
Synergistic Action of Antimicrobial Lung Proteins against Klebsiella pneumoniae



Abstract
:
1. Introduction
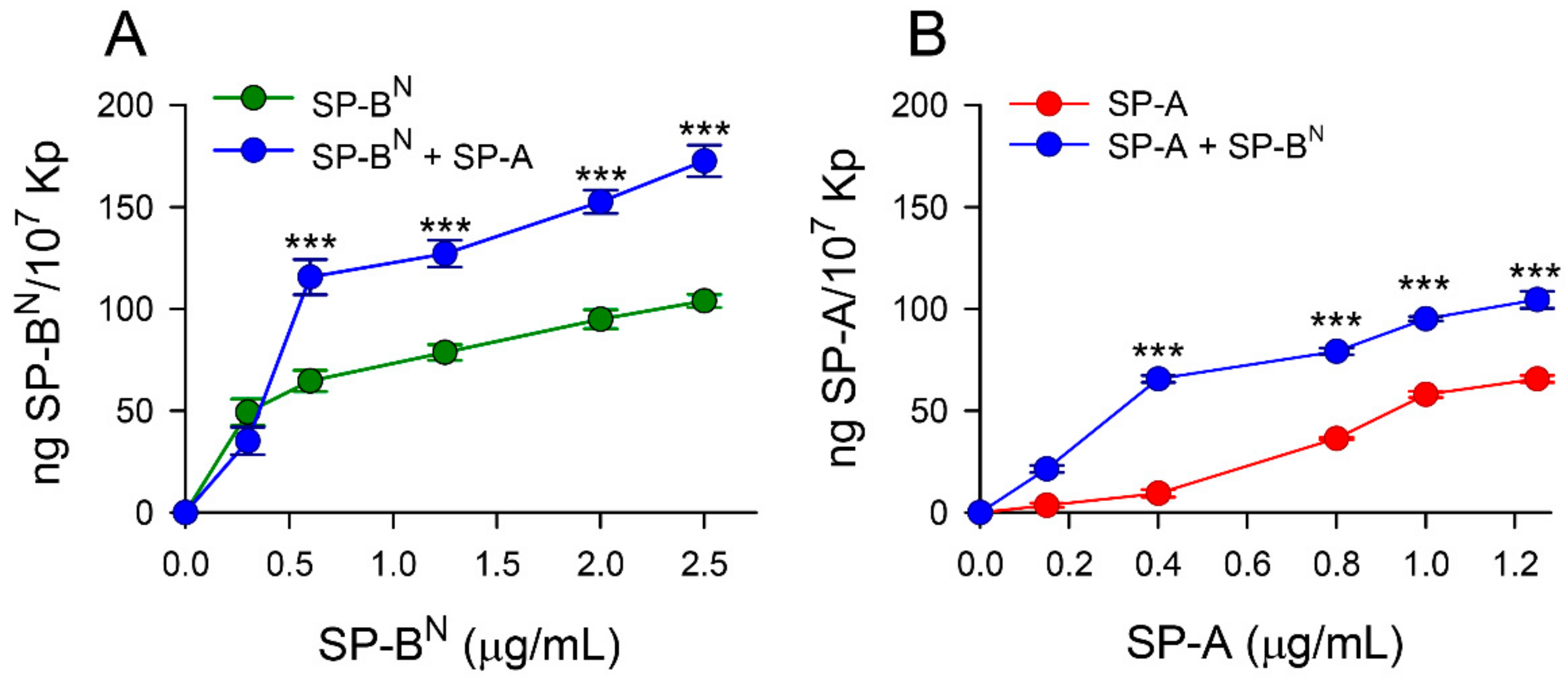
2. Results
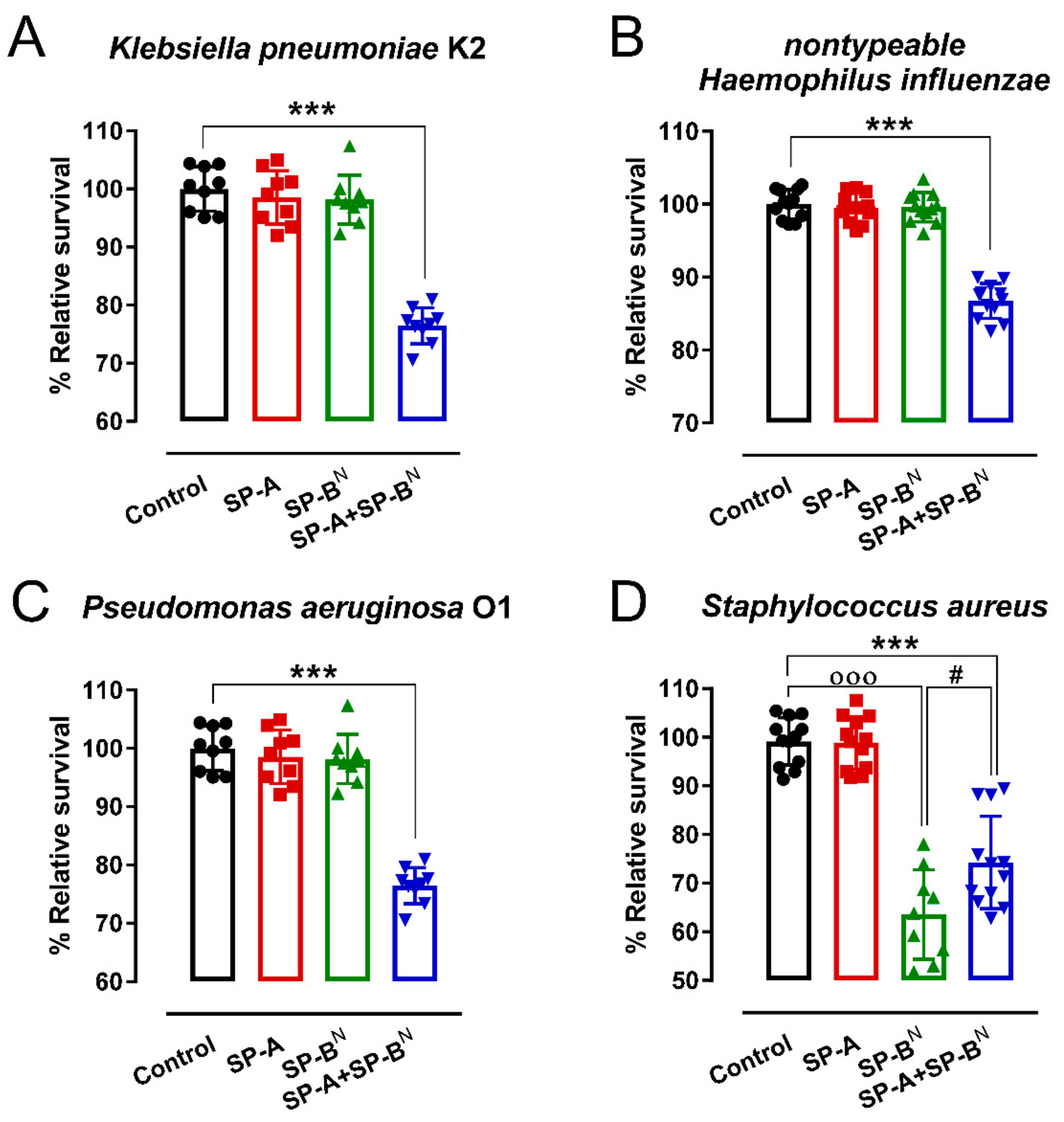
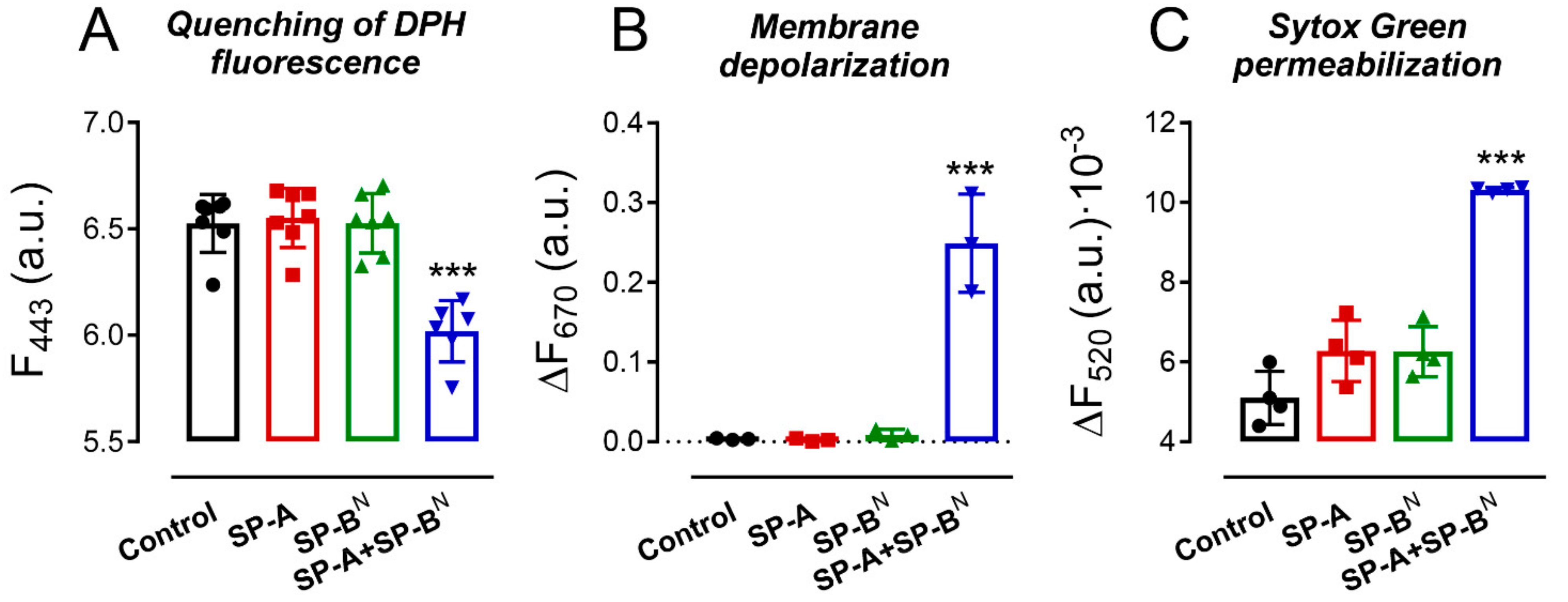
2.1. Effect of SP-A and SP-BN on the Integrity of K. pneumoniae Membranes
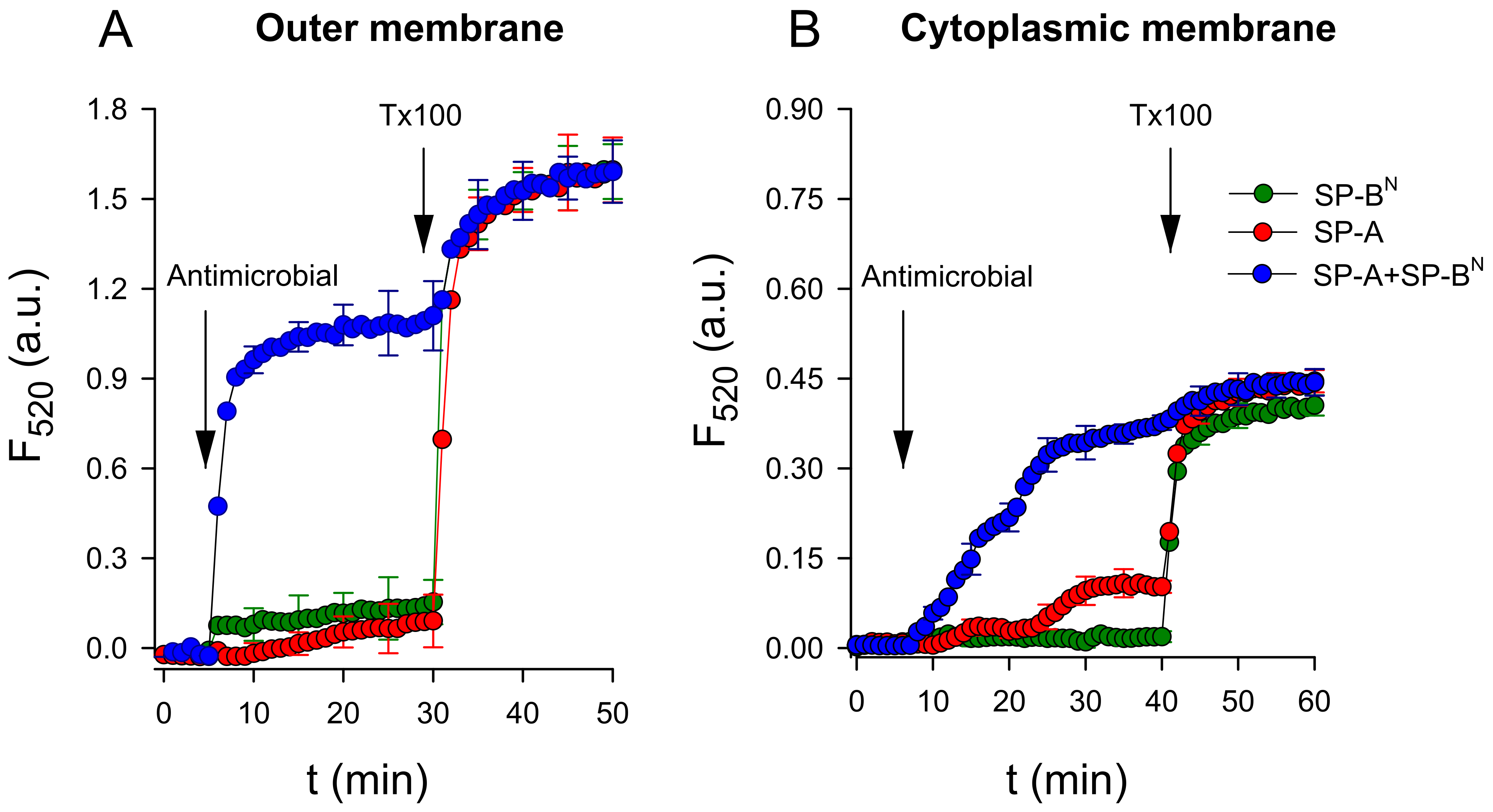
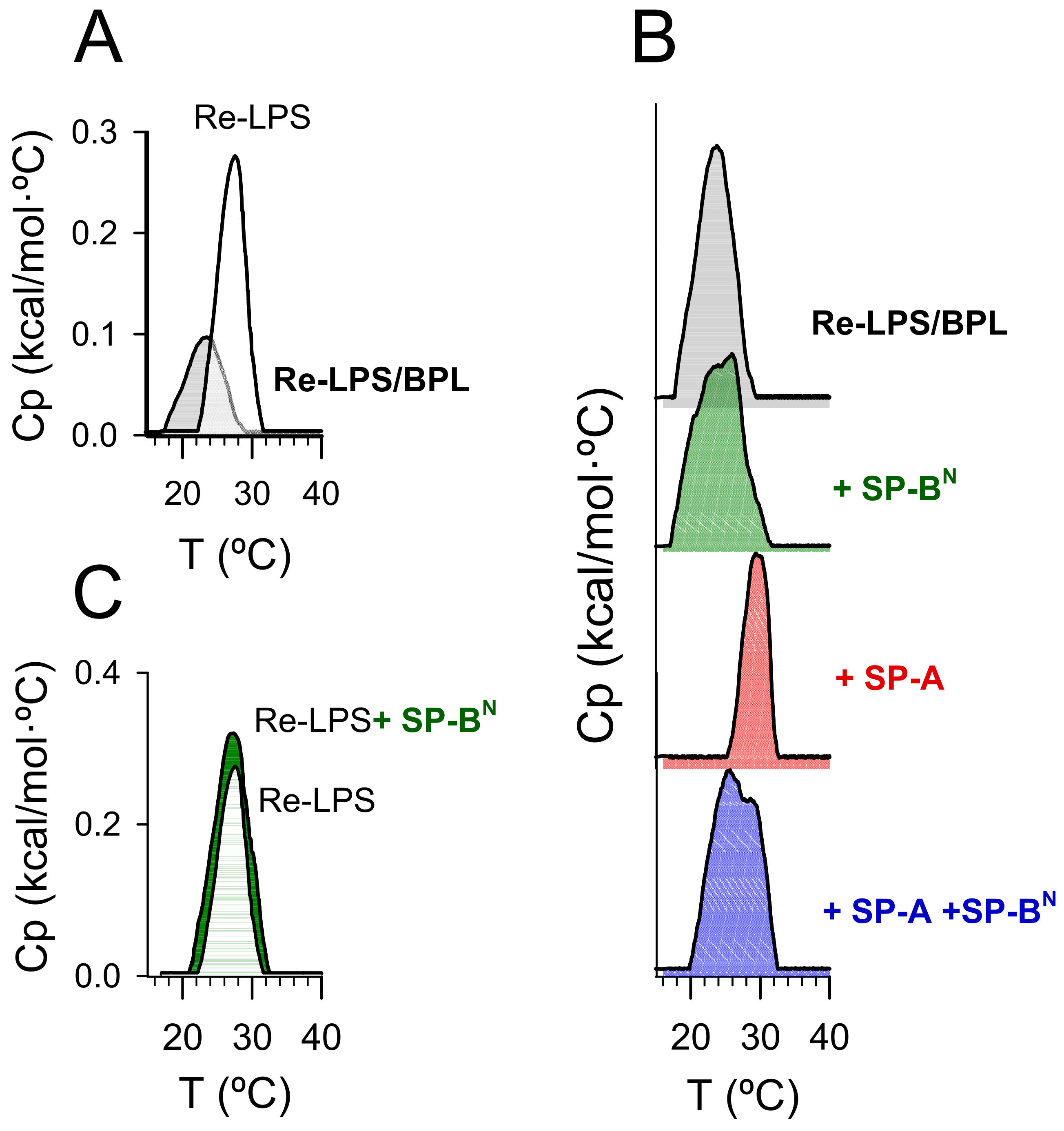
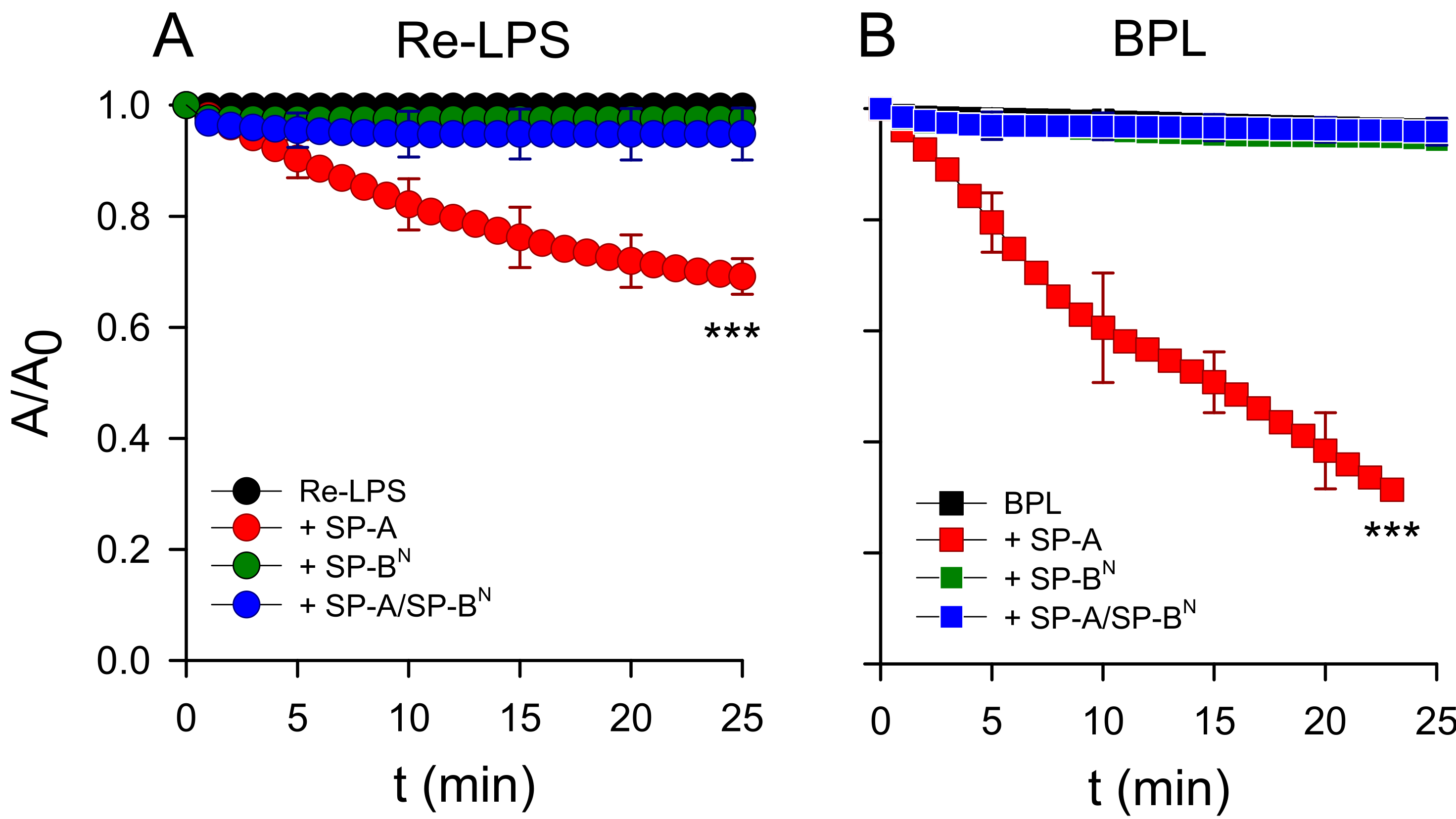
2.2. Effect of SP-A and SP-BN on Bacterial Model Membranes
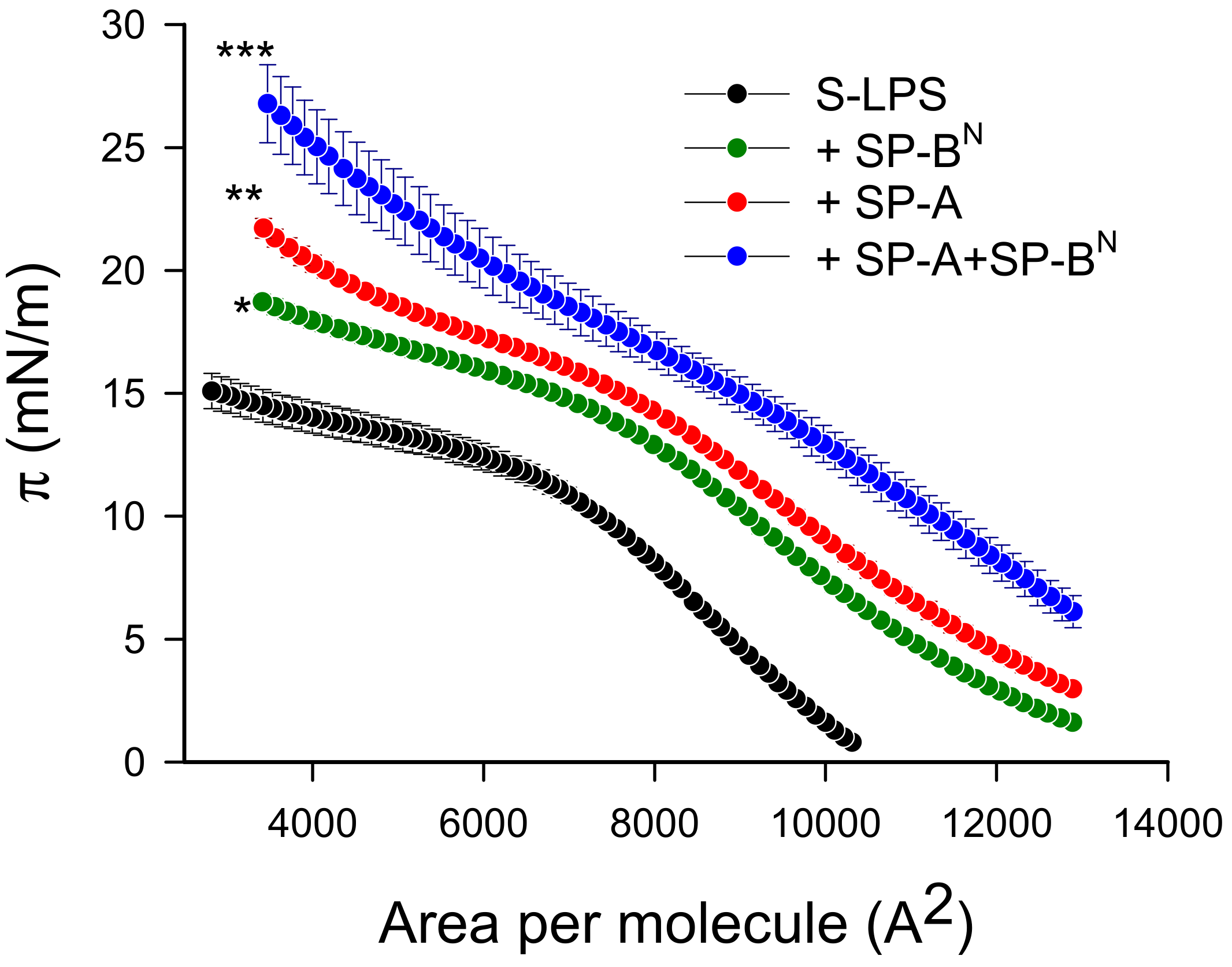
2.2.1. The Outer Membrane
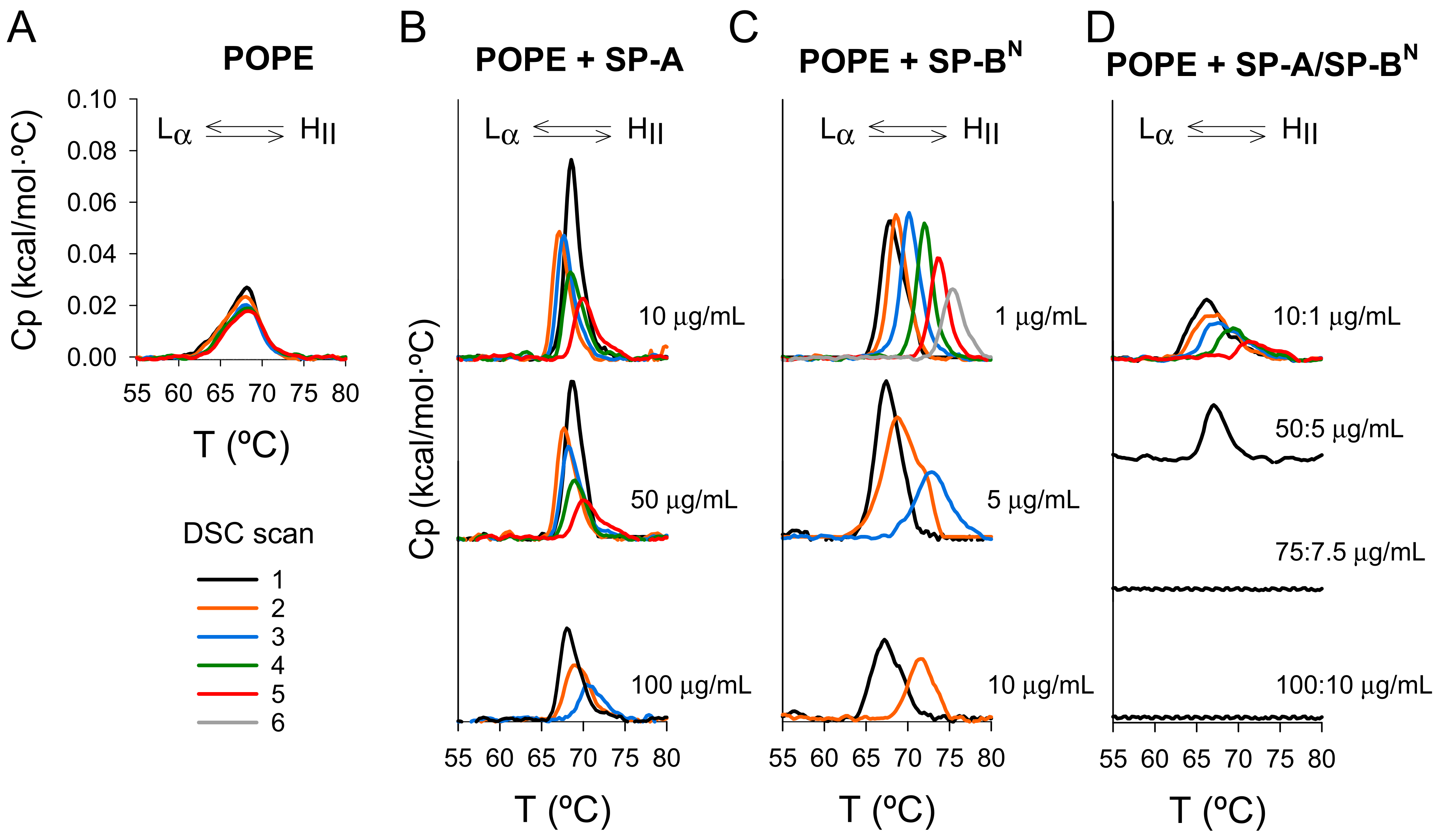
2.2.2. The Inner Membrane
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Bacteria
4.3. SP-A Isolation
4.4. Bacterial Killing Assay
4.5. Binding Assay of SP-A and SP-BN to Bacteria
4.6. Bacterial Membrane Permeabilization Assays
4.7. Cytoplasmic Membrane Depolarization
4.8. Transmision Electron Microscopy
4.9. Liposome Preparation
4.10. Membrane Leakage
4.11. Differential Scanning Calorimetry (DSC)
4.12. Relaxation Kinetics of the Re-LPS and BPL Monolayers
4.13. Pressure–Area Isotherms of S-LPS Monolayers
4.14. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). The Top 10 Causes of Death. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 28 July 2021).
- Pendleton, J.N.; Gorman, S.P.; Gilmore, B.F. Clinical relevance of the ESKAPE pathogens. Expert Rev. Anti. Infect. Ther. 2013, 11, 297–308. [Google Scholar] [CrossRef]
- Fair, R.J.; Tor, Y. Antibiotics and bacterial resistance in the 21st century. Perspect. Medicin. Chem. 2014, 6, 25–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podschun, R.; Ullmann, U. Klebsiella spp. as nosocomial pathogens: Epidemiology, taxonomy, typing methods, and pathogenicity factors. Clin. Microbiol. Rev. 1998, 11, 589–603. [Google Scholar] [CrossRef] [Green Version]
- Struve, C.; Krogfelt, K.A. Pathogenic potential of environmental Klebsiella pneumoniae isolates. Environ. Microbiol. 2004, 6, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.H.; McDonald, G.; Alton, H.; Gordon, S.B. Pneumonia in the immunocompetent patient. Br. J. Radiol. 2010, 83, 998–1009. [Google Scholar] [CrossRef] [Green Version]
- Kalanuria, A.A.; Zai, W.; Mirski, M. Ventilator-associated pneumonia in the ICU. Crit. Care 2014, 18, 208. [Google Scholar] [CrossRef] [Green Version]
- Paczosa, M.K.; Mecsas, J. Klebsiella pneumoniae: Going on the offense with a strong defense. Microbiol. Mol. Biol. Rev. 2016, 80, 629–661. [Google Scholar] [CrossRef] [Green Version]
