
Deconstructing Alzheimer’s Disease: How to Bridge the Gap between Experimental Models and the Human Pathology?



Abstract
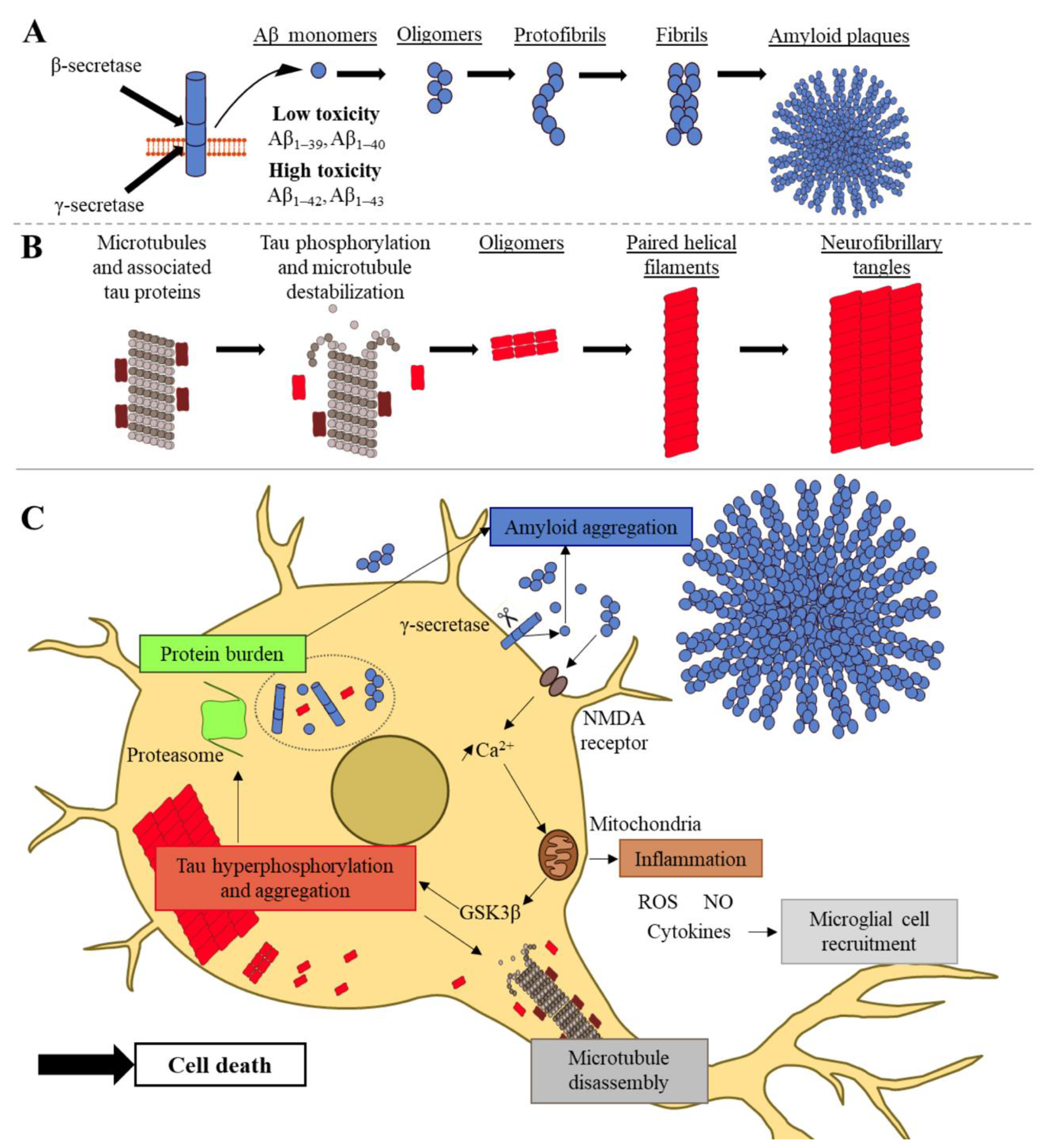
:1. Introduction
2. Molecular Features of Alzheimer’s Disease: Aβ Peptides and Tau Proteins
3. Cell-Free In Vitro and In Silico Models of Alzheimer’s Disease
3.1. In Vitro Aβ Models of Fibrillization—Monitoring the Kinetics of Fibril Formation Using Biochemical and Biophysical Methods
3.1.1. Dye-Based Methods
3.1.2. Antibody-Based Methods
3.1.3. Microscopy and Spectroscopy Techniques
3.2. Structural Models of Aβ Amyloid Peptides
3.3. Pharmacological Development Targeting Aβ Peptides
3.4. In Silico Studies of Aβ Peptides
3.5. In Vitro and In Silico Models of the Structure and Aggregation of Tau
3.5.1. Fibrillization of Tau Proteins and Pharmacological Studies
3.5.2. Structural Models of Tau Proteins
3.5.3. In Silico Studies of Tau Proteins
4. In Cellulo Models of AD
4.1. Primary Cells
4.1.1. Tissues
4.1.2. Neurons
4.1.3. Astrocytes
4.1.4. Microglia
4.1.5. Oligodendrocytes
4.1.6. Endothelial Cells and Pericytes—The Blood–Brain Barrier Model
4.2. Cell Lines
4.2.1. Cell Lines Derived from Tumors
4.2.2. Immortalized Cells
4.3. Reprogrammed and Differentiated Cells
5. In Vivo Models of AD
5.1. Caenorhabditis elegans, Drosophila melanogaster, and Danio rerio
5.1.1. Caenorhabditis elegans
5.1.2. Drosophila melanogaster
5.1.3. Danio rerio
5.2. Mouse and Rat Models
5.2.1. Transgenic Mouse and Rat Models
5.2.2. Interventional Mouse and Rat Models
5.2.3. Natural Mouse and Rat Models
5.3. Other Mammals as Interventional or Natural Models
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Prince, M.; Wimo, A.; Guerchet, M.; Ali, G.-C.; Wu, Y.-T.; Prina, M. World Alzheimer Report 2015. The Global Impact of Dementia. An Analysis of Prevalence, Incidence, Cost and Trends; Alzheimer’s Disease International: London, UK, 2015. [Google Scholar]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 118. [Google Scholar] [CrossRef] [Green Version]
- Ranjan, V.D.; Qiu, L.; Tan, E.K.; Zeng, L.; Zhang, Y. Modelling Alzheimer’s disease: Insights fromin vivotoin vitrothree-dimensional culture platforms. J. Tissue Eng. Regen. Med. 2018, 12, 1944–1958. [Google Scholar] [CrossRef]
- Mendez, M.F. Early-Onset Alzheimer Disease. Neurol. Clin. 2017, 35, 263–281. [Google Scholar] [CrossRef] [Green Version]
- Dubey, S.K.; Ram, M.S.; Krishna, K.V.; Saha, R.N.; Singhvi, G.; Agrawal, M.; Uddin, A.; Saraf, S.; Saraf, S.; Alexander, A. Recent Expansions on Cellular Models to Uncover the Scientific Barriers Towards Drug Development for Alzheimer’s Disease. Cell. Mol. Neurobiol. 2019, 39, 181–209. [Google Scholar] [CrossRef]
- Tanaka, M.; Toldi, J.; Vécsei, L. Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines. Int. J. Mol. Sci. 2020, 21, 2431. [Google Scholar] [CrossRef] [Green Version]
- Wenk, G.L. Neuropathologic changes in Alzheimer’s disease. J. Clin. Psychiatry 2003, 64 (Suppl. 9), 7–10. [Google Scholar] [PubMed]
- Surguchov, A. Caveolin: A New Link Between Diabetes and AD. Cell. Mol. Neurobiol. 2020, 40, 1059–1066. [Google Scholar] [CrossRef]
- Berchtold, N.; Cotman, C. Evolution in the Conceptualization of Dementia and Alzheimer’s Disease: Greco-Roman Period to the 1960s. Neurobiol. Aging 1998, 19, 173–189. [Google Scholar] [CrossRef]
- Liu, P.-P.; Xie, Y.; Meng, X.-Y.; Kang, J.-S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target. Ther. 2019, 4, 292. [Google Scholar] [CrossRef]
- Davis, K.; Powchick, P. Tacrine. Lancet 1995, 345, 625–630. [Google Scholar] [CrossRef]
- Bullock, R.; Touchon, J.; Bergman, H.; Gambina, G.; He, Y.; Rapatz, G.; Nagel, J.; Lane, R. Rivastigmine and donepezil treatment in moderate to moderately-severe Alzheimer’s disease over a 2-year period. Curr. Med. Res. Opin. 2005, 21, 1317–1327. [Google Scholar] [CrossRef]
- Reisberg, B.; Doody, R.; Stöffler, A.; Schmitt, F.; Ferris, S.; Möbius, H.J. Memantine in Moderate-to-Severe Alzheimer’s Disease. N. Engl. J. Med. 2003, 348, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Török, N.; Vécsei, L. Novel Pharmaceutical Approaches in Dementia. In NeuroPsychopharmacotherapy; Riederer, P., Laux, G., Nagatsu, T., Le, W., Riederer, C., Eds.; Springer: Cham, Switzerland, 2020; pp. 1–18. ISBN 978-3-319-56015-1. [Google Scholar]
- Frost, G.; Li, Y.-M. The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol. 2017, 7, 170228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Liu, F.; Huang, C.; Shentu, J.; Wang, M.; Sun, C.; Chen, L.; Yan, S.; Fang, F.; Wang, Y.; et al. 5-Hydroxycyclopenicillone Inhibits β-Amyloid Oligomerization and Produces Anti-β-Amyloid Neuroprotective Effects In Vitro. Molecules 2017, 22, 1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, V.W.; Mattson, M.P.; Wong, P.C.; Gleichmann, M. An Overview of APP Processing Enzymes and Products. Neuro Mol. Med. 2010, 12, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-Secretase Cleavage of Alzheimer’s Amyloid Precursor Protein by the Transmembrane Aspartic Protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef] [Green Version]
- Sakono, M.; Zako, T. Amyloid oligomers: Formation and toxicity of Aβ oligomers. FEBS J. 2010, 277, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.; Bateman, R.J. Decreased Clearance of CNS -Amyloid in Alzheimer’s Disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef] [Green Version]
- Bates, K.; Verdile, G.; Li, Q.; Ames, D.; Hudson, P.J.; Masters, C.L.; Martins, R.N. Clearance mechanisms of Alzheimer’s amyloid-β peptide: Implications for therapeutic design and diagnostic tests. Mol. Psychiatry 2008, 14, 469–486. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The Blood–Brain Barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Falcon, B.; Zhang, W.; Schweighauser, M.; Murzin, A.G.; Vidal, R.; Garringer, H.J.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Tau filaments from multiple cases of sporadic and inherited Alzheimer’s disease adopt a common fold. Acta Neuropathol. 2018, 136, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M. Tau filaments in neurodegenerative diseases. FEBS Lett. 2018, 592, 2383–2391. [Google Scholar] [CrossRef]
- Keck, S.; Nitsch, R.; Grune, T.; Ullrich, O. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer’s disease. J. Neurochem. 2003, 85, 115–122. [Google Scholar] [CrossRef]
- Barrio, R.; Sutherland, J.D.; Rodriguez, M.S. Proteostasis and Disease: From Basic Mechanisms to Clinics. In Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2020; Volume 1233, ISBN 978-3-030-38265-0. [Google Scholar]
- Esposito, Z.; Belli, L.; Toniolo, S.; Sancesario, G.; Bianconi, C.; Martorana, A. Amyloid β, Glutamate, Excitotoxicity in Alzheimer’s Disease: Are We on the Right Track? CNS Neurosci. Ther. 2013, 19, 549–555. [Google Scholar] [CrossRef]
- Schmitz, C.; Rutten, B.P.F.; Pielen, A.; Schäfer, S.; Wirths, O.; Tremp, G.; Czech, C.; Blanchard, V.; Multhaup, G.; Rezaie, P.; et al. Hippocampal Neuron Loss Exceeds Amyloid Plaque Load in a Transgenic Mouse Model of Alzheimer’s Disease. Am. J. Pathol. 2004, 164, 1495–1502. [Google Scholar] [CrossRef]
- Forloni, G.; Balducci, C. Alzheimer’s Disease, Oligomers, and Inflammation. J. Alzheimer’s Dis. 2018, 62, 1261–1276. [Google Scholar] [CrossRef] [Green Version]
- Nisbet, R.M.; Polanco, J.C.; Ittner, L.; Götz, J. Tau aggregation and its interplay with amyloid-β. Acta Neuropathol. 2015, 129, 207–220. [Google Scholar] [CrossRef] [Green Version]
- Bloom, G.S. Amyloid-β, and Tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yakupova, E.I.; Bobyleva, L.G.; Vikhlyantsev, I.M.; Bobylev, A.G. Congo Red and amyloids: History and relationship. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassar, P.S.; Culling, C.F. Fluorescent stains, with special reference to amyloid and connective tissues. Arch. Pathol. 1959, 68, 487–498. [Google Scholar]
- Malmos, K.G.; Blancas-Mejia, L.M.; Weber, B.; Buchner, J.; Ramirez-Alvarado, M.; Naiki, H.; Otzen, D. ThT 101: A primer on the use of thioflavin T to investigate amyloid formation. Amyloid 2017, 24, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, A.D.F.-; Robbs, B.; Moreau, V.H.; Ferreira, A.; Juliano, L.; Valente, A.P.; Almeida, F.C.; Silva, J.; Foguel, D. Controlling β-Amyloid Oligomerization by the Use of Naphthalene Sulfonates: Trapping low molecular weight oligomeric species. J. Biol. Chem. 2005, 280, 34747–34754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulatsky, M.I.; Sulatskaya, A.I.; Povarova, O.I.; Antifeeva, I.A.; Kuznetsova, I.M.; Turoverov, K.K. Effect of the fluorescent probes ThT and ANS on the mature amyloid fibrils. Prion 2020, 14, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Bruggink, K.A.; Müller, M.; Kuiperij, H.B.; Verbeek, M.M. Methods for Analysis of Amyloid-β Aggregates. J. Alzheimer’s Dis. 2012, 28, 735–758. [Google Scholar] [CrossRef]
