
Endoglin Is an Endothelial Housekeeper against Inflammation: Insight in ECFC-Related Permeability through LIMK/Cofilin Pathway

 , , , , , , , ,
, , , , , , , ,  and
and 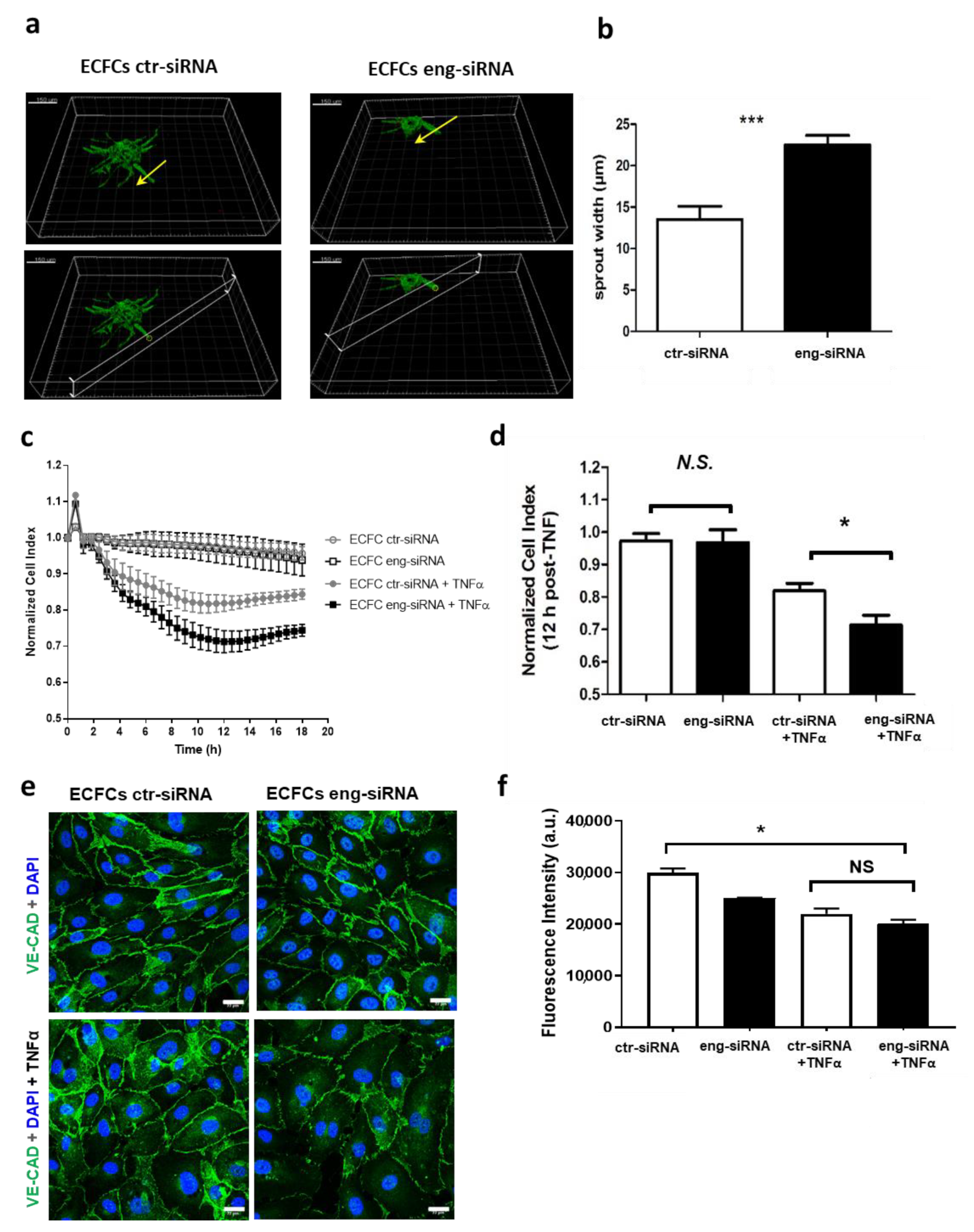{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
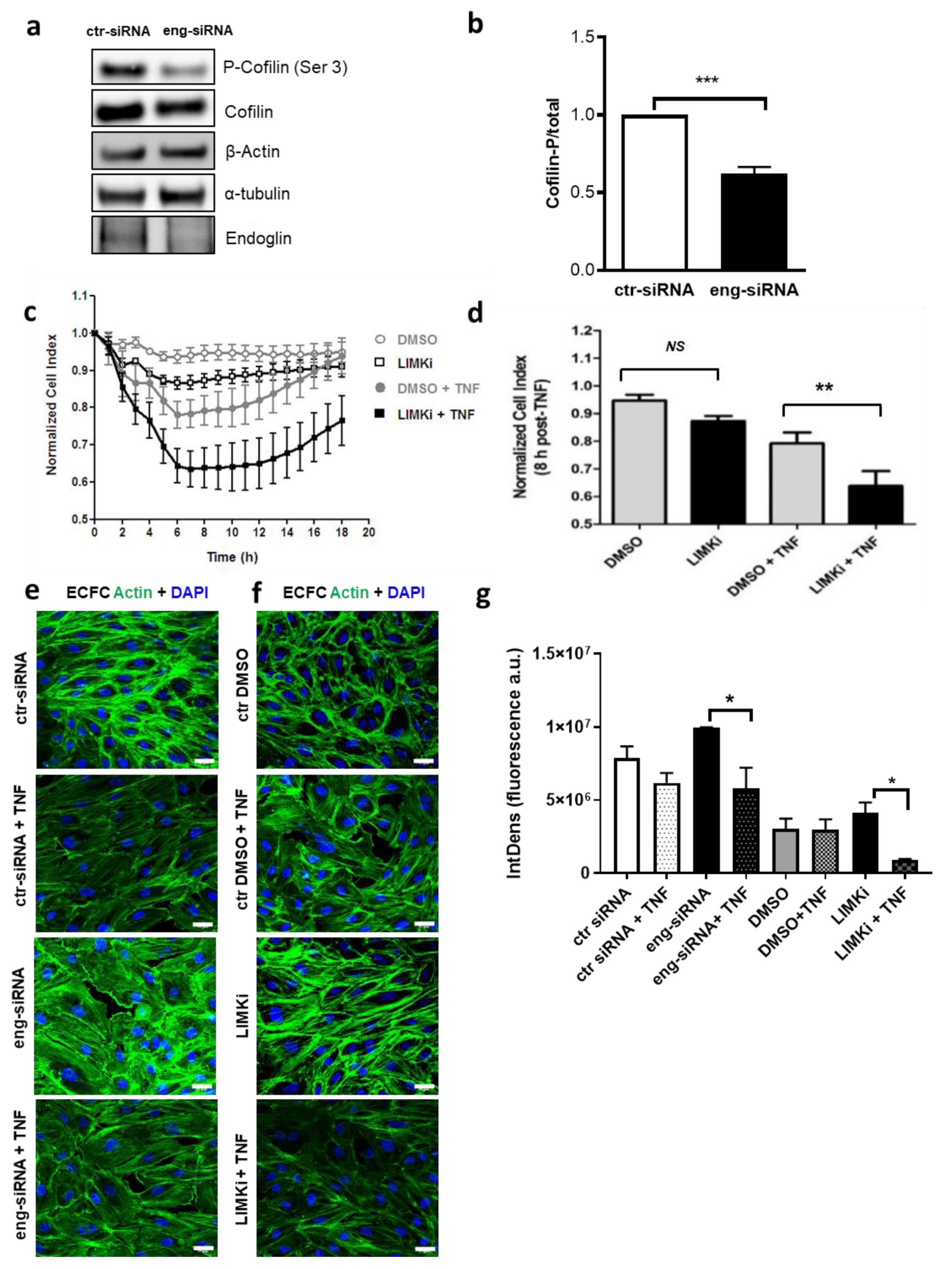
2.1. Endoglin Regulates ECFC Tubulogenesis and Permeability under Inflammatory Conditions
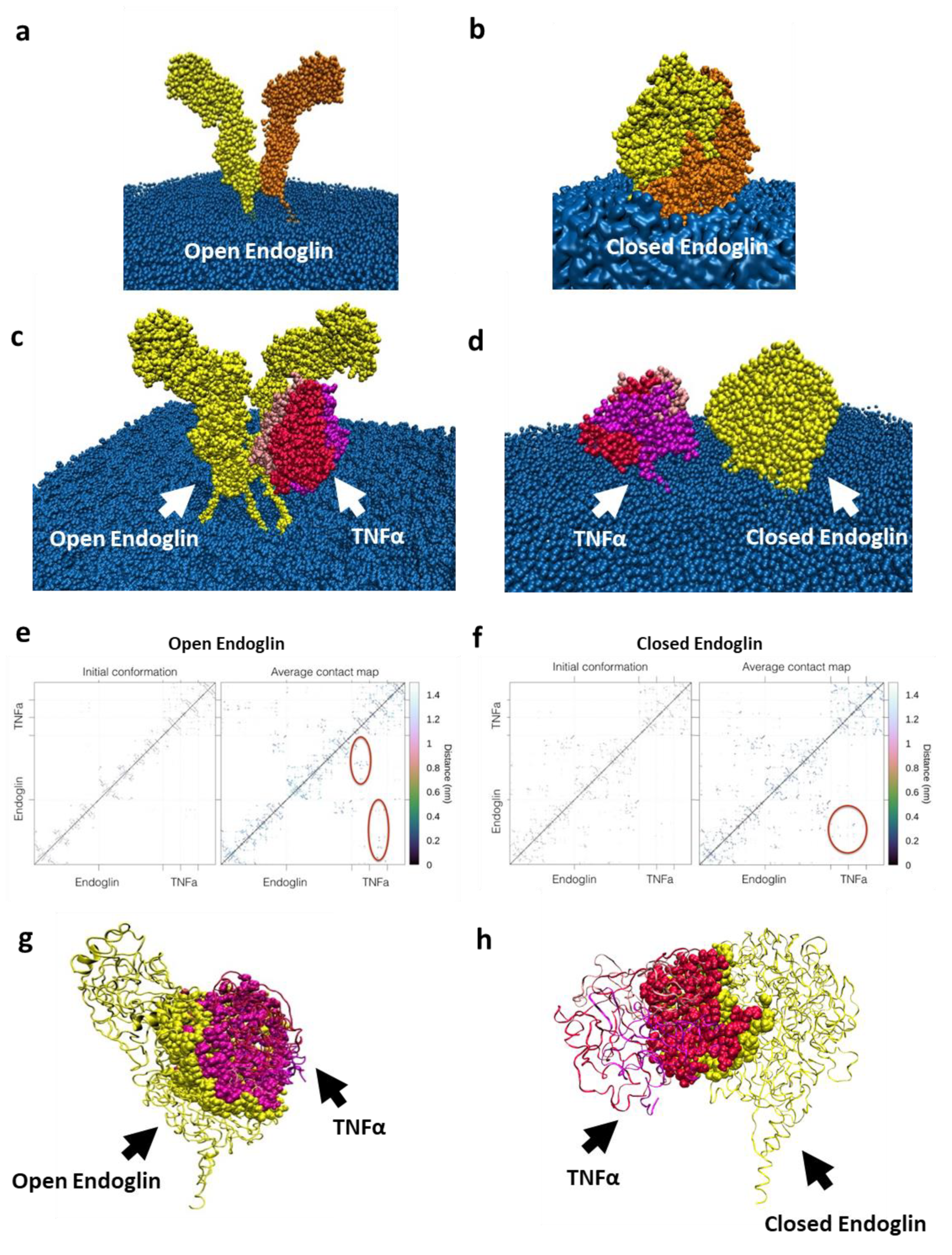
2.2. In Silico Interaction between Endoglin and TNFα
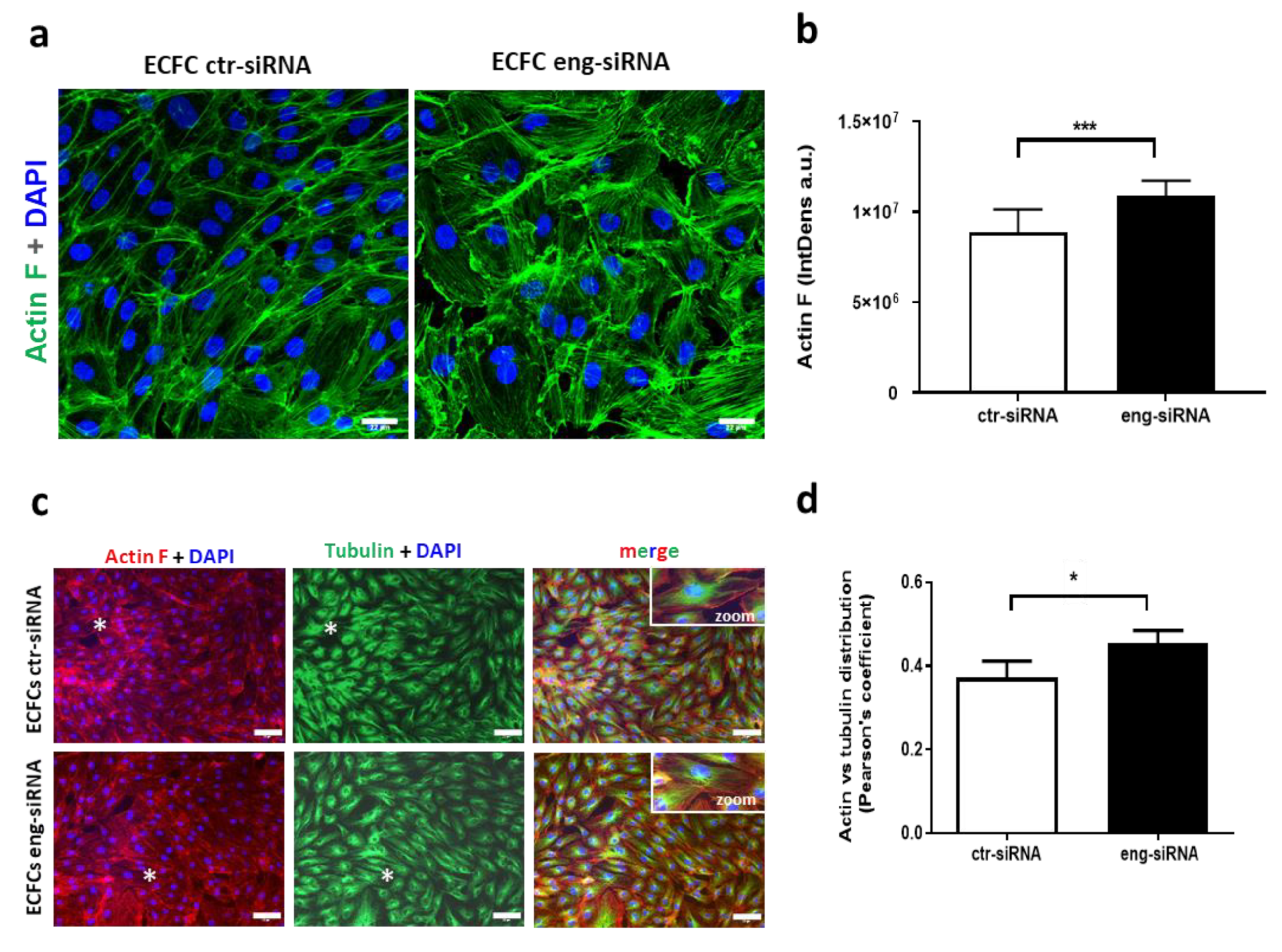
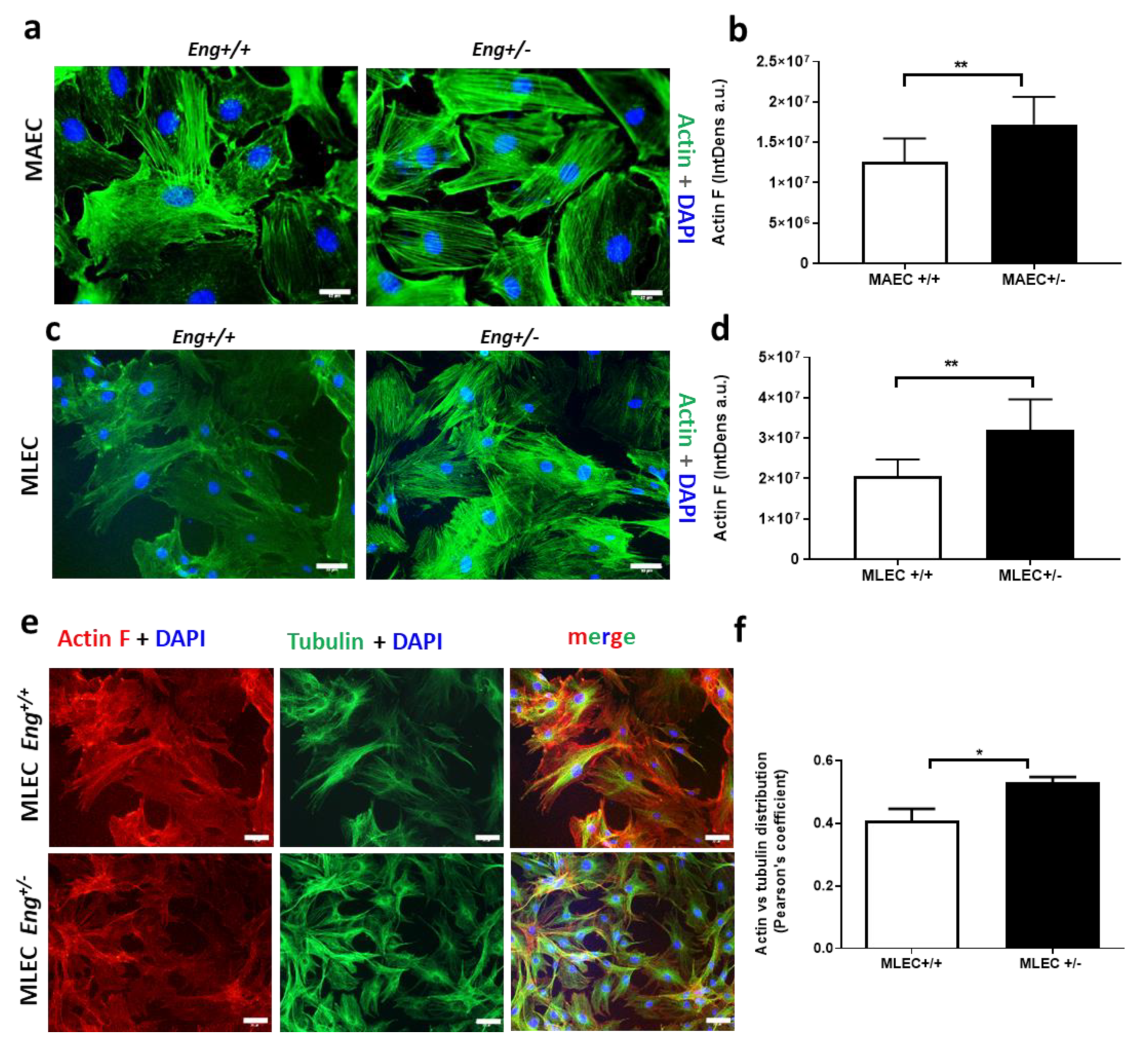
2.3. Endoglin Regulates Actin Dynamics
3. Discussion
4. Materials and Methods
4.1. ECFC Isolation, Culture, and Transfection
4.2. MAEC and MLEC Isolation from Mouse Model of HHT1 (Eng+/− Mice)
4.3. Immunofluorescence Microscopy
4.4. Immunofluorescence Flow Cytometry
4.5. Angiogenesis Assays Using Cytodex
4.6. ECFC Barrier Permeability Assay
4.7. Measurement of Intracellular Ca2+