- Sierra, J.M.; Fusté, E.; Rabanal, F.; Vinuesa, T.; Viñas, M. An overview of antimicrobial peptides and the latest advances in their development. Expert Opin. Biol. Ther. 2017, 17, 663–676. [Google Scholar] [CrossRef]
- Zhang, G.; Meredith, T.C.; Kahne, D. On the essentiality of lipopolysaccharide to Gram-negative bacteria. Curr. Opin. Microbiol. 2013, 16, 779–785. [Google Scholar] [CrossRef] [Green Version]
- Raetz, C.R.H.; Ulevitch, R.I.; Wright, S.D.; Sibley, C.H.; Ding, A.; Nathan, C.F. Gram-negative endotoxin: An extraordinary lipid with profound effects on eukaryotic signal transduction 1. FASEB J. 1991, 5, 2652–2660. [Google Scholar] [CrossRef] [Green Version]
- Morrison, D.C.; Danner, R.L.; Dinarello, C.A.; Munford, R.S.; Natanson, C.; Pollack, M.; Spitzer, J.J.; Ulevitch, R.J.; Vogel, S.N.; McSweegan, E. Bacterial endotoxins and pathogenesis of Gram-negative infections: Current status and future direction. J. Endotoxin Res. 1994, 1, 71–83. [Google Scholar] [CrossRef]
- Raetz, C.R.H.; Whitfield, C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nan, Y.H.; Bang, J.K.; Jacob, B.; Park, I.S.; Shin, S.Y. Prokaryotic selectivity and LPS-neutralizing activity of short antimicrobial peptides designed from the human antimicrobial peptide LL-37. Peptides 2012, 35, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Seydel, U.; Oikawa, M.; Fukase, K.; Kusumoto, S.; Brandenburg, K. Intrinsic conformation of lipid A is responsible for agonistic and antagonistic activity. Eur. J. Biochem. 2000, 267, 3032–3039. [Google Scholar] [CrossRef]
- Hornef, M.W.; Wick, M.J.; Rhen, M.; Normark, S. Bacterial strategies for overcoming host innate and adaptive immune responses. Nat. Immunol. 2002, 3, 1033–1040. [Google Scholar] [CrossRef]
- Delcour, A.H. Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta—Proteins Proteom. 2009, 1794, 808–816. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Zhang, L.; Wang, J.; Ma, Z.; Xu, W.; Li, J.; Shan, A. Characterization of antimicrobial activity and mechanisms of low amphipathic peptides with different α-helical propensity. Acta Biomater. 2015, 18, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Moravej, H.; Moravej, Z.; Yazdanparast, M.; Heiat, M.; Mirhosseini, A.; Moosazadeh Moghaddam, M.; Mirnejad, R. Antimicrobial Peptides: Features, Action, and Their Resistance Mechanisms in Bacteria. Microb. Drug Resist. 2018, 24, 747–767. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, J.; Vilcinskas, A. Antimicrobial peptides: The ancient arm of the human immune system. Virulence 2010, 1, 440–464. [Google Scholar] [CrossRef] [PubMed]