- Siddiqi, M.K.; Majid, N.; Malik, S.; Alam, P.; Khan, R.H. Amyloid Oligomers, Protofibrils and Fibrils. In Macromolecular Protein Complexes II: Structure and Function; Harris, J.R., Marles-Wright, J., Eds.; Subcellular Biochemistry; Springer: Cham, Switzerland, 2019; pp. 471–503. ISBN 978-3-030-28151-9. [Google Scholar]
- Kumar, E.K.; Haque, N.; Prabhu, N.P. Kinetics of protein fibril formation: Methods and mechanisms. Int. J. Biol. Macromol. 2017, 100, 3–10. [Google Scholar] [CrossRef]
- Alghazwi, M.; Smid, S.; Musgrave, I.; Zhang, W. In vitro studies of the neuroprotective activities of astaxanthin and fucoxanthin against amyloid β (Aβ1-42) toxicity and aggregation. Neurochem. Int. 2019, 124, 215–224. [Google Scholar] [CrossRef] [Green Version]
- Bana, L.; Minniti, S.; Salvati, E.; Sesana, S.; Zambelli, V.; Cagnotto, A.; Orlando, A.; Cazzaniga, E.; Zwart, R.; Scheper, W.; et al. Liposomes bi-functionalized with phosphatidic acid and an ApoE-derived peptide affect Aβ aggregation features and cross the blood–brain-barrier: Implications for therapy of Alzheimer disease. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 1583–1590. [Google Scholar] [CrossRef]
- Scheidt, T.; Łapińska, U.; Kumita, J.R.; Whiten, D.R.; Klenerman, D.; Wilson, M.R.; Cohen, S.I.A.; Linse, S.; Vendruscolo, M.; Dobson, C.M.; et al. Secondary nucleation and elongation occur at different sites on Alzheimer’s amyloid-β aggregates. Sci. Adv. 2019, 5, eaau3112. [Google Scholar] [CrossRef] [Green Version]
- Chaudhury, S.S.; Sannigrahi, A.; Nandi, M.; Mishra, V.K.; De, P.; Chattopadhyay, K.; Mishra, S.; Sil, J.; Das Mukhopadhyay, C. A Novel PEGylated Block Copolymer in New Age Therapeutics for Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 6551–6565. [Google Scholar] [CrossRef]
- Clarke, E.E.; Shearman, M.S. Quantitation of amyloid-β peptides in biological milieu using a novel homogeneous time-resolved fluorescence (HTRF) assay. J. Neurosci. Methods 2000, 102, 61–68. [Google Scholar] [CrossRef]
- Lewis, H.; Beher, D.; Cookson, N.; Oakley, A.; Piggott, M.; Morris, C.M.; Jaros, E.; Perry, R.; Ince, P.; Kenny, R.A.; et al. Quantification of Alzheimer pathology in ageing and dementia: Age-related accumulation of amyloid-β(42) peptide in vascular dementia. Neuropathol. Appl. Neurobiol. 2006, 32, 103–118. [Google Scholar] [CrossRef]
- Maulthaup, G.; Mechler, H.; Masters, C.L. Characterization of the high affinity heparin binding site of the Alzheimer’s disease βA4 amyloid precursor protein (APP) and its enhancement by zinc(II). J. Mol. Recognit. 1995, 8, 247–257. [Google Scholar] [CrossRef]
- Palladino, P.; Aura, A.M.; Spoto, G. Surface plasmon resonance for the label-free detection of Alzheimer’s β-amyloid peptide aggregation. Anal. Bioanal. Chem. 2015, 408, 849–854. [Google Scholar] [CrossRef]
- Patching, S.G. Surface plasmon resonance spectroscopy for characterisation of membrane protein–ligand interactions and its potential for drug discovery. Biochim. Biophys. Acta Biomembr. 2014, 1838, 43–55. [Google Scholar] [CrossRef] [Green Version]
- Yi, X.; Feng, C.; Hu, S.; Li, H.; Wang, J. Surface plasmon resonance biosensors for simultaneous monitoring of amyloid-β oligomers and fibrils and screening of select modulators. Analyst 2015, 141, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Bjijrling, S.; Thyberg, P.; Thyberg, J.; Berndt, K.D.; Tereniusl, L. Amyloid B-Peptide Polymerization Studied Using Fluorescence Correlation Spectroscopy. Chem. Biol. 1999, 6, 53–62. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, P.; Garai, K.; Sahoo, B.; Shi, Y.; Callaway, D.J.E.; Maiti, S. The Amyloid β Peptide (Aβ1-40) Is Thermodynamically Soluble at Physiological Concentrations. Biochemistry 2003, 42, 10506–10513. [Google Scholar] [CrossRef]
- Krichevsky, O.; Bonnet, G. Fluorescence correlation spectroscopy: The technique and its applications. Rep. Prog. Phys. 2002, 65, 251–297. [Google Scholar] [CrossRef]
- Alghamdi, A.; Vyshemirsky, V.; Birch, D.; Rolinski, O.J. Detecting β-amyloid aggregation from time-resolved emission spectra. Methods Appl. Fluoresc. 2017, 6, 024002. [Google Scholar] [CrossRef] [Green Version]
- Alghamdi, A.; Wellbrock, T.; Birch, D.; Vyshemirsky, V.; Rolinski, O.J. Cu 2+ Effects on Beta-Amyloid Oligomerisation Monitored by the Fluorescence of Intrinsic Tyrosine. ChemPhysChem 2019, 20, 3181–3185. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Scott, G.; Merchant, F.; Murphy, R. Light scattering analysis of fibril growth from the amino-terminal fragment β(1–28) of β-amyloid peptide. Biophys. J. 1993, 65, 2383–2395. [Google Scholar] [CrossRef] [Green Version]
- Tomski, S.J.; Murphy, R.M. Kinetics of aggregation of synthetic β-amyloid peptide. Arch. Biochem. Biophys. 1992, 294, 630–638. [Google Scholar] [CrossRef]
- Gremer, L.; Schölzel, D.; Schenk, C.; Reinartz, E.; Labahn, J.; Ravelli, R.B.G.; Tusche, M.; Lopez-Iglesias, C.; Hoyer, W.; Heise, H.; et al. Fibril structure of amyloid-β(1–42) by cryo–electron microscopy. Science 2017, 358, 116–119. [Google Scholar] [CrossRef] [Green Version]
- Popov, K.I.; Makepeace, K.A.; Petrotchenko, E.V.; Dokholyan, N.V.; Borchers, C.H. Insight into the Structure of the “Unstructured” Tau Protein. Structure 2019, 27, 1710–1715.e4. [Google Scholar] [CrossRef]
- Weickenmeier, J.; Kuhl, E.; Goriely, A. Multiphysics of Prionlike Diseases: Progression and Atrophy. Phys. Rev. Lett. 2018, 121, 158101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Chowdhury, S.R.; Jacobs, H.; Johnson, K.A.; Dutta, J. A Longitudinal Model for Tau Aggregation in Alzheimer’s Disease Based on Structural Connectivity. Process. Med. Imaging 2019, 11492, 384–393. [Google Scholar] [CrossRef]
- Arosio, P.; Cukalevski, R.; Frohm, B.; Knowles, T.; Linse, S. Quantification of the Concentration of Aβ42 Propagons during the Lag Phase by an Amyloid Chain Reaction Assay. J. Am. Chem. Soc. 2014, 136, 219–225. [Google Scholar] [CrossRef]
- Nick, M.; Wu, Y.; Schmidt, N.W.; Prusiner, S.B.; Stöhr, J.; De Grado, W.F. A long-lived Aβ oligomer resistant to fibrillization. Biopolymer 2018, 109, e23096. [Google Scholar] [CrossRef]
- Inouye, H.; Fraser, P.E.; Kirschner, D. Structure of β-crystallite assemblies formed by Alzheimer β-amyloid protein analogues: Analysis by x-ray diffraction. Biophys. J. 1993, 64, 502–519. [Google Scholar] [CrossRef] [Green Version]
- Kollmer, M.; Close, W.; Funk, L.; Rasmussen, J.; Bsoul, A.; Schierhorn, A.; Schmidt, M.; Sigurdson, C.J.; Jucker, M.; Fändrich, M. Cryo-EM structure and polymorphism of Aβ amyloid fibrils purified from Alzheimer’s brain tissue. Nat. Commun. 2019, 10, 4760. [Google Scholar] [CrossRef] [Green Version]
- Qiang, W.; Yau, W.-M.; Lu, J.-X.; Collinge, J.; Tycko, R. Structural variation in amyloid-β fibrils from Alzheimer’s disease clinical subtypes. Nature 2017, 541, 217–221. [Google Scholar] [CrossRef] [Green Version]
- He, K.-C.; Chen, Y.-R.; Liang, C.-T.; Huang, S.-J.; Tzeng, C.-Y.; Chang, C.-F.; Huang, S.-J.; Huang, H.-B.; Lin, T.-H. Conformational Characterization of Native and L17A/F19A-Substituted Dutch-Type β-Amyloid Peptides. Int. J. Mol. Sci. 2020, 21, 2571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Nehru, B.; Saini, A. Inhibition of Alzheimer’s amyloid-β aggregation in-vitro by carbenoxolone: Insight into mechanism of action. Neurochem. Int. 2017, 108, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Syarifah-Noratiqah, S.-B.; Mohamed, Z.; Zulfarina, M.S.; Qodriyah, H. Natural Polyphenols in the Treatment of Alzheimer’s Disease. Curr. Drug Targets 2018, 19, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Witter, S.; Witter, R.; Vilu, R.; Samoson, A. Medical Plants and Nutraceuticals for Amyloid-β Fibrillation Inhibition. J. Alzheimer’s Dis. Rep. 2018, 2, 239–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, J.; Takahashi, T.; Oshima, H.; Matsumura, S.; Mihara, H. Design of Peptides That Form Amyloid-Like Fibrils Capturing Amyloid β1–42 Peptides. Chem. A Eur. J. 2007, 13, 7745–7752. [Google Scholar] [CrossRef] [PubMed]
- Nardo, L.; Re, F.; Brioschi, S.; Cazzaniga, E.; Orlando, A.; Minniti, S.; Lamperti, M.; Gregori, M.; Cassina, V.; Brogioli, D.; et al. Fluorimetric detection of the earliest events in amyloid β oligomerization and its inhibition by pharmacologically active liposomes. Biochim. Biophys. Acta Gen. Subj. 2016, 1860, 746–756. [Google Scholar] [CrossRef]
- Morris, G.M.; Lim-Wilby, M. Molecular Docking. Obes. Cancer 2008, 443, 365–382. [Google Scholar] [CrossRef]