4.8. Computational Analysis of Endoglin Structure
4.9. Coarse-Grain Molecular Dynamic Simulations for TNFα and Endoglin
4.10. Immunoblotting
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ctr-siRNA | scrambled siRNA used as control |
| EC | Endothelial cells |
| ECFC | Endothelial colony forming cells |
| Eng | Endoglin |
| Eng-siRNA | Endoglin-specific siRNA |
| HHT | Hereditary Hemorrhagic Telangiectasia |
| LIMKi | LIM Kinase inhibitors |
| MAEC | Mouse aortic endothelial cells |
| MLEC | Mouse lung endothelial cells |
| PTM | Post-translational modifications |
| TGF-β | Transforming growth factor-β |
| TNFα | Tumor necrosis factor-α |
References
- Ricard, N.; Ciais, D.; Levet, S.; Subileau, M.; Mallet, C.; Zimmers, T.A.; Lee, S.J.; Bidart, M.; Feige, J.J.; Bailly, S. BMP9 and BMP10 are critical for postnatal retinal vascular remodeling. Blood 2012, 119, 6162–6171. [Google Scholar] [CrossRef] [Green Version]
- Castonguay, R.; Werner, E.D.; Matthews, R.G.; Presman, E.; Mulivor, A.W.; Solban, N.; Sako, D.; Pearsall, R.S.; Underwood, K.W.; Seehra, J.; et al. Soluble endoglin specifically binds bone morphogenetic proteins 9 and 10 via its orphan domain, inhibits blood vessel formation, and suppresses tumor growth. J. Biol. Chem. 2011, 286, 30034–30046. [Google Scholar] [CrossRef] [Green Version]
- Alt, A.; Miguel-Romero, L.; Donderis, J.; Aristorena, M.; Blanco, F.J.; Round, A.; Rubio, V.; Bernabeu, C.; Marina, A. Structural and functional insights into endoglin ligand recognition and binding. PLoS ONE 2012, 7, e29948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Bokhove, M.; Croci, R.; Zamora-Caballero, S.; Han, L.; Letarte, M.; de Sanctis, D.; Jovine, L. Structural Basis of the Human Endoglin-BMP9 Interaction: Insights into BMP Signaling and HHT1. Cell Rep. 2017, 19, 1917–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Novoa, J.M.; Bernabeu, C. The physiological role of endoglin in the cardiovascular system. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H959–H974. [Google Scholar] [CrossRef] [Green Version]