- Coya, J.M.; Akinbi, H.T.; Sáenz, A.; Yang, L.; Weaver, T.E.; Casals, C. Natural Anti-Infective Pulmonary Proteins: In Vivo Cooperative Action of Surfactant Protein SP-A and the Lung Antimicrobial Peptide SP-BN. J. Immunol. 2015, 195, 1628–1636. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Johansson, J.; Ridsdale, R.; Willander, H.; Fitzen, M.; Akinbi, H.T.; Weaver, T.E. Surfactant Protein B Propeptide Contains a Saposin-Like Protein Domain with Antimicrobial Activity at Low pH. J. Immunol. 2010, 184, 975–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, J.R. Immunoregulatory functions of surfactant proteins. Nat. Rev. Immunol. 2005, 5, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Casals, C.; Campanero-Rhodes, M.A.; García-Fojeda, B.; Solís, D. The role of collectins and galectins in lung innate immune defense. Front. Immunol. 2018, 9, 1998. [Google Scholar] [CrossRef] [PubMed]
- Casals, C.; García-Fojeda, B.; Minutti, C.M. Soluble defense collagens: Sweeping up immune threats. Mol. Immunol. 2019, 112, 291–304. [Google Scholar] [CrossRef]
- Giannoni, E.; Sawa, T.; Allen, L.; Wiener-Kronish, J.; Hawgood, S. Surfactant proteins A and D enhance pulmonary clearance of Pseudomonas aeruginosa. Am. J. Respir. Cell Mol. Biol. 2006, 34, 704–710. [Google Scholar] [CrossRef] [Green Version]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Roth, B.L.; Poot, M.; Yue, S.T.; Millard, P.J. Bacterial viability and antibiotic susceptibility testing with SYTOX green nucleic acid stain. Appl. Environ. Microbiol. 1997, 63, 2421–2431. [Google Scholar] [CrossRef] [Green Version]
- Cañadas, O.; Casals, C. Differential scanning calorimetry of protein-lipid interactions. Methods Mol. Biol. 2013, 974, 55–71. [Google Scholar] [CrossRef]
- Cañadas, O.; García-Verdugo, I.; Keough, K.M.W.; Casals, C. SP-A permeabilizes lipopolysaccharide membranes by forming protein aggregates that extract lipids from the membrane. Biophys. J. 2008, 95, 3287–3294. [Google Scholar] [CrossRef] [Green Version]
- García-Verdugo, I.; Cañadas, O.; Taneva, S.G.; Keough, K.M.W.; Casals, C. Surfactant protein A forms extensive lattice-like structures on 1,2-dipalmitoylphosphatidylcholine/rough-lipopolysaccharide-mixed monolayers. Biophys. J. 2007, 93, 3529–3540. [Google Scholar] [CrossRef] [Green Version]
- Cañadas, O.; Keough, K.M.W.; Casals, C. Bacterial lipopolysaccharide promotes destabilization of lung surfactant-like films. Biophys. J. 2011, 100, 108–116. [Google Scholar] [CrossRef] [Green Version]
- Hallock, K.J.; Lee, D.K.; Ramamoorthy, A. MSI-78, an analogue of the magainin antimicrobial peptides, disrupts lipid bilayer structure via positive curvature strain. Biophys. J. 2003, 84, 3052–3060. [Google Scholar] [CrossRef] [Green Version]
- Henzler Wildman, K.A.; Lee, D.K.; Ramamoorthy, A. Mechanism of lipid bilayer disruption by the human antimicrobial peptide, LL-37. Biochemistry 2003, 42, 6545–6558. [Google Scholar] [CrossRef]
- Haney, E.F.; Nathoo, S.; Vogel, H.J.; Prenner, E.J. Induction of non-lamellar lipid phases by antimicrobial peptides: A potential link to mode of action. Chem. Phys. Lipids 2010, 163, 82–93. [Google Scholar] [CrossRef]
- Epand, R.M.; Epand, R.F. Lipid domains in bacterial membranes and the action of antimicrobial agents. Biochim. Biophys. Acta—Biomembr. 2009, 1788, 289–294. [Google Scholar] [CrossRef] [Green Version]
- Ruano, M.L.F.; Nag, K.; Worthman, L.A.; Casals, C.; Pérez-Gil, J.; Keough, K.M.W. Differential partitioning of pulmonary surfactant protein SP-A into regions of monolayers of dipalmitoylphosphatidylcholine and dipalmitoylphosphatidylcholine/dipalmitoylphosphatidylglycerol. Biophys. J. 1998, 74, 1101–1109. [Google Scholar] [CrossRef] [Green Version]
- Worthman, L.A.D.; Nag, K.; Rich, N.; Ruano, M.L.F.; Casals, C.; Pérez-Gil, J.; Keough, K.M.W. Pulmonary surfactant protein A interacts with gel-like regions in monolayers of pulmonary surfactant lipid extract. Biophys. J. 2000, 79, 2657–2666. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, K.; Sugishita, K.I.; Ishibe, N.; Ueha, M.; Nakata, S.; Miyajima, K.; Epand, R.M. Relationship of membrane curvature to the formation of pores by magainin 2. Biochemistry 1998, 37, 11856–11863. [Google Scholar] [CrossRef] [PubMed]
- Mahlapuu, M.; Björn, C.; Ekblom, J. Antimicrobial peptides as therapeutic agents: Opportunities and challenges. Crit. Rev. Biotechnol. 2020, 40, 978–992. [Google Scholar] [CrossRef]
- Ahn, V.E.; Faull, K.F.; Whitelegge, J.P.; Fluharty, A.L.; Privé, G.G. Crystal structure of saposin B reveals a dimeric shell for lipid binding. Proc. Natl. Acad. Sci. USA 2003, 100, 38–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruhn, H. A short guided tour through functional and structural features of saposin-like proteins. Biochem. J. 2005, 389, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Barbero, F.; Strassner, J.; García-Cañero, R.; Steinhilber, W.; Casals, C. Role of the degree of oligomerization in the structure and function of human surfactant protein A. J. Biol. Chem. 2005, 280, 7659–7670. [Google Scholar] [CrossRef] [Green Version]
- Mahalka, A.K.; Kinnunen, P.K.J. Binding of amphipathic α-helical antimicrobial peptides to lipid membranes: Lessons from temporins B and L. Biochim. Biophys. Acta—Biomembr. 2009, 1788, 1600–1609. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.; Arce, F.T.; Mustata, M.; Ramachandran, S.; Capone, R.; Nussinov, R.; Lal, R. Antimicrobial protegrin-1 forms amyloid-like fibrils with rapid kinetics suggesting a functional link. Biophys. J. 2011, 100, 1775–1783. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, M.; Berditsch, M.; Hawecker, J.; Ardakani, M.F.; Gerthsen, D.; Ulrich, A.S. Damage of the bacterial cell envelope by antimicrobial peptides gramicidin S and PGLa as revealed by transmission and scanning electron microscopy. Antimicrob. Agents Chemother. 2010, 54, 3132–3142. [Google Scholar] [CrossRef] [Green Version]
- Nikaido, H.; Vaara, M. Outer membrane. In Escherichia Coli and Salmonella Typhimurium. Cellular and Molecular Biology; Neidhardt, C., Ingraham, J., Brooks Low, K., Magasanik, B., Schaechter, M., Umbarger, H., Eds.; American Society for Microbiology: Washington, DC, USA, 1987; pp. 7–22. [Google Scholar]
- Pogliano, J.; Pogliano, N.; Silverman, J.A. Daptomycin-mediated reorganization of membrane architecture causes mislocalization of essential cell division proteins. J. Bacteriol. 2012, 194, 4494–4504. [Google Scholar] [CrossRef]
- Scheinpflug, K.; Krylova, O.; Nikolenko, H.; Thurm, C.; Dathe, M. Evidence for a novel mechanism of antimicrobial action of a cyclic R-,W-rich hexapeptide. PLoS ONE 2015, 10, e0125056. [Google Scholar] [CrossRef] [PubMed]
- Den Hertog, A.L.; Van Marle, J.; Veerman, E.C.I.; Valentijn-Benz, M.; Nazmi, K.; Kalay, H.; Grün, C.H.; Van’t Hof, W.; Bolscher, J.G.M.; Amerongen, A.V.N. The human cathelicidin peptide LL-37 and truncated variants induce segregation of lipids and proteins in the plasma membrane of Candida albicans. Biol. Chem. 2006, 387, 1495–1502. [Google Scholar] [CrossRef] [PubMed]
- Arouri, A.; Dathe, M.; Blume, A. Peptide induced demixing in PG/PE lipid mixtures: A mechanism for the specificity of antimicrobial peptides towards bacterial membranes? Biochim. Biophys. Acta—Biomembr. 2009, 1788, 650–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epand, R.F.; Wang, G.; Berno, B.; Epand, R.M. Lipid segregation explains selective toxicity of a series of fragments derived from the human cathelicidin LL-37. Antimicrob. Agents Chemother. 2009, 53, 3705–3714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teixeira, V.; Feio, M.J.; Bastos, M. Role of lipids in the interaction of antimicrobial peptides with membranes. Prog. Lipid Res. 2012, 51, 149–177. [Google Scholar] [CrossRef]
- Domanov, Y.A.; Kinnunen, P.K.J. Antimicrobial peptides temporins B and L induce formation of tubular lipid protrusions from supported phospholipid bilayers. Biophys. J. 2006, 91, 4427–4439. [Google Scholar] [CrossRef] [Green Version]
- Koller, D.; Lohner, K. The role of spontaneous lipid curvature in the interaction of interfacially active peptides with membranes. Biochim. Biophys. Acta—Biomembr. 2014, 1838, 2250–2259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchoux, S.; Lai-Kee-Him, J.; Garnier, M.; Tsan, P.; Besson, F.; Brisson, A.; Dufourc, E.J. Surfactin-triggered small vesicle formation of negatively charged membranes: A novel membrane-lysis mechanism. Biophys. J. 2008, 95, 3840–3849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, F.; Dennison, S.; Phoenix, D. Anionic Antimicrobial Peptides from Eukaryotic Organisms. Curr. Protein Pept. Sci. 2009, 10, 585–606. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.-H.; Hall, N.K.; Aguilar, M.-I. Antimicrobial Peptide Structure and Mechanism of Action: A Focus on the Role of Membrane Structure. Curr. Top. Med. Chem. 2015, 16, 25–39. [Google Scholar] [CrossRef]
- Cushnie, T.P.T.; O’Driscoll, N.H.; Lamb, A.J. Morphological and ultrastructural changes in bacterial cells as an indicator of antibacterial mechanism of action. Cell. Mol. Life Sci. 2016, 73, 4471–4492. [Google Scholar] [CrossRef]
- Wu, H.; Kuzmenko, A.; Wan, S.; Schaffer, L.; Weiss, A.; Fisher, J.H.; Kim, K.S.; McCormack, F.X. Surfactant proteins A and D inhibit the growth of Gram-negative bacteria by increasing membrane permeability. J. Clin. Investig. 2003, 111, 1589–1602. [Google Scholar] [CrossRef] [Green Version]
- Graf, M.; Mardirossian, M.; Nguyen, F.; Seefeldt, A.C.; Guichard, G.; Scocchi, M.; Innis, C.A.; Wilson, D.N. Proline-rich antimicrobial peptides targeting protein synthesis. Nat. Prod. Rep. 2017, 34, 702–711. [Google Scholar] [CrossRef]
- Mardirossian, M.; Barrière, Q.; Timchenko, T.; Müller, C.; Pacor, S.; Mergaert, P.; Scocchi, M.; Wilson, D.N. Fragments of the Nonlytic Proline-Rich Antimicrobial Peptide Bac5 Kill Escherichia coli Cells by Inhibiting Protein Synthesis. Antimicrob. Agents Chemother. 2018, 62, e00534–e00618. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Ross, C.R.; Chengappa, M.M.; Sylte, M.J.; McVey, D.S.; Blecha, F. Antibacterial activity of a synthetic peptide (PR-26) derived from PR-39, a proline-arginine-rich neutrophil antimicrobial peptide. Antimicrob. Agents Chemother. 1996, 40, 115–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Barbero, F.; Rivas, G.; Steinhilber, W.; Casals, C. Structural and functional differences among human surfactant proteins SP-A1, SP-A2 and co-expressed SP-A1/SP-A2: Role of supratrimeric oligomerization. Biochem. J. 2007, 406, 479–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saenz, A.; Lopez-Sanchez, A.; Mojica-Lazaro, J.; Martinez-Caro, L.; Nin, N.; Bagatolli, L.A.; Casals, C. Fluidizing effects of C-reactive protein on lung surfactant membranes: Protective role of surfactant protein A. FASEB J. 2010, 24, 3662–3673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Te Winkel, J.D.; Gray, D.A.; Seistrup, K.H.; Hamoen, L.W.; Strahl, H. Analysis of antimicrobial-triggered membrane depolarization using voltage sensitive dyes. Front. Cell Dev. Biol. 2016, 4, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yusook, K.; Weeranantanapan, O.; Hua, Y.; Kumkrai, P.; Chudapongse, N. Lupinifolin from Derris reticulata possesses bactericidal activity on Staphylococcus aureus by disrupting bacterial cell membrane. J. Nat. Med. 2017, 71, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Cañadas, O.; Guerrero, R.; García-Cañero, R.; Orellana, G.; Menéndez, M.; Casals, C. Characterization of liposomal tacrolimus in lung surfactant-like phospholipids and evaluation of its immunosuppressive activity. Biochemistry 2004, 43, 9926–9938. [Google Scholar] [CrossRef]
- Hristova, K.; Selsted, M.E.; White, S.H. Critical role of lipid composition in membrane permeabilization by rabbit neutrophil defensins. J. Biol. Chem. 1997, 272, 24224–24233. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Bacterial Length (µM) |
|---|---|
| Untreated | 1.4 ± 0.2 |
| +SP-A 100 µg/mL | 1.3 ± 0.2 |
| +SP-BN 10 µg/mL | 1.4 ± 0.2 |
| +SP-A/SP-BN (population 1) | 1.4 ± 0.2 |
| +SP-A/SP-BN (population 2) | 2.1 ± 0.3 *** |
| Sample | ∆H (kcal/mol) | (°C) | T1/2 (°C) |
|---|---|---|---|
| Re-LPS/BPL | 0.6 ± 0.1 | 23.5 ± 0.2 | 5.9 ± 0.6 |