- Safarizadeh, H.; Garkani-Nejad, Z. Molecular docking, molecular dynamics simulations and QSAR studies on some of 2-arylethenylquinoline derivatives for inhibition of Alzheimer’s amyloid-β aggregation: Insight into mechanism of interactions and parameters for design of new inhibitors. J. Mol. Graph. Model. 2019, 87, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Nirmalraj, P.N.; List, J.; Battacharya, S.; Howe, G.; Xu, L.; Thompson, D.; Mayer, M. Complete aggregation pathway of amyloid β (1-40) and (1-42) resolved on an atomically clean interface. Sci. Adv. 2020, 6, eaaz6014. [Google Scholar] [CrossRef] [Green Version]
- Acosta, D.M.Á.V.; Vega, B.C.; Basurto, J.C.; Morales, L.G.F.; Hernandez, M.C.R. Recent Advances by In Silico and In Vitro Studies of Amyloid-β 1-42 Fibril Depicted a S-Shape Conformation. Int. J. Mol. Sci. 2018, 19, 2415. [Google Scholar] [CrossRef] [Green Version]
- Nie, R.-Z.; Huo, Y.-Q.; Yu, B.; Liu, C.-J.; Zhou, R.; Bao, H.-H.; Tang, S.-W. Molecular insights into the inhibitory mechanisms of gallate moiety on the Aβ1–40 amyloid aggregation: A molecular dynamics simulation study. Int. J. Biol. Macromol. 2020, 156, 40–50. [Google Scholar] [CrossRef]
- Berriman, J.; Serpell, L.; Oberg, K.A.; Fink, A.L.; Goedert, M.; Crowther, R.A. Tau filaments from human brain and from in vitro assembly of recombinant protein show cross- structure. Proc. Natl. Acad. Sci. USA 2003, 100, 9034–9038. [Google Scholar] [CrossRef] [Green Version]
- Crespo, R.; Koudstaal, W.; Apetri, A. In Vitro Assay for Studying the Aggregation of Tau Protein and Drug Screening. J. Vis. Exp. 2018, 58570, e58570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Despres, C.; Di, J.; Cantrelle, F.-X.; Li, Z.; Huvent, I.; Chambraud, B.; Zhao, J.; Chen, J.; Chen, S.; Lippens, G.; et al. Major Differences between the Self-Assembly and Seeding Behavior of Heparin-Induced and in Vitro Phosphorylated Tau and Their Modulation by Potential Inhibitors. ACS Chem. Biol. 2019, 14, 1363–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedert, M.; Jakes, R.; Spillantini, M.G.; Hasegawa, M.; Smith, M.J.; Crowther, R.A. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nat. Cell Biol. 1996, 383, 550–553. [Google Scholar] [CrossRef]
- Mutreja, Y.; Gamblin, T.C. Optimization of in vitro conditions to study the arachidonic acid induction of 4R isoforms of the microtubule-associated protein tau. Micropatterning Cell Biol. Part B 2017, 141, 65–88. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.; Cairns, N.; Crowther, R. Tau proteins of alzheimer paired helical filaments: Abnormal phosphorylation of all six brain isoforms. Neuron 1992, 8, 159–168. [Google Scholar] [CrossRef]
- Pickhardt, M.; Gazova, Z.; von Bergen, M.; Khlistunova, I.; Wang, Y.; Hascher, A.; Mandelkow, E.-M.; Biernat, J.; Mandelkow, E. Anthraquinones Inhibit Tau Aggregation and Dissolve Alzheimer’s Paired Helical Filaments in Vitro and in Cells. J. Biol. Chem. 2005, 280, 3628–3635. [Google Scholar] [CrossRef] [Green Version]
- Peterson, D.W.; George, R.C.; Scaramozzino, F.; LaPointe, N.E.; Anderson, R.A.; Graves, D.J.; Lew, J. Cinnamon Extract Inhibits Tau Aggregation Associated with Alzheimer’s Disease In Vitro. J. Alzheimer’s Dis. 2009, 17, 585–597. [Google Scholar] [CrossRef] [Green Version]
- Rane, J.S.; Bhaumik, P.; Panda, D. Curcumin Inhibits Tau Aggregation and Disintegrates Preformed Tau Filaments in vitro. J. Alzheimer’s Dis. 2017, 60, 999–1014. [Google Scholar] [CrossRef] [PubMed]
- Seidler, P.M.; Boyer, D.R.; Rodriguez, J.A.; Sawaya, M.R.; Cascio, D.; Murray, K.; Gonen, T.; Eisenberg, D.S. Structure-based inhibitors of tau aggregation. Nat. Chem. 2018, 10, 170–176. [Google Scholar] [CrossRef]
- Pir, G.J.; Choudhary, B.; Kaniyappan, S.; Chandupatla, R.R.; Mandelkow, E.; Mandelkow, E.-M.; Wang, Y. Suppressing Tau Aggregation and Toxicity by an Anti-Aggregant Tau Fragment. Mol. Neurobiol. 2018, 56, 3751–3767. [Google Scholar] [CrossRef] [Green Version]
- Dregni, A.J.; Mandala, V.S.; Wu, H.; Elkins, M.R.; Wang, H.K.; Hung, I.; DeGrado, W.F.; Hong, M. In vitro 0N4R tau fibrils contain a monomorphic β-sheet core enclosed by dynamically heterogeneous fuzzy coat segments. Proc. Natl. Acad. Sci. USA 2019, 116, 16357–16366. [Google Scholar] [CrossRef] [Green Version]
- Sato, R.; Vohra, S.; Yamamoto, S.; Suzuki, K.; Pavel, K.; Shulga, S.; Blume, Y.; Kurita, N. Specific interactions between tau protein and curcumin derivatives: Molecular docking and ab initio molecular orbital simulations. J. Mol. Graph. Model. 2020, 98, 107611. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, G.K.; Shwartz, D.; Losev, Y.; Arad, E.; Shemesh, C.; Pichinuk, E.; Engel, H.; Raveh, A.; Jelinek, R.; Cooper, I.; et al. Purpurin modulates Tau-derived VQIVYK fibrillization and ameliorates Alzheimer’s disease-like symptoms in animal model. Cell. Mol. Life Sci. 2020, 77, 2795–2813. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, O.; Huo, Y.; Wang, G.; Man, H.-Y. Amyloid-β Induces AMPA Receptor Ubiquitination and Degradation in Primary Neurons and Human Brains of Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1789–1801. [Google Scholar] [CrossRef] [PubMed]
- Drummond, E.; Wisniewski, T. Alzheimer’s disease: Experimental models and reality. Acta Neuropathol. 2017, 133, 155–175. [Google Scholar] [CrossRef]
- Julia, Tcw. Human iPSC application in Alzheimer’s disease and Tau-related neurodegenerative diseases. Neurosci. Lett. 2019, 699, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Shepardson, N.; Yang, T.; Chen, G.; Walsh, D.; Selkoe, D.J. Soluble amyloid -protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 5819–5824. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Lee, B.K.; Jeong, G.S.; Hyun, J.K.; Lee, C.J.; Lee, S.-H. Three-dimensional brain-on-a-chip with an interstitial level of flow and its application as an in vitro model of Alzheimer’s disease. Lab Chip 2014, 15, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wu, Z.-G.; Shi, W.-J.; Gao, H.; Wu, H.-H.; Bian, F.; Jia, P.-P.; Hou, Y.-N. Effects of progesterone on glucose uptake in neurons of Alzheimer’s disease animals and cell models. Life Sci. 2019, 238, 116979. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Park, J.; Lee, S.-H. Size-controllable networked neurospheres as a 3D neuronal tissue model for Alzheimer’s disease studies. Biomaterials 2013, 34, 2938–2946. [Google Scholar] [CrossRef]
- Kunze, A.; Meissner, R.; Brando, S.; Renaud, P. Co-pathological connected primary neurons in a microfluidic device for alzheimer studies. Biotechnol. Bioeng. 2011, 108, 2241–2245. [Google Scholar] [CrossRef] [PubMed]
- Jorda, A.; Aldasoro, M.; Aldasoro, C.; Guerra-Ojeda, S.; Iradi, A.; Vila, J.M.; Campos-Campos, J.; Valles, S.L. Action of low doses of Aspirin in Inflammation and Oxidative Stress induced by aβ1-42 on Astrocytes in primary culture. Int. J. Med Sci. 2020, 17, 834–843. [Google Scholar] [CrossRef]
- Limbad, C.; Oron, T.R.; Alimirah, F.; Davalos, A.R.; Tracy, T.E.; Gan, L.; Desprez, P.-Y.; Campisi, J. Astrocyte senescence promotes glutamate toxicity in cortical neurons. PLoS ONE 2020, 15, e0227887. [Google Scholar] [CrossRef]
- Hopp, S.C.; Lin, Y.; Oakley, D.; Roe, A.D.; Devos, S.L.; Hanlon, D.; Hyman, B.T. The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 269. [Google Scholar] [CrossRef] [Green Version]
- Stansley, B.; Post, J.; Hensley, K. A comparative review of cell culture systems for the study of microglial biology in Alzheimer’s disease. J. Neuroinflamm. 2012, 9, 115. [Google Scholar] [CrossRef] [Green Version]
- Horiuchi, M.; Maezawa, I.; Itoh, A.; Wakayama, K.; Jin, L.-W.; Itoh, T.; DeCarli, C. Amyloid β1–42 oligomer inhibits myelin sheet formation in vitro. Neurobiol. Aging 2012, 33, 499–509. [Google Scholar] [CrossRef] [Green Version]
- Roth, A.D.; Ramírez, G.; Alarcón, R.; VON Bernhardi, R. Oligodendrocytes damage in Alzheimer’s disease: Beta amyloid toxicity and inflammation. Biol. Res. 2005, 38, 381–387. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Liu, G.; Wang, Y.; Yao, Y.; Wang, G.; Lei, X.; Zhang, N.; Dong, X. Protective Effect of Buyang Huanwu Decoction on Neurovascular Unit in Alzheimer’s Disease Cell Model via Inflammation and RAGE/LRP1 Pathway. Med. Sci. Monit. 2019, 25, 7813–7825. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, P.N.; Kruck, T.P.; Lukiw, W.J. Nanomolar aluminum induces expression of the inflammatory systemic biomarker C-reactive protein (CRP) in human brain microvessel endothelial cells (hBMECs). J. Inorg. Biochem. 2015, 152, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.-C.; Tsao, C.-W. Neuroprotection against Apoptosis of SK-N-MC Cells Using RMP-7- and Lactoferrin-Grafted Lipo-somes Carrying Quercetin. Available online: https://www.dovepress.com/neuroprotection-against-apoptosis-of-sk-n-mc-cells-using-rmp-7--and-la-peer-reviewed-fulltext-article-IJN (accessed on 20 May 2020).
- McCarthy, R.C.; Kosman, D.J. Iron transport across the Blood-Brain barrier: Development, neurovascular regulation and cerebral amyloid angiopathy. Cell. Mol. Life Sci. 2015, 72, 709–727. [Google Scholar] [CrossRef] [Green Version]
- Sun, R.; He, T.; Pan, Y.; Katusic, Z.S. Effects of senescence and angiotensin II on expression and processing of amyloid precursor protein in human cerebral microvascular endothelial cells. Aging 2018, 10, 100–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spampinato, S.F.; Merlo, S.; Fagone, E.; Fruciano, M.; Barbagallo, C.; Kanda, T.; Sano, Y.; Purrello, M.; Vancheri, C.; Ragusa, M.; et al. Astrocytes Modify Migration of PBMCs Induced by β-Amyloid in a Blood-Brain Barrier in vitro Model. Front. Cell. Neurosci. 2019, 13, 337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, N.; Brännström, K.; Byman, E.; Moussaud, S.; Nielsen, H.M.; Olofsson, A.; Wennstrom, M. Amyloid-β 1-40 is associated with alterations in NG2+ pericyte population ex vivo and in vitro. Aging Cell 2018, 17, e12728. [Google Scholar] [CrossRef] [Green Version]
- Burkhart, A.; Thomsen, L.B.; Thomsen, M.S.; Lichota, J.; Fazakas, C.; Krizbai, I.; Moos, T. Transfection of brain capillary endothelial cells in primary culture with defined blood-brain barrier properties. Fluids Barriers CNS 2015, 12, 19. [Google Scholar] [CrossRef] [Green Version]
- Stoppelkamp, S.; Bell, H.S.; Palacios-Filardo, J.; Shewan, D.A.; Riedel, G.; Platt, B. In vitro modelling of Alzheimer’s disease: Degeneration and cell death induced by viral delivery of amyloid and tau. Exp. Neurol. 2011, 229, 226–237. [Google Scholar] [CrossRef] [Green Version]
- Hargus, G.; Ehrlich, M.; Hallmann, A.-L.; Kuhlmann, T. Human stem cell models of neurodegeneration: A novel approach to study mechanisms of disease development. Acta Neuropathol. 2013, 127, 151–173. [Google Scholar] [CrossRef]
- Devinsky, O.; Vezzani, A.; Najjar, S.; De Lanerolle, N.C.; Rogawski, M. Glia and epilepsy: Excitability and inflammation. Trends Neurosci. 2013, 36, 174–184. [Google Scholar] [CrossRef]
- Jäkel, S.; Dimou, L. Glial Cells and Their Function in the Adult Brain: A Journey through the History of Their Ablation. Front. Cell. Neurosci. 2017, 11, 24. [Google Scholar] [CrossRef] [Green Version]
- Colin, M.; Dujardin, S.; Schraen-Maschke, S.; Meno-Tetang, G.; Duyckaerts, C.; Courade, J.-P.; Buée, L. From the prion-like propagation hypothesis to therapeutic strategies of anti-tau immunotherapy. Acta Neuropathol. 2020, 139, 3–25. [Google Scholar] [CrossRef] [Green Version]
- Zandl-Lang, M.; Fanaee-Danesh, E.; Sun, Y.; Albrecher, N.M.; Gali, C.; Čančar, I.; Kober, A.; Tam-Amersdorfer, C.; Stracke, A.; Storck, S.; et al. Regulatory effects of simvastatin and apoJ on APP processing and amyloid-β clearance in blood-brain barrier endothelial cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 40–60. [Google Scholar] [CrossRef]
- He, Y.; Yao, Y.; Tsirka, S.E.; Cao, Y. Cell-Culture Models of the Blood–Brain Barrier. Stroke 2014, 45, 2514–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cecchelli, R.; Aday, S.; Sevin, E.; Almeida, C.; Culot, M.; Dehouck, L.; Coisne, C.M.; Engelhardt, B.; Dehouck, M.-P.; Ferreira, L. A Stable and Reproducible Human Blood-Brain Barrier Model Derived from Hematopoietic Stem Cells. PLoS ONE 2014, 9, e99733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labour, M.-N.; Vigier, S.; Lerner, D.A.; Marcilhac, A.; Belamie, E. 3D compartmented model to study the neurite-related toxicity of Aβ aggregates included in collagen gels of adaptable porosity. Acta Biomater. 2016, 37, 38–49. [Google Scholar] [CrossRef]
- Atluri, V.S.R.; Tiwari, S.; Rodriguez, M.; Kaushik, A.; Yndart, A.; Kolishetti, N.; Yatham, M.; Nair, M. Inhibition of Amyloid-Beta Production, Associated Neuroinflammation, and Histone Deacetylase 2-Mediated Epigenetic Modifications Prevent Neuropathology in Alzheimer’s Disease in vitro Model. Front. Aging Neurosci. 2020, 11, 342. [Google Scholar] [CrossRef] [Green Version]
- Arslan, M.E.; Türkez, H.; Mardinoğlu, A. In vitro neuroprotective effects of farnesene sesquiterpene on alzheimer’s disease model of differentiated neuroblastoma cell line. Int. J. Neurosci. 2020, 1–10. [Google Scholar] [CrossRef] [PubMed]
- De Medeiros, L.M.; De Bastiani, M.A.; Rico, E.P.; Schonhofen, P.; Pfaffenseller, B.; Wollenhaupt-Aguiar, B.; Grun, L.; Barbé-Tuana, F.; Zimmer, E.R.; Castro, M.A.A.; et al. Cholinergic Differentiation of Human Neuroblastoma SH-SY5Y Cell Line and Its Potential Use as an In vitro Model for Alzheimer’s Disease Studies. Mol. Neurobiol. 2019, 56, 7355–7367. [Google Scholar] [CrossRef]
- Seidel, D.; Krinke, D.; Jahnke, H.-G.; Hirche, A.; Kloß, D.; Mack, T.G.A.; Striggow, F.; Robitzki, A. Induced Tauopathy in a Novel 3D-Culture Model Mediates Neurodegenerative Processes: A Real-Time Study on Biochips. PLoS ONE 2012, 7, e49150. [Google Scholar] [CrossRef]
- Gu, R.; Wang, L.; Tang, M.; Li, S.-R.; Liu, R.; Hu, X. LncRNA Rpph1 protects amyloid-β induced neuronal injury in SK-N-SH cells via miR-122/Wnt1 axis. Int. J. Neurosci. 2020, 130, 443–453. [Google Scholar] [CrossRef]
- Su, B.; Wang, X.; Lee, H.-G.; Tabaton, M.; Perry, G.; Smith, M.A.; Zhu, X. Chronic oxidative stress causes increased tau phosphorylation in M17 neuroblastoma cells. Neurosci. Lett. 2010, 468, 267–271. [Google Scholar] [CrossRef]
- Heo, H.J.; Yang, H.C.; Cho, H.Y.; Hong, B.; Lim, S.T.; Park, H.J.; Kim, K.H.; Kim, H.K.; Shin, D.H. Inhibitory effect of Artemisia asiatica alkaloids on acetylcholinesterase activity from rat PC12 cells. Mol. Cells 2000, 10, 253–262. [Google Scholar] [PubMed]
- Spilman, P.; Descamps, O.; Gorostiza, O.; Peters-Libeu, C.; Poksay, K.S.; Matalis, A.; Campagna, J.; Patent, A.; Rao, R.; John, V.; et al. The multi-functional drug tropisetron binds APP and normalizes cognition in a murine Alzheimer’s model. Brain Res. 2014, 1551, 25–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uemura, K.; Farner, K.C.; Hashimoto, T.; Nasser-Ghodsi, N.; Wolfe, M.S.; Koo, E.H.; Hyman, B.T.; Berezovska, O. Substrate docking to γ-secretase allows access of γ-secretase modulators to an allosteric site. Nat. Commun. 2010, 1, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Shao, X.; Zhang, C.; Tan, Y.; Liu, Q.; Wan, X.; Zhang, Q.; Xu, S.; Jiang, X. Intranasal H102 Peptide-Loaded Liposomes for Brain Delivery to Treat Alzheimer’s Disease. Pharm. Res. 2015, 32, 3837–3849. [Google Scholar] [CrossRef]
- Srinivasan, B.; Kolli, A.R.; Esch, M.B.; Abaci, H.E.; Shuler, M.L.; Hickman, J.J. TEER Measurement Techniques for In Vitro Barrier Model Systems. J. Lab. Autom. 2015, 20, 107–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Wetzel, I.; Marriott, I.; Dreau, D.; D’Avanzo, C.; Kim, D.Y.; Tanzi, R.E.; Cho, H. A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer’s disease. Nat. Neurosci. 2018, 21, 941–951. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, Y.H.; Hebisch, M.; Sliwinski, C.; Lee, S.; D’Avanzo, C.; Chen, J.; Hooli, B.; Asselin, C.; Muffat, J.; et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nat. Cell Biol. 2014, 515, 274–278. [Google Scholar] [CrossRef]
- Biemans, E.A.; Jäkel, L.; de Waal, R.M.; Kuiperij, B.; Verbeek, M.M. Limitations of the hCMEC/D3 cell line as a model for Aβ clearance by the human blood-brain barrier. J. Neurosci. Res. 2017, 95, 1513–1522. [Google Scholar] [CrossRef]
- Weksler, B.; Romero, I.; Couraud, P.-O. The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids Barriers CNS 2013, 10, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qosa, H.; Mohamed, L.A.; Al Rihani, S.B.; Batarseh, Y.S.; Duong, Q.-V.; Keller, J.N.; Kaddoumi, A. High-Throughput Screening for Identification of Blood-Brain Barrier Integrity Enhancers: A Drug Repurposing Opportunity to Rectify Vascular Amyloid Toxicity. J. Alzheimer’s Dis. 2016, 53, 1499–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waxman, E.; Giasson, B.I. Induction of Intracellular Tau Aggregation Is Promoted by -Synuclein Seeds and Provides Novel Insights into the Hyperphosphorylation of Tau. J. Neurosci. 2011, 31, 7604–7618. [Google Scholar] [CrossRef] [Green Version]
- Houck, A.L.; Hernández, F.; Ávila, J. A Simple Model to Study Tau Pathology. J. Exp. Neurosci. 2016, 10, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.H.; Kim, Y.H.; Quinti, L.; Tanzi, R.E.; Kim, D.Y. 3D culture models of Alzheimer’s disease: A road map to a “cure-in-a-dish. Mol. Neurodegener. 2016, 11, 75. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arber, C.; Lovejoy, C.; Wray, S. Stem cell models of Alzheimer’s disease: Progress and challenges. Alzheimer’s Res. Ther. 2017, 9, 117. [Google Scholar] [CrossRef] [PubMed]
- Auboyer, L.; Monzo, C.; Wallon, D.; Rovelet-Lecrux, A.; Gabelle, A.; Gazagne, I.; Cacheux, V.; Lehmann, S.; Crozet, C. Generation of induced pluripotent stem cells (IRMBi001-A) from an Alzheimer’s disease patient carrying a G217D mutation in the PSEN1 gene. Stem Cell Res. 2019, 34, 101381. [Google Scholar] [CrossRef]
- Auboyer, L.; Monzo, C.; Wallon, D.; Rovelet-Lecrux, A.; Gabelle, A.; Gazagne, I.; Cacheux, V.; Lehmann, S.; Crozet, C. Generation of induced pluripotent stem cells (iPSCs) IRMBi002-A from an Alzheimer’s disease patient carrying a D694N mutation in the APP gene. Stem Cell Res. 2019, 37, 101438. [Google Scholar] [CrossRef]
- Lee, H.-K.; Sanchez, C.V.; Chen, M.; Morin, P.J.; Wells, J.M.; Hanlon, E.B.; Xia, W. Three Dimensional Human Neuro-Spheroid Model of Alzheimer’s Disease Based on Differentiated Induced Pluripotent Stem Cells. PLoS ONE 2016, 11, e0163072. [Google Scholar] [CrossRef] [Green Version]
- Raja, W.K.; Mungenast, A.E.; Lin, Y.-T.; Ko, T.; Abdurrob, F.; Seo, J.; Tsai, L.-H. Self-Organizing 3D Human Neural Tissue Derived from Induced Pluripotent Stem Cells Recapitulate Alzheimer’s Disease Phenotypes. PLoS ONE 2016, 11, e0161969. [Google Scholar] [CrossRef] [Green Version]
- Rouleau, N.; Cantley, W.L.; Liaudanskaya, V.; Berk, A.; Du, C.; Rusk, W.; Peirent, E.; Koester, C.; Nieland, T.J.F.; Kaplan, D.L. A Long-Living Bioengineered Neural Tissue Platform to Study Neurodegeneration. Macromol. Biosci. 2020, 20, e2000004. [Google Scholar] [CrossRef] [PubMed]
- Yagi, T.; Ito, D.; Okada, Y.; Akamatsu, W.; Nihei, Y.; Yoshizaki, T.; Yamanaka, S.; Okano, H.; Suzuki, N. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum. Mol. Genet. 2011, 20, 4530–4539. [Google Scholar] [CrossRef]
- Atkinson-Dell, R.; Mohamet, L. Induced Pluripotent Stem Cell-Derived Astroglia: A New Tool for Research Towards the Treatment of Alzheimer’s Disease. Adv. Exp. Med. Biol. 2019, 1175, 383–405. [Google Scholar] [CrossRef] [PubMed]
- Katt, M.E.; Xu, Z.S.; Gerecht, S.; Searson, P.C. Human Brain Microvascular Endothelial Cells Derived from the BC1 iPS Cell Line Exhibit a Blood-Brain Barrier Phenotype. PLoS ONE 2016, 11, e0152105. [Google Scholar] [CrossRef] [PubMed]
- Oikari, L.; Pandit, R.; Stewart, R.; Cuni-Lopez, C.; Quek, H.; Sutharsan, R.; Rantanen, L.M.; Oksanen, M.; Lehtonen, S.; de Boer, C.M.; et al. Altered Brain Endothelial Cell Phenotype from a Familial Alzheimer Mutation and Its Potential Implications for Amyloid Clearance and Drug Delivery. Stem Cell Rep. 2020, 14, 924–939. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Chen, X.; Gong, S.; Yu, P.; Yau, S.S.Y.; Su, Z.; Zhou, L.; Yu, J.; Pan, G.; Shi, L. Characteristic analyses of a neural differentiation model from iPSC-derived neuron according to morphology, physiology, and global gene expression pattern. Sci. Rep. 2017, 7, 12233. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.; Cutts, J.; Kimura, A.; Varun, D.; Brafman, D.A. A chemically defined substrate for the expansion and neuronal differentiation of human pluripotent stem cell-derived neural progenitor cells. Stem Cell Res. 2015, 15, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Arendt, T.; Stieler, J.; Ueberham, U. Is sporadic Alzheimer′s disease a developmental disorder? J. Neurochem. 2017, 143, 396–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, J.J.; Jones, V.; Tabuchi, M.; Allan, S.; Knight, E.; La Ferla, F.M.; Oddo, S.; Verkhratsky, A. Impaired Adult Neurogenesis in the Dentate Gyrus of a Triple Transgenic Mouse Model of Alzheimer’s Disease. PLoS ONE 2008, 3, e2935. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Lafon, P.-A.; Salvador-Prince, L.; Gines, A.R.; Trousse, F.; Torrent, J.; Prevostel, C.; Crozet, C.; Liu, J.; Perrier, V. Prenatal exposure to low doses of fungicides corrupts neurogenesis in neonates. Environ. Res. 2021, 195, 110829. [Google Scholar] [CrossRef] [PubMed]
- Amin, N.; Tan, X.; Ren, Q.; Zhu, N.; Botchway, B.O.; Hu, Z.; Fang, M. Recent advances of induced pluripotent stem cells application in neurodegenerative diseases. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2019, 95, 109674. [Google Scholar] [CrossRef]
- Gonzalez, C.; Armijo, E.; Bravo-Alegria, J.; Calixto, A.B.; Mays, C.E.; Soto, C. Modeling amyloid β and tau pathology in human cerebral organoids. Mol. Psychiatry 2018, 23, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Song, L.; Bejoy, J.; Zhao, J.; Kanekiyo, T.; Bu, G.; Zhou, Y.; Li, Y. Modeling Neurodegenerative Microenvironment Using Cortical Organoids Derived from Human Stem Cells. Tissue Eng. Part A 2018, 24, 1125–1137. [Google Scholar] [CrossRef]
- Mertens, J.; Paquola, A.C.; Ku, M.; Hatch, E.; Böhnke, L.; Ladjevardi, S.; McGrath, S.; Campbell, B.; Lee, H.; Herdy, J.R.; et al. Directly Reprogrammed Human Neurons Retain Aging-Associated Transcriptomic Signatures and Reveal Age-Related Nucleocytoplasmic Defects. Cell Stem Cell 2015, 17, 705–718. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Qiu, B.; Guan, W.; Wang, Q.; Wang, M.; Li, W.; Gao, L.; Shen, L.; Huang, Y.; Xie, G.; et al. Direct Conversion of Normal and Alzheimer’s Disease Human Fibroblasts into Neuronal Cells by Small Molecules. Cell Stem Cell 2015, 17, 204–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez, M.J.; Ivanyuk, D.; Panagiotakopoulou, V.; Di Napoli, G.; Kalb, S.; Brunetti, D.; Al-Shaana, R.; Kaeser, S.A.; Fraschka, S.A.-K.; Jucker, M.; et al. Loss of function of the mitochondrial peptidase PITRM1 induces proteotoxic stress and Alzheimer’s disease-like pathology in human cerebral organoids. Mol. Psychiatry 2020, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, P.; Payab, M.; Alavi-Moghadam, S.; Larijani, B.; Rahim, F.; Bana, N.; Sarvari, M.; Adibi, H.; Heravani, N.F.; Hadavandkhani, M.; et al. Development and validation of Alzheimer’s Disease Animal Model for the Purpose of Regenerative Medicine. Cell Tissue Bank. 2019, 20, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Zhou, Q. How efficient are rodent models for Alzheimer’s disease drug discovery? Expert Opin. Drug Discov. 2017, 13, 113–115. [Google Scholar] [CrossRef] [Green Version]
- Shen, P.; Yue, Y.; Zheng, J.; Park, Y. Caenorhabditis elegans: A Convenient In Vivo Model for Assessing the Impact of Food Bioactive Compounds on Obesity, Aging, and Alzheimer’s Disease. Annu. Rev. Food Sci. Technol. 2018, 9, 1–22. [Google Scholar] [CrossRef]
- Brandt, R.; Gergou, A.; Wacker, I.; Fath, T.; Hutter, H. A Caenorhabditis elegans model of tau hyperphosphorylation: Induction of developmental defects by transgenic overexpression of Alzheimer’s disease-like modified tau. Neurobiol. Aging 2009, 30, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Eidenmüller, J.; Fath, T.; Maas, T.; Pool, M.; Sontag, E.; Brandt, R. Phosphorylation-mimicking glutamate clusters in the proline-rich region are sufficient to simulate the functional deficiencies of hyperphosphorylated tau protein. Biochem. J. 2001, 357, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Link, C.D.; Taft, A.; Kapulkin, V.; Duke, K.; Kim, S.; Fei, Q.; Wood, D.E.; Sahagan, B.G. Gene expression analysis in a transgenic Caenorhabditis elegans Alzheimer’s disease model. Neurobiol. Aging 2003, 24, 397–413. [Google Scholar] [CrossRef]
- Mango, S. Stop making nonSense: The C. elegans smg genes. Trends Genet. 2001, 17, 646–653. [Google Scholar] [CrossRef]
- Sultana, R.; Perluigi, M.; Butterfield, D.A. Oxidatively modified proteins in Alzheimer’s disease (AD), mild cognitive impairment and animal models of AD: Role of Aβ in pathogenesis. Acta Neuropathol. 2009, 118, 131–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haque, R.; Nazir, A. SMAD Transcription Factor, Sma-9, Attunes TGF-β Signaling Cascade Towards Modulating Amyloid Beta Aggregation and Associated Outcome in Transgenic C. elegans. Mol. Neurobiol. 2014, 53, 109–119. [Google Scholar] [CrossRef]
- Chakraborty, R.; Vepuri, V.; Mhatre, S.D.; Paddock, B.E.; Miller, S.; Michelson, S.J.; Delvadia, R.; Desai, A.; Vinokur, M.; Melicharek, D.J.; et al. Characterization of a Drosophila Alzheimer’s Disease Model: Pharmacological Rescue of Cognitive Defects. PLoS ONE 2011, 6, e20799. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Funez, P.; de Mena, L.; Rincon-Limas, D.E. Modeling the complex pathology of Alzheimer’s disease in Drosophila. Exp. Neurol. 2015, 274, 58–71. [Google Scholar] [CrossRef] [Green Version]
- Tsuda, L.; Lim, Y.-M. Alzheimer’s Disease Model System Using Drosophila. In Drosophila Models for Human Diseases; Advances in Experimental Medicine and Biology; Yamaguchi, M., Ed.; Springer: Singapore, 2018; Volume 1076, pp. 25–40. ISBN 978-981-1-305-283. [Google Scholar]
- Huang, J.-K.; Ma, P.-L.; Ji, S.-Y.; Zhao, X.; Tan, J.-X.; Sun, X.-J.; Huang, F.-D. Age-dependent alterations in the presynaptic active zone in a Drosophila model of Alzheimer’s Disease. Neurobiol. Dis. 2013, 51, 161–167. [Google Scholar] [CrossRef]
- Crowther, D.; Kinghorn, K.; Miranda, E.; Page, R.; Curry, J.; Duthie, F.; Gubb, D.; Lomas, D. Intraneuronal Aβ, non-amyloid aggregates and neurodegeneration in a Drosophila model of Alzheimer’s disease. Neuroscience 2005, 132, 123–135. [Google Scholar] [CrossRef]
- Liu, Q.F.; Lee, J.H.; Kim, Y.-M.; Lee, S.; Hong, Y.K.; Hwang, S.; Oh, Y.; Lee, K.; Yun, H.S.; Lee, I.-S.; et al. In Vivo Screening of Traditional Medicinal Plants for Neuroprotective Activity against Aβ42 Cytotoxicity by Using Drosophila Models of Alzheimer’s Disease. Biol. Pharm. Bull. 2015, 38, 1891–1901. [Google Scholar] [CrossRef] [Green Version]
- Xia, W. Exploring Alzheimer’s Disease in Zebrafish. J. Alzheimer’s Dis. 2010, 20, 981–990. [Google Scholar] [CrossRef]
- Paquet, D.; Bhat, R.; Sydow, A.; Mandelkow, E.-M.; Berg, S.; Hellberg, S.; Fälting, J.; Distel, M.; Köster, R.W.; Schmid, B.; et al. A zebrafish model of tauopathy allows in vivo imaging of neuronal cell death and drug evaluation. J. Clin. Investig. 2009, 119, 1382–1395. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, D.B.; Soria, J.P.; Tomasiewicz, H.G.; Wood, J.G. Phosphorylation of Human Tau Protein by Microtubule-Associated Kinases: GSK3β and Cdk5 Are Key Participants. J. Neurosci. Res. 2000, 62, 463–472. [Google Scholar] [CrossRef]
- Rubin, G.; Yandell, M.D.; Wortman, J.; Gabor, G.L.; Miklos, G.L.G.; Nelson, C.R.; Hariharan, I.K.; Fortini, M.E.; Li, P.W.; Apweiler, R.; et al. Comparative Genomics of the Eukaryotes. Science 2000, 287, 2204–2215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talmat-Amar, Y.; Arribat, Y.; Parmentier, M.-L. Vesicular Axonal Transport is Modified In Vivo by Tau Deletion or Overexpression in Drosophila. Int. J. Mol. Sci. 2018, 19, 744. [Google Scholar] [CrossRef] [Green Version]
- Lv, F.; Yang, X.; Cui, C.; Su, C. Exogenous expression of Drp1 plays neuroprotective roles in the Alzheimer’s disease in the Aβ42 transgenic Drosophila model. PLoS ONE 2017, 12, e0176183. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Chiang, H.-C.; Wu, W.; Liang, B.; Xie, Z.; Yao, X.; Ma, W.; Du, S.; Zhong, Y. Epidermal growth factor receptor is a preferred target for treating Amyloid- induced memory loss. Proc. Natl. Acad. Sci. USA 2012, 109, 16743–16748. [Google Scholar] [CrossRef] [Green Version]
- Kalueff, A.V.; Stewart, A.M.; Gerlai, R. Zebrafish as an emerging model for studying complex brain disorders. Trends Pharmacol. Sci. 2014, 35, 63–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huc-Brandt, S.; Hieu, N.; Imberdis, T.; Cubedo, N.; Silhol, M.; Leighton, P.L.A.; Domaschke, T.; Allison, T.; Perrier, V.; Rossel, M. Zebrafish Prion Protein PrP2 Controls Collective Migration Process during Lateral Line Sensory System Development. PLoS ONE 2014, 9, e113331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaser, R.; Gerlai, R. Behavioral phenotyping in zebrafish: Comparison of three behavioral quantification methods. Behav. Res. Methods 2006, 38, 456–469. [Google Scholar] [CrossRef] [PubMed]
- Gerlai, R. Zebra Fish: An Uncharted Behavior Genetic Model. Behav. Genet. 2003, 33, 461–468. [Google Scholar] [CrossRef]
- Guo, S. Linking genes to brain, behavior and neurological diseases: What can we learn from zebrafish? Genes Brain Behav. 2004, 3, 63–74. [Google Scholar] [CrossRef]
- Vernier, P.; Kyzar, E.J.; Maximino, C.; Tierney, K.; Gebhardt, M.; Lange, M.; Jesuthasan, S.; Stewart, A.M.; Neuhauss, S.C.; Robinson, K.; et al. Time to recognize zebrafish ‘affective’ behavior. Behavior 2012, 149, 1019–1036. [Google Scholar] [CrossRef] [Green Version]
- Musa, A.; Lehrach, H.; Russo, V.E. Distinct expression patterns of two zebrafish homologues of the human APP gene during embryonic development. Dev. Genes Evol. 2001, 211, 563–567. [Google Scholar] [CrossRef]
- Folwell, J.; Cowan, C.M.; Ubhi, K.K.; Shiabh, H.; Newman, T.; Shepherd, D.; Mudher, A. Aβ exacerbates the neuronal dysfunction caused by human tau expression in a Drosophila model of Alzheimer’s disease. Exp. Neurol. 2010, 223, 401–409. [Google Scholar] [CrossRef]
- Cosacak, M.I.; Bhattarai, P.; Bocova, L.; Dzewas, T.; Mashkaryan, V.; Papadimitriou, C.; Brandt, K.; Hollak, H.; Antos, C.L.; Kizil, C. Human TAUP301L overexpression results in TAU hyperphosphorylation without neurofibrillary tangles in adult zebrafish brain. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Moussaed, M.; Huc-Brandt, S.; Cubedo, N.; Silhol, M.; Murat, S.; Lebart, M.-C.; Kovacs, G.G.; Verdier, J.-M.; Trousse, F.; Rossel, M.; et al. Regenerating islet-derived 1α (REG-1α) protein increases tau phosphorylation in cell and animal models of tauopathies. Neurobiol. Dis. 2018, 119, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Mucke, L.; Masliah, E.; Yu, G.Q.; Mallory, M.; Rockenstein, E.M.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Johnson-Wood, K.; McConlogue, L. High-Level Neuronal Expression of Aβ 1-42 in Wild-Type Human Amyloid Protein Precursor Transgenic Mice: Synaptotoxicity without Plaque Formation. J. Neurosci. 2000, 20, 4050–4058. [Google Scholar] [CrossRef] [Green Version]
- Wright, A.; Zinn, R.; Hohensinn, B.; Konen, L.M.; Beynon, S.B.; Tan, R.P.; Clark, I.A.; Abdipranoto, A.; Vissel, B. Neuroinflammation and Neuronal Loss Precede Aβ Plaque Deposition in the hAPP-J20 Mouse Model of Alzheimer’s Disease. PLoS ONE 2013, 8, e59586. [Google Scholar] [CrossRef] [Green Version]
- Radde, R.; Bolmont, T.; Kaeser, S.A.; Coomaraswamy, J.; Lindau, D.; Stoltze, L.; Calhoun, M.E.; Jäggi, F.; Wolburg, H.; Gengler, S.; et al. Aβ42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006, 7, 940–946. [Google Scholar] [CrossRef] [Green Version]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal β-Amyloid Aggregates, Neurodegeneration, and Neuron Loss in Transgenic Mice with Five Familial Alzheimer’s Disease Mutations: Potential Factors in Amyloid Plaque Formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-Transgenic Model of Alzheimer’s Disease with Plaques and Tangles: Intracellular Aβ and Synaptic Dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Matsuba, Y.; Mihira, N.; Takano, J.; Nilsson, P.; Itohara, S.; Iwata, N.; Saido, T.C. Single App knock-in mouse models of Alzheimer’s disease. Nat. Neurosci. 2014, 17, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zuo, P. Effects of Aβ25–35on neurogenesis in the adult mouse subventricular zone and dentate gyrus. Neurol. Res. 2005, 27, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Salkovic-Petrisic, M.; Knezovic, A.; Hoyer, S.; Riederer, P. What have we learned from the streptozotocin-induced animal model of sporadic Alzheimer’s disease, about the therapeutic strategies in Alzheimer’s research. J. Neural Transm. 2012, 120, 233–252. [Google Scholar] [CrossRef] [Green Version]
- Aleksandrova, I.Y.; Kuvichkin, V.V.; Kashparov, I.A.; Medvinskaya, N.I.; Nesterova, I.V.; Lunin, S.; Samokhin, A.; Bobkova, N.V. Increased Level of β-Amyloid in the Brain of Bulbectomized Mice. Biochemistry 2004, 69, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Yagi, H.; Katoh, S.; Akiguchi, I.; Takeda, T. Age-related deterioration of ability of acquisition in memory and learning in senescence accelerated mouse: SAM-P/8 as an animal model of disturbances in recent memory. Brain Res. 1988, 474, 86–93. [Google Scholar] [CrossRef]
- Cohen, R.M.; Rezai-Zadeh, K.; Weitz, T.M.; Rentsendorj, A.; Gate, D.; Spivak, I.; Bholat, Y.; Vasilevko, V.; Glabe, C.G.; Breunig, J.; et al. A Transgenic Alzheimer Rat with Plaques, Tau Pathology, Behavioral Impairment, Oligomeric A and Frank Neuronal Loss. J. Neurosci. 2013, 33, 6245–6256. [Google Scholar] [CrossRef]
- Tambini, M.D.; D’Adamio, L. Trem2 Splicing and Expression are Preserved in a Human Aβ-producing, Rat Knock-in Model of Trem2-R47H Alzheimer’s Risk Variant. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Stefanova, N.A.; Korbolina, E.E.; Ershov, N.I.; Rogaev, E.I.; Kolosova, N.G. Changes in the transcriptome of the prefrontal cortex of OXYS rats as the signs of Alzheimer’s disease development. Vavilov J. Genet. Breed. 2015, 19, 445. [Google Scholar] [CrossRef] [Green Version]
- Alzheimer’s Disease Research Models|ALZFORUM. Available online: https://www.alzforum.org/research-models/alzheimers-disease (accessed on 24 March 2021).
- Hutton, M.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nat. Cell Biol. 1998, 393, 702–705. [Google Scholar] [CrossRef]
- Billings, L.M.; Oddo, S.; Green, K.N.; McGaugh, J.L.; LaFerla, F.M. Intraneuronal Aβ Causes the Onset of Early Alzheimer’s Disease-Related Cognitive Deficits in Transgenic Mice. Neuron 2005, 45, 675–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulyaeva, N.V.; Bobkova, N.V.; Kolosova, N.G.; Samokhin, A.; Stepanichev, M.Y.; Stefanova, N.A. Molecular and cellular mechanisms of sporadic Alzheimer’s disease: Studies on rodent models in vivo. Biochemistry 2017, 82, 1088–1102. [Google Scholar] [CrossRef]
- Sotthibundhu, A.; Li, Q.-X.; Thangnipon, W.; Coulson, E.J. Aβ1–42 stimulates adult SVZ neurogenesis through the p75 neurotrophin receptor. Neurobiol. Aging 2009, 30, 1975–1985. [Google Scholar] [CrossRef] [PubMed]
- Bobkova, N.V.; Nesterova, I.V.; Nesterov, V.V. The state of cholinergic structures in forebrain of bulbectomized mice. Bull. Exp. Biol. Med. 2001, 131, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Gerschütz, A.; Heinsen, H.; Grünblatt, E.; Wagner, A.K.; Bartl, J.; Meissner, C.; Fallgatter, A.J.; Al-Sarraj, S.; Troakes, C.; Ferrer, I.; et al. Neuron-Specific Mitochondrial DNA Deletion Levels in Sporadic Alzheimer’s Disease. Available online: https://www.eurekaselect.com/116820/article (accessed on 6 July 2020).
- Savory, J. Intracellular mechanisms underlying aluminum-induced apoptosis in rabbit brain. J. Inorg. Biochem. 2003, 97, 151–154. [Google Scholar] [CrossRef]
- Song, J. Animal Model of Aluminum-Induced Alzheimer’s Disease. In Neurotoxicity of Aluminum; Advances in Experimental Medicine and Biology; Niu, Q., Ed.; Springer: Singapore, 2018; Volume 1091, pp. 113–127. ISBN 978-981-1-313-691. [Google Scholar]
- Woodruff-Pak, D.S.; Agelan, A.; Del Valle, L. A Rabbit Model of Alzheimer’s Disease: Valid at Neuropathological, Cognitive, and Therapeutic Levels. J. Alzheimer’s Dis. 2007, 11, 371–383. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Reyes, A.E.; Chacón, M.A.; Cerpa, W.; Villalón, A.; Montiel, J.F.; Merabachvili, G.; Aldunate, R.; Bozinovic, F.; Aboitiz, F. Human-like rodent amyloid-β-peptide determines Alzheimer pathology in aged wild-type Octodon degu. Neurobiol. Aging 2005, 26, 1023–1028. [Google Scholar] [CrossRef]
- Van Groen, T.; Kadish, I.; Popović, N.; Popovic, M.; Caballero-Bleda, M.; Baño-Otálora, B.; Vivanco, P.; Rol, A.; Madrid, J.A. Age-related brain pathology in Octodon degu: Blood vessel, white matter and Alzheimer-like pathology. Neurobiol. Aging 2011, 32, 1651–1661. [Google Scholar] [CrossRef]
- Braidy, N.; Poljak, A.; Jayasena, T.; Mansour, H.; Inestrosa, N.C.; Sachdev, P. Accelerating Alzheimerʼs research through ‘natural’ animal models. Curr. Opin. Psychiatry 2015, 28, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Heuer, E.; Rosen, R.; Cintron, A.; Walker, L.C. Nonhuman Primate Models of Alzheimer-Like Cerebral Proteopathy. Curr. Pharm. Des. 2012, 18, 1159–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gearing, M.; Tigges, J.; Mori, H.; Mirra, S. β-Amyloid (Aβ) Deposition in the Brains of Aged Orangutans. Neurobiol. Aging 1997, 18, 139–146. [Google Scholar] [CrossRef]
- Gearing, M.; Rebeck, G.W.; Hyman, B.T.; Tigges, J.; Mirra, S.S. Neuropathology and apolipoprotein E profile of aged chimpanzees: Implications for Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 9382–9386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, N.; Nakamura, S.; Goto, N.; Narushima, E.; Hara, I.; Shichiri, S.; Saitou, K.; Nose, M.; Hayashi, T.; Kawamura, S.; et al. Senile Plaques in an Aged Western Lowland Gorilla. Exp. Anim. 2001, 50, 77–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finch, C.E.; Austad, S.N. Commentary: Is Alzheimer’s disease uniquely human? Neurobiol. Aging 2015, 36, 553–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Chen, B.; Lu, J.; Wu, Y.; Wang, S.; Yao, Z.; Zhu, L.; Qiao, Y.; Sun, Q.; Qin, W.; et al. Brains of rhesus monkeys display Aβ deposits and glial pathology while lacking Aβ dimers and other Alzheimer’s pathologies. Aging Cell 2019, 18, e12978. [Google Scholar] [CrossRef] [PubMed]
- Härtig, W.; Brückner, G.; Schmidt, C.; Brauer, K.; Bodewitz, G.; Turner, J.D.; Bigl, V. Co-localization of β-amyloid peptides, apolipoprotein E and glial markers in senile plaques in the prefrontal cortex of old rhesus monkeys. Brain Res. 1997, 751, 315–322. [Google Scholar] [CrossRef]
- Heilbroner, P.L.; Kemper, T.L. The cytoarchitectonic distribution of senile plaques in three aged monkeys. Acta Neuropathol. 1990, 81, 60–65. [Google Scholar] [CrossRef]
- Rosen, R.F.; Walker, L.C.; Levine, H. PIB binding in aged primate brain: Enrichment of high-affinity sites in humans with Alzheimer’s disease. Neurobiol. Aging 2011, 32, 223–234. [Google Scholar] [CrossRef] [Green Version]
- Shah, P.; Lal, N.; Leung, E.; Traul, D.E.; Gonzalo-Ruiz, A.; Geula, C. Neuronal and Axonal Loss Are Selectively Linked to Fibrillar Amyloid-β within Plaques of the Aged Primate Cerebral Cortex. Am. J. Pathol. 2010, 177, 325–333. [Google Scholar] [CrossRef]
- Chambers, J.; Kuribayashi, H.; Ikeda, S.-I.; Une, Y. Distribution of neprilysin and deposit patterns of Aβ subtypes in the brains of aged squirrel monkeys (Saimiri sciureus). Amyloid 2010, 17, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Elfenbein, H.; Rosen, R.; Stephens, S.; Switzer, R.; Smith, Y.; Pare, J.; Mehta, P.; Warzok, R.; Walker, L. Cerebral β-Amyloid Angiopathy in Aged Squirrel Monkeys. Histol. Histopathol. 2007, 22, 155–167. [Google Scholar] [CrossRef]
- Bartus, R.T.; Dean, R.L.; Beer, B. Memory Deficits in Aged Cebus Monkeys and Facilitation with Central Cholinomimetics. Neurobiol. Aging 1981, 1, 145–152. [Google Scholar] [CrossRef]
- Lyons, D.M.; Yang, C.; Eliez, S.; Reiss, A.L.; Schatzberg, A.F. Cognitive Correlates of White Matter Growth and Stress Hormones in Female Squirrel Monkey Adults. J. Neurosci. 2004, 24, 3655–3662. [Google Scholar] [CrossRef] [Green Version]
- Dhenain, M.; Michot, J.; Privat, N.; Picq, J.; Boller, F.; Duyckaerts, C.; Volk, A. MRI description of cerebral atrophy in mouse lemur primates. Neurobiol. Aging 2000, 21, 81–88. [Google Scholar] [CrossRef]
- Languille, S.; Blanc, S.; Blin, O.; Canale, C.; Dal-Pan, A.; Devau, G.; Dhenain, M.; Dorieux, O.; Epelbaum, J.; Gomez, D.; et al. The grey mouse lemur: A non-human primate model for ageing studies. Ageing Res. Rev. 2012, 11, 150–162. [Google Scholar] [CrossRef]
- Silhol, S.; Calenda, A.; Jallageas, V.; Mestre-Francés, N.; Bellis, M.; Bons, N. β-Amyloid Protein Precursor in Microcebus murinus: Genotyping and Brain Localization. Neurobiol. Dis. 1996, 3, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, D.; Zimmermann, E.; Trouche, S.G.; Fontès, P.; Verdier, J.-M.; Mestre-Francés, N. Linking cognition to age and amyloid-β burden in the brain of a nonhuman primate (Microcebus murinus). Neurobiol. Aging 2020, 94, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Bons, N.; Mestre-Frances, N.; Petter, A. Senile plaques and neurofibrillary changes in the brain of an aged lemurian primate, Microcebus murinus. Neurobiol. Aging 1992, 13, 99–105. [Google Scholar] [CrossRef]
- Kraska, A.; Dorieux, O.; Picq, J.-L.; Petit, F.; Bourrin, E.; Chenu, E.; Volk, A.; Perret, M.; Hantraye, P.; Mestre-Frances, N.; et al. Age-associated cerebral atrophy in mouse lemur primates. Neurobiol. Aging 2011, 32, 894–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perret, M.; Aujard, F. Aging and season affect plasma dehydroepiandrosterone sulfate (DHEA-S) levels in a primate. Exp. Gerontol. 2005, 40, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Seita, Y.; Morimura, T.; Watanabe, N.; Iwatani, C.; Tsuchiya, H.; Nakamura, S.; Suzuki, T.; Yanagisawa, D.; Tsukiyama, T.; Nakaya, M.; et al. Generation of Transgenic Cynomolgus Monkeys Overexpressing the Gene for Amyloid-β Precursor Protein. J. Alzheimer’s Dis. 2020, 75, 45–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, J.C.; Aisen, P.S.; Bateman, R.J.; Benzinger, T.L.; Cairns, N.J.; Fagan, A.M.; Ghetti, B.; Goate, A.M.; Holtzman, D.M.; Klunk, W.E.; et al. Developing an international network for Alzheimer’s research: The Dominantly Inherited Alzheimer Network. Clin. Investig. 2012, 2, 975–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bateman, R.J.; Benzinger, T.; Berry, S.; Clifford, D.B.; Duggan, C.; Fagan, A.M.; Fanning, K.; Farlow, M.R.; Hassenstab, J.; McDade, E.M.; et al. The DIAN-TU Next Generation Alzheimer’s prevention trial: Adaptive design and disease progression model. Alzheimer’s Dement. 2017, 13, 8–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortea, J.; Carmona-Iragui, M.; Benejam, B.; Fernández, S.; Videla, L.; Barroeta, I.; Alcolea, D.; Pegueroles, J.; Muñoz, L.; Belbin, O.; et al. Plasma and CSF biomarkers for the diagnosis of Alzheimer’s disease in adults with Down syndrome: A cross-sectional study. Lancet Neurol. 2018, 17, 860–869. [Google Scholar] [CrossRef]
- Lehmann, S.; Delaby, C.; Touchon, J.; Hirtz, C.; Gabelle, A. Biomarkers of Alzheimer’s disease: The present and the future. Rev. Neurol. 2013, 169, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Magnin, E.; Dumurgier, J.; Bouaziz-Amar, E.; Bombois, S.; Wallon, D.; Gabelle, A.; Lehmann, S.; Blanc, F.; Bousiges, O.; Hannequin, D.; et al. Les biomarqueurs du liquide cérébro-spinal dans la maladie d’Alzheimer: Un outil de recherche utile dans la pratique clinique courante des consultations mémoire pour les cas complexes. Rev. Méd. Interne 2017, 38, 250–255. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Aβ Fibrillization Models and Associated Techniques | ||||
|---|---|---|---|---|
| Associated Methods | Examples | Advantages | Pitfalls | References |
| Additive models | ||||
| Dye-based methods | Congo Red (CR) dye | Historical dye. Can also be used on tissues. | The short beta-sheet structures are not bound. The oligomers and protofibrils are not detected. | [32] |
| Thioflavin T (ThT) assays | Best tool to study amyloid fibril formation: does not affect fibril formation, linearity, availability, robustness; easy to use. | ThT does not bind specifically to fibrils but also to DNA, cyclodextrin and SDS micelles. Need to use protein-pure samples. Cannot detect early aggregates (oligomers and protofibrils). The binding affinity depends on the fibril type. Need to use complementary techniques to confirm the results. | [33,34,40,41,42,43] | |
| ANS dye | Useful to characterize protein folding and aggregation intermediates. | Not specific to fibrils. Any protein with a hydrophobic region folded in the protein core has a fluorescent intensity. | [35,36,38] | |
| Antibody-based methods | Time-resolved fluorescence (HTRF) immunoassay | Aβ peptide quantification. Sensitivity. Simple, rapid and robust method. Real-time kinetic study. | This technique requires specific antibody non cross-reacting with the different Aβ peptides. | [44,45] |
| Surface plasmon resonance (SPR) | Real-time method. Study short-term or long-term aggregation kinetics (from second to hours). Study of aggregation modulators and potential drug inhibitors. | Need to know precisely which oligomer species or fibrils are bound by the antibody. | [46,47,48,49] | |
| Microscopy and spectroscopy | Fluorescence microscopy and Fluorescence correlation spectroscopy (FCS) | Sensitivity. Real-time imaging. Small samples are sufficient. Can be used with fluorophore-coupled antibody (specificity gain). Also used to observe samples stained with Thioflavin T or ANS dyes. | Labeling can change aggregation. Autofluorescence interference. | [37,50,51,52] |
| Pure models | ||||
| Microscopy and spectroscopy | Time-resolved emission spectra (TRES) | Non-invasive and label-free technique. Nanosecond timescale and nanometer spatial resolution. | Difficulties for data treatment and interpretation. | [53,54] |
| Turbidity, multiangle laser light scattering (MALLS), dynamic light scattering (DLS) | Label-free methods. Light scattering is very sensitive. Real-time detection. | Turbidity is not a very reliable technique. Cannot differentiate oligomer intermediates. Low resolution of light scattering techniques. | [37,38,55,56] | |
| Primary Cells | ||
|---|---|---|
| Cell Types | Detailed Example | References |
| Tissues: all the brain cell types | In AD patient brains, Aβ downregulates the neuronal receptor AMPA by increasing its ubiquitination [91]. | [91,94] |
| Neurons | Park et al. (2015) developed an AD model based on 3D cell culture. Cultured neurons form neurospheroids in a microfluidic chip. Neurospheroids mimic a tissue with a complex neural network better than 2D-cultured neurons. Treatment with Aβ induces cell death and damages the neurospheroid network [95]. | 2D culture: [91,94,96] 3D culture: [95,97,98] |
| Astrocytes | Aβ1-42-exposed primary astrocytes better survive with a low dose of aspirin, probably because of a decrease in inflammation and oxidative stress [99]. | [99,100] |
| Microglial cells | During AD, microglial cells take up tau seeds to clear the aggregates, but, because of an incomplete clearance mechanism, these cells also propagate tau seeds in other brain regions after migration [101]. | [101,102] |
| Oligodendrocytes | Aβ prevents the myelin sheet formation in vitro, inducing oligodendrocyte damages and cell death [103]. | [103,104] |