- Schoonderwoerd, M.J.A.; Goumans, M.J.T.H.; Hawinkels, L.J.A.C. Endoglin: Beyond the endothelium. Biomolecules 2020, 10, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Llorente, L.; Gallardo-Vara, E.; Rossi, E.; Smadja, D.M.; Botella, L.M.; Bernabeu, C. Endoglin and alk1 as therapeutic targets for hereditary hemorrhagic telangiectasia. Expert Opin. Ther. Targets 2017, 21, 933–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shovlin, C.L. Hereditary haemorrhagic telangiectasia: Pathophysiology, diagnosis and treatment. Blood Rev. 2010, 24, 203–219. [Google Scholar] [CrossRef] [Green Version]
- Young, K.; Tweedie, E.; Conley, B.; Ames, J.; FitzSimons, M.L.; Brooks, P.; Liaw, L.; Vary, C.P.H. BMP9 crosstalk with the hippo pathway regulates endothelial cell matricellular and chemokine responses. PLoS ONE 2015, 10, e0122892. [Google Scholar] [CrossRef] [Green Version]
- Rossi, E.; Sanz-Rodriguez, F.; Eleno, N.; Düwell, A.; Blanco, F.J.; Langa, C.; Botella, L.M.; Cabañas, C.; Lopez-Novoa, J.M.; Bernabeu, C. Endothelial endoglin is involved in inflammation: Role in leukocyte adhesion and transmigration. Blood 2013, 121, 403–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, E.; Smadja, D.M.; Boscolo, E.; Langa, C.; Arevalo, M.A.; Pericacho, M.; Gamella-Pozuelo, L.; Kauskot, A.; Botella, L.M.; Gaussem, P.; et al. Endoglin regulates mural cell adhesion in the circulatory system. Cell. Mol. Life Sci. 2016, 73, 1715–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, E.; Pericacho, M.; Bachelot-Loza, C.; Pidard, D.; Gaussem, P.; Poirault-Chassac, S.; Blanco, F.J.; Langa, C.; González-Manchón, C.; Novoa, J.M.L.; et al. Human endoglin as a potential new partner involved in platelet–endothelium interactions. Cell. Mol. Life Sci. 2018, 75, 1269–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jerkic, M.; Letarte, M. Increased endothelial cell permeability in endoglin-deficient cells. FASEB J. 2015, 29, 3678–3688. [Google Scholar] [CrossRef] [Green Version]