| Re-LPS/BPL+ SP-A | 0.35 ± 0.03 *** | 30.1 ± 0.4 *** | 4.2 ± 0.2 *** |
| Re-LPS/BPL + SP-BN | 0.68 ± 0.02 | 25.6 ± 0.5 *** | 8.6 ± 0.1 *** |
| Re-LPS/BPL + SP-A/SP-BN | 0.70 ± 0.08 | 25.4 ± 0.3 *** | 8.6 ± 0.4 *** |
| Sample | Cycle Number | ∆H (kcal/mol) | (°C) | T1/2 (°C) |
|---|---|---|---|---|
| POPE | 0.14 ± 0.01 | 67.9 ± 0.8 | 3.7 ± 0.6 | |
| + SP-A 10 µg/mL | 1 | 0.13 ± 0.02 | 68.5 ± 0.5 | 1.5 ± 0.4 |
| + SP-A 10 µg/mL | 5 | 0.07 ± 0.03 | 69.8 ± 0.9 | 2.5 ± 0.3 |
| + SP-A 50 µg/mL | 1 | 0.15 ± 0.02 | 68.6 ± 0.4 | 2.0 ± 0.3 |
| + SP-A 50 µg/mL | 5 | 0.05 ± 0.03 | 69.8 ± 0.7 | 2.5 ± 0.4 |
| + SP-A 100 µg/mL | 1 | 0.11 ± 0.02 | 68.5 ± 0.4 | 2.2 ± 0.4 |
| + SP-A 100 µg/mL | 3 | 0.04 ± 0.02 | 71.0 ± 1.0 | 3.2 ± 0.9 |
| + SP-BN 1 µg/mL | 1 | 0.18 ± 0.01 | 68.3 ± 0.4 | 2.5 ± 0.9 |
| + SP-BN 1 µg/mL | 6 | 0.07 ± 0.02 | 76.0 ± 1.0 | 2.5 ± 0.5 |
| + SP-BN 5 µg/mL | 1 | 0.21 ± 0.01 | 67.5 ± 0.1 | 4.1 ± 1.2 |
| + SP-BN 5 µg/mL | 3 | 0.10 ± 0.01 | 74.0 ± 2.0 | 3.6 ± 0.5 |
| + SP-BN 10 µg/mL | 1 | 0.12 ± 0.02 | 67.5 ± 0.4 | 4.0 ± 0.4 |
| + SP-BN 10 µg/mL | 2 | 0.09 ± 0.01 | 72.0 ± 1.0 | 3.5 ± 0.9 |
| + SP-A/SP-BN 10:1 µg/mL | 1 | 0.11 ± 0.02 | 66.3 ± 0.4 | 4.4 ± 0.3 |
| + SP-A/SP-BN 10:1 µg/mL | 4 | 0.06 ± 0.01 | 70.0 ± 1.0 | 3.6 ± 0.7 |
| + SP-A/SP-BN 50:5 µg/mL | 1 | 0.07 ± 0.02 | 67.5 ± 0.2 | 3.0 ± 0.5 |
| + SP-A/SP-BN 75:7.5 µg/mL | 1 | No transition | ||
| + SP-A/SP-BN 100:10 µg/mL | 1 | No transition | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fraile-Ágreda, V.; Cañadas, O.; Weaver, T.E.; Casals, C. Synergistic Action of Antimicrobial Lung Proteins against Klebsiella pneumoniae. Int. J. Mol. Sci. 2021, 22, 11146. https://doi.org/10.3390/ijms222011146
Fraile-Ágreda V, Cañadas O, Weaver TE, Casals C. Synergistic Action of Antimicrobial Lung Proteins against Klebsiella pneumoniae. International Journal of Molecular Sciences. 2021; 22(20):11146. https://doi.org/10.3390/ijms222011146
Chicago/Turabian StyleFraile-Ágreda, Víctor, Olga Cañadas, Timothy E. Weaver, and Cristina Casals. 2021. "Synergistic Action of Antimicrobial Lung Proteins against Klebsiella pneumoniae" International Journal of Molecular Sciences 22, no. 20: 11146. https://doi.org/10.3390/ijms222011146
APA StyleFraile-Ágreda, V., Cañadas, O., Weaver, T. E., & Casals, C. (2021). Synergistic Action of Antimicrobial Lung Proteins against Klebsiella pneumoniae. International Journal of Molecular Sciences, 22(20), 11146. https://doi.org/10.3390/ijms222011146