| BBB: endothelial cells and pericytes | The Buyang Huanwu decoction inhibits the Aβ25–35-induced endothelial inflammation and RAGE/LRP1 dysregulation [105]. | Endothelial cells: [105,106,107,108,109] Pericytes: [110,111,112] |
| Cell Lines | |||
|---|---|---|---|
| Cell Lines | Associated Cell Type and Tumor | Detailed Example | References |
| Derived from tumor | |||
| SH-SY5Y (also SH-SY6Y) cells | Neurons (cholinergic neurons after differentiation), derived from a neuroblastoma | In 3D culture, SH-SY5Y cells were used to model an AD-like tauopathy, induced with okadaic acid and the recombinant mutated human tau [121]. | 2D: [16,113,122,123,124] 3D: [121,125] |
| SK-N-MC cells | Neurons, derived from a neuroepithelioma | Aβ-treated SK-N-MC cells were used to find efficient drugs able to cross the BBB and rescue the degenerating neurons from apoptosis. | [107] |
| SK-N-SH cells | Neurons, derived from a neuroblastoma | Treatment of SK-N-SH with Aβ25-35 peptides was used to model AD in vitro. With this model, Gu et al. (2020) investigated genes and proteins involved in cell death during AD, identifying pathways to improve cell viability. | [126] |
| BE(2)-M17 cells | Neurons, derived from a neuroblastoma | Su et al. (2010) studied the role of chronic oxidative stress on tau hyperphosphorylation with a M17-based cellular stress model. They showed that stress increases tau phosphorylation in vitro and suggested a role in neurofibrillary pathology in vivo. | [127] |
| PC-12 cells | Chromaffin cells (modified neurons), derived from a pheochromocytoma | The neuroprotective effects of two marine-derived carotenoids was assessed with Aβ1-42-treated PC-12 cells [40]. | [40,121,128] |
| 7W-CHO cells | Chinese ovary cells overexpressing the human APP gene | 7W-CHO cells were used to screen drugs able to increase the ratio between sAPPα, a neurite extending fragment, and Aβ peptides, which are neurite retractive [129]. | [129,130] |
| CALU-3 cells | Epithelial cells, from an adenocarcinoma | CALU-3 cells were used to measure drug delivery through epithelium of a β-sheet breaker [131]. | [131,132] |
| Immortalized with a viral vector | |||
| ReN cells and immortalized microglial cells | Neural stem cells and microglial cells | Park et al. (2018) engineered a 3D triculture system as a model of AD neurodegeneration and neuroinflammation. They notably cultured fAD-mutated ReN cells, which are neural progenitor cells, and induced their differentiation into Aβ-overexpressing neurons and astrocytes. They also added immortalized microglial cells, completing the triculture system [133]. | [133,134] |
| Immortalized brain endothelial cells | Endothelial cells | Endothelial cells were used to model Aβ clearance through BBB [135]. | Human cells: [135,136] Mouse cells: [137] |
| HEK293 cells | Embryonic kidney cells | Waxman and Giasson (2011) developed a cellular model to study the induction of tau aggregation with preformed α-synuclein fibrils, another protein involved in Parkinson’s disease [138]. | [138,139] |
| Reprogrammed Cells | ||
|---|---|---|
| Derived Cell Type or Tissue | Detailed Example | References |
| iPSCs | ||
| Neurons | Rouleau et al. (2020) developed a 3D neural tissue with human iPSC from healthy or AD donors [147]. | 2D culture, iPCS from fAD patients: [93,148] 3D culture, sAD patients: [145] 3D culture, fAD patients: [146] iPSC development from AD patients: [143,144,147] |
| Others: astroglia, endothelial cells, NPCs | iPSC-derived human brain endothelial cells with the PSEN1 mutation show altered tight and adherent junctions and efflux properties compared to cells derived from healthy donors [151]. | Astroglia: [149] Endothelial cell: [150,151] NPC: [152,153] |
| Organoids | Gonzalez et al. (2018) developed iPSC-derived cerebral organoids, which show a cortical organization. When the used iPSC comes from an AD patient, the developed cerebral organoid exhibits AD features such as Aβ deposition and accumulation of hyperphosphorylated tau [158]. | Examples of iPSC-derived organoid with fAD mutations: [158,159] Organoid with an AD-like pathology: [162]. Reviews: [5,157] |
| iNs | ||
| Neurons | Hu et al. (2015) derived fibroblasts from control and AD patients into functional neurons with chemicals [161]. | [93,161] |
| Non-Mammalian AD Models | ||||
|---|---|---|---|---|
| Models | Model Type | Advantages | Pitfalls | References |
| Caenorhabditis elegans | Transgenic | Small, easy to breed, lots of progenies. Characterized nervous system, short lifespan. Sequenced genome. Transgenic C. elegans can express human hyperphosphorylated tau mutant or Aβ peptides and develop some AD features. Used to study molecular interactions and cellular pathways. | Do not naturally have Aβ and β-secretase, and so, do not have amyloid aggregates. Do not naturally have tau aggregates, either. Need to be used in combination with other models. | [165,166,170] |
| Drosophila melanogaster | Transgenic | Small, easy to breed. Characterized nervous system. Sequenced genome. Have AD-related genes. Behavioral tests. Availability of genetic tools to do transgenic or knockdown models. Used for high-throughput drug screening. Transgenic flies develop AD hallmarks, such as overexpression of amyloid peptides, amyloid aggregate formation, tau hyperphosphorylation, synaptic impairments, neurodegeneration, and reduction of memory and lifespan. | AD genes are not well-characterized. Homology with human proteins but not sufficient to naturally develop the disease. Need to do transgenic models, but they do not clearly recapitulate the disease. Need to be used in combination with other models such as mouse models. Invertebrate model is very different from human than all other vertebrate models. | [172,173,174,175,176,177] |
| Danio rerio (zebrafish) | Transgenic | Small, easy to breed, lots of progenies. Characterized nervous system. Entirely sequenced genome. Have AD-related genes. Behavioral tests. Used for high-throughput drug screening. Available genetic tools for transgenic or knockdown models. | AD genes are not all well-characterized. Homology with human proteins but not sufficient to naturally develop the disease. Need to do transgenic models. Lack of data due to its recent development. | [178,179,180] |
| Mouse and Rat AD Models | |||||
|---|---|---|---|---|---|
| Model Type | Model Name | Associated Mutation(s) | AD Characteristics | Discrepancies with AD | References |
| Mice | |||||
| Transgenic | J20 | APP KM670/671NL (Swedish), APP V717F (Indiana) | Amyloid aggregation, neurodegeneration, neuroinflammation, cognitive impairments. | No NFTs, overexpression of mutated APP and associated fragments, deposition of amyloid plaques at 4–6 months. | [195,196] |
| APPPS1 | APP KM670/671NL (Swedish)PSEN1 L166P | Amyloid aggregation, neurodegeneration, neuroinflammation, cognitive impairments. | No NFTs, overexpression of mutated PSEN1 as well as mutated APP and associated fragments, deposition of amyloid plaques at 2–4 months. | [197] | |
| 5xFAD | APP KM670/671NL (Swedish), APP I716V (Florida), APP V717I (London) PSEN1 M146L (A > C), PSEN1 L286V | Amyloid aggregation, neurodegeneration, neuroinflammation, cognitive impairments. | No NFTs, overexpression of mutated PSEN1 as well as mutated APP and associated fragments, very aggressive form, deposition of amyloid plaques at 2 months. | [198] | |
| 3xTg | APP KM670/671NL (Swedish) MAPT P301L PSEN1 M146V | Amyloid aggregation and NFT formation, neurodegeneration, neuroinflammation, cognitive impairments. | Overexpression of mutated APP, tau, and PSEN1, amyloid plaques at 6 months, development of cognitive impairments before protein aggregation. | [199] | |
| APPNL-F KI | APP KM670/671NL (Swedish), APP I716F (Iberian) | Amyloid aggregation, neurodegeneration, neuroinflammation, cognitive impairments, chronology of symptom development. | no NFTs. | [200] | |
| Interventional | Aβ-injected | - | Neurodegeneration, neuroinflammation, cognitive impairments | No amyloid plaques, no NFTs. | [201] |
| Receptor antagonist-injected | - | Neurodegeneration, neuroinflammation, cognitive impairments. | No amyloid plaques, no NFTs. | [202] | |
| Olfactory bulbectomy | - | Increase in Aβ level, neurodegeneration, neuroinflammation, cognitive impairments. | No amyloid plaques, no NFTs. | [203] | |
| Natural | SAMP8 | - | Neurodegeneration, neuroinflammation, cognitive impairments. | No amyloid plaques, no NFTs. | [204] |
| Rats | |||||
| Transgenic | Tg F344-AD | APP KM670/671NL (Swedish) PSEN deltaE9 | Amyloid aggregation and NFT formation, neurodegeneration, neuroinflammation, cognitive impairments. | Overexpression of mutated APP and associated fragments, deposition of amyloid plaques at 6 months. | [205] |
| TREM2 KI (in Human App background) | TREM2 R47H | Physiological expression of a sAD risk factor, production of human Aβ, only. | No amyloid plaques, no NFTs, no neurodegeneration, no neuroinflammation, no cognitive impairments. | [206] | |
| Natural | OXYS | - | Increase in Aβ level, neurodegeneration, neuroinflammation, cognitive impairments. | No amyloid plaques, no NFTs. | [207] |
| Non-Mammalian AD Models | ||||
|---|---|---|---|---|
| Models | Model Type | Advantages | Pitfalls | References |
| Rabbits | Interventional | Induction of AD-pathology after brain injection of aluminum maltolate (features: amyloid aggregation, NFT formation and neurodegeneration). Non aggressive animal. | The structure of NFTs is different from human. | [215,216] |
| Octodon Degus | Natural | Development of AD-like disease with age. Aβ accumulation and plaque formation, with age. Tau accumulation. Memory impairments. | Inconsistensy from one study to another. Lack of appropriate brain map. | [92,218,219,220] |
| Dogs | Natural | Development of AD-like disease with age (Aβ plaques and cognitive deficits). | Diffuse plaques contrary to compact human plaques. No NFTs but pretangles. No cholinergic deficit. Long and variable lifespan. Lack of consistency. | [92,220] |
| Non human Primates (NHPs) | Natural | Development of AD-like disease with age. Genetically and anatomically closest animal to human (example: 100% homology in Aβ sequence). Well-characterized, complex, and quantifiable behaviors. Four groups of NHPs with different specificities. Similar AD symptoms: Aβ accumulation and amyloid plaque formation in the brain. | Ethical concerns. Long lifespan. Costly, few animals. Do not perfectly reproduce the human disease (often develop diffuse amyloid plaques instead of compact plaques, some primates have NFTs and others do not, mild cognitive deficits rather similar to normal ageing than to AD-induced cognitive impairments). Inter individual variability. | [3,92] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vignon, A.; Salvador-Prince, L.; Lehmann, S.; Perrier, V.; Torrent, J. Deconstructing Alzheimer’s Disease: How to Bridge the Gap between Experimental Models and the Human Pathology? Int. J. Mol. Sci. 2021, 22, 8769. https://doi.org/10.3390/ijms22168769
Vignon A, Salvador-Prince L, Lehmann S, Perrier V, Torrent J. Deconstructing Alzheimer’s Disease: How to Bridge the Gap between Experimental Models and the Human Pathology? International Journal of Molecular Sciences. 2021; 22(16):8769. https://doi.org/10.3390/ijms22168769
Chicago/Turabian StyleVignon, Anaïs, Lucie Salvador-Prince, Sylvain Lehmann, Véronique Perrier, and Joan Torrent. 2021. "Deconstructing Alzheimer’s Disease: How to Bridge the Gap between Experimental Models and the Human Pathology?" International Journal of Molecular Sciences 22, no. 16: 8769. https://doi.org/10.3390/ijms22168769
APA StyleVignon, A., Salvador-Prince, L., Lehmann, S., Perrier, V., & Torrent, J. (2021). Deconstructing Alzheimer’s Disease: How to Bridge the Gap between Experimental Models and the Human Pathology? International Journal of Molecular Sciences, 22(16), 8769. https://doi.org/10.3390/ijms22168769