- Uemura, A.; Fukushima, Y. Rho gtpases in retinal vascular diseases. Int. J. Mol. Sci. 2021, 22, 3684. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, M.; Allinson, K.R.; Zhai, Z.; Oakenfull, R.; Ghandi, P.; Adams, R.H.; Fruttiger, M.; Arthur, H.M. Pathogenesis of arteriovenous malformations in the absence of endoglin. Circ. Res. 2010, 106, 1425–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnett, J.M.; Suarez, S.; McCollum, G.W.; Penn, J.S. Endoglin promotes angiogenesis in cell- and animal- based models of retinal neovascularization. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6490–6498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallardo-Vara, E.; Tual-Chalot, S.; Botella, L.M.; Arthur, H.M.; Bernabeu, C. Soluble endoglin regulates expression of angiogenesis-related proteins and induction of arteriovenous malformations in a mouse model of hereditary hemorrhagic telangiectasia. Dis. Model. Mech. 2018, 11, dmm034397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, R.A.; Li, C.; Aziz, W.; Olson, J.A.; Vohra, A.; McHardy, K.C.; Forrester, J.V.; Boulton, A.J.M.; Wilson, P.B.; Liu, D.; et al. Elevated plasma CD105 and vitreous VEGF levels in diabetic retinopathy. J. Cell. Mol. Med. 2005, 9, 692–697. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, K.; Li, J.; Shen, J.; Xu, W. Elevated Levels of Endoglin, Endostatin, FGF-α, HGF, and Thrombospondin-2 in Aqueous Humor of nAMD Patients. Ocul. Immunol. Inflamm. 2021, 1, 1–8, Online ahead of print. [Google Scholar] [CrossRef]
- Shen, W.; Lee, S.R.; Yam, M.; Zhu, L.; Zhang, T.; Pye, V.; Mathai, A.E.; Shibagaki, K.; Zhang, J.Z.; Matsugi, T.; et al. A combination therapy targeting endoglin and VEGF-A prevents subretinal fibro-neovascularization caused by induced müller cell disruption. Investig. Ophthalmol. