai, T.; Turiák, L.; Drahos, L.; Wacha, A.; Bóta, A.; Varga, Z. Nanoerythrosomes tailoring: Lipid induced protein scaffolding in ghost membrane derived vesicles. Mater. Sci. Eng. C 2020, 109, 110428. [Google Scholar] [CrossRef]
- Singh, P.; Szigyártó, I.C.; Ricci, M.; Zsila, F.; Juhász, T.; Mihály, J.; Bősze, S.; Bulyáki, E.; Kardos, J.; Kitka, D.; et al. Membrane active peptides remove surface adsorbed protein corona from extracellular vesicles of red blood cells. Front. Chem. 2020, 8, 703. [Google Scholar] [CrossRef] [PubMed]
- Szigyártó, I.C.; Deák, R.; Mihály, J.; Rocha, S.; Zsila, F.; Varga, Z.; Beke-Somfai, T. Flow Alignment of Extracellular Vesicles: Structure and Orientation of Membrane-Associated Bio-macromolecules Studied with Polarized Light. ChemBioChem 2018, 19, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K. Why and how are peptide–lipid interactions utilized for self-defense? Magainins and tachyplesins as archetypes. Biochim. Biophys. Acta Biomembr. 1999, 1462, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Quemé-Peña, M.; Juhász, T.; Mihály, J.; Szigyártó, I.C.; Horváti, K.; Bősze, S.; Henczkó, J.; Pályi, B.; Németh, C.; Varga, Z.; et al. Manipulating Active Structure and Function of Cationic Antimicrobial Peptide CM15 by the Polysulfonated Drug Suramin: A Step Closer to in vivo Complexity. ChemBioChem 2019, 20, 1578. [Google Scholar] [CrossRef]
- Locock, K.E.S. Bioinspired Polymers: Antimicrobial Polymethacrylates. Aust. J. Chem. 2016, 69, 717–724. [Google Scholar] [CrossRef]
- Van den Bergen, G.; Stroet, M.; Caron, B.; Poger, D.; Mark, A.E. Curved or linear? Predicting the 3-dimensional structure of α-helical antimicrobial peptides in an amphipathic environment. FEBS Lett. 2020, 594, 1062–1080. [Google Scholar] [CrossRef] [PubMed]
- Pathak, N.; Salas-Auvert, R.; Ruche, G.; Janna, M.h.; McCarthy, D.; Harrison, R.G. Comparison of the effects of hydrophobicity, amphiphilicity, and α-helicity on the activities of antimicrobial peptides. Proteins Struct. Funct. Bioinform. 1995, 22, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238. [Google Scholar] [CrossRef]
- Sato, H.; Feix, J.B. Peptide–membrane interactions and mechanisms of membrane destruction by amphipathic α-helical antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1245–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porcelli, F.; Verardi, R.; Shi, L.; Henzler-Wildman, K.A.; Ramamoorthy, A.; Veglia, G. NMR structure of the cathelicidin-derived human antimicrobial peptide LL-37 in dodecylphosphocholine micelles. Biochemistry 2008, 47, 5565–5572. [Google Scholar] [CrossRef] [Green Version]
- Battista, F.; Oliva, R.; Del Vecchio, P.; Winter, R.; Petraccone, L. Insights into the Action Mechanism of the Antimicrobial Peptide Lasioglossin III. Int. J. Mol. Sci. 2021, 22, 2857. [Google Scholar] [CrossRef]
- Caputo, G.A.; London, E. Cumulative effects of amino acid substitutions and hydrophobic mismatch upon the transmembrane stability and conformation of hydrophobic α-helices. Biochemistry 2003, 42, 3275–3285. [Google Scholar] [CrossRef]
- Jafari, M.; Mehrnejad, F.; Doustdar, F. Insight into the interactions, residue snorkeling, and membrane disordering potency of a single antimicrobial peptide into different lipid bilayers. PLoS ONE 2017, 12, e0187216. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Yan, C.; Guo, J.; Xu, C. Ionic protein-lipid interactions at the plasma membrane regulate the structure and function of immunoreceptors. Adv. Immunol. 2019, 144, 65–85. [Google Scholar]
- Mishra, V.; Palgunachari, M.; Segrest, J.; Anantharamaiah, G. Interactions of synthetic peptide analogs of the class A amphipathic helix with lipids. Evidence for the snorkel hypothesis. J. Biol. Chem. 1994, 269, 7185–7191. [Google Scholar] [CrossRef]
- Hazam, P.K.; Akhil, R.; Jerath, G.; Saikia, J.; Ramakrishnan, V. Topological effects on the designability and bactericidal potency of antimicrobial peptides. Biophys. Chem. 2019, 248, 1–8. [Google Scholar] [CrossRef]
- Pandidan, S.; Mechler, A. Membrane morphology effects in quartz crystal microbalance characterization of antimicrobial peptide activity. Biophys. Chem. 2020, 262, 106381. [Google Scholar] [CrossRef] [PubMed]
- Travers, W.; Kelleher, F. Studies of the highly potent lantibiotic peptide nisin Z in aqueous solutions of salts and biological buffer components. Biophys. Chem. 2021, 274, 106603. [Google Scholar] [CrossRef]
- Santana, H.J.A.; Caseli, L. A bactericide peptide changing the static and dilatational surface elasticity properties of zwitterionic lipids at the air-water interface: Relationship with the thermodynamic, structural and morphological properties. Biophys. Chem. 2021, 277, 106638. [Google Scholar] [CrossRef] [PubMed]
- Svensson, F.R.; Lincoln, P.; Nordén, B.; Esbjörner, E.K. Retinoid chromophores as probes of membrane lipid order. J. Phys. Chem. B 2007, 111, 10839–10848. [Google Scholar] [CrossRef]
- Ardhammar, M.; Mikati, N.; Nordén, B. Chromophore orientation in liposome membranes probed with flow dichroism. J. Am. Chem. Soc. 1998, 120, 9957–9958. [Google Scholar] [CrossRef]
- Berendsen, H.J.; van der Spoel, D.; van Drunen, R. Gromacs: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. Gromacs: High. performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Jo, S.; Lim, J.B.; Klauda, J.B.; Im, W. CHARMM-GUI Membrane Builder for mixed bilayers and its application to yeast membranes. Biophys. J. 2009, 97, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.; Postma, J.v.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Crystal structure and pair potentials: A molecular-dynamics study. Phys. Rev. Lett. 1980, 45, 1196. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅ log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quemé-Peña, M.; Juhász, T.; Kohut, G.; Ricci, M.; Singh, P.; Szigyártó, I.C.; Papp, Z.I.; Fülöp, L.; Beke-Somfai, T. Membrane Association Modes of Natural Anticancer Peptides: Mechanistic Details on Helicity, Orientation, and Surface Coverage. Int. J. Mol. Sci. 2021, 22, 8613. https://doi.org/10.3390/ijms22168613
Quemé-Peña M, Juhász T, Kohut G, Ricci M, Singh P, Szigyártó IC, Papp ZI, Fülöp L, Beke-Somfai T. Membrane Association Modes of Natural Anticancer Peptides: Mechanistic Details on Helicity, Orientation, and Surface Coverage. International Journal of Molecular Sciences. 2021; 22(16):8613. https://doi.org/10.3390/ijms22168613
Chicago/Turabian StyleQuemé-Peña, Mayra, Tünde Juhász, Gergely Kohut, Maria Ricci, Priyanka Singh, Imola Cs. Szigyártó, Zita I. Papp, Lívia Fülöp, and Tamás Beke-Somfai. 2021. "Membrane Association Modes of Natural Anticancer Peptides: Mechanistic Details on Helicity, Orientation, and Surface Coverage" International Journal of Molecular Sciences 22, no. 16: 8613. https://doi.org/10.3390/ijms22168613
APA StyleQuemé-Peña, M., Juhász, T., Kohut, G., Ricci, M., Singh, P., Szigyártó, I. C., Papp, Z. I., Fülöp, L., & Beke-Somfai, T. (2021). Membrane Association Modes of Natural Anticancer Peptides: Mechanistic Details on Helicity, Orientation, and Surface Coverage. International Journal of Molecular Sciences, 22(16), 8613. https://doi.org/10.3390/ijms22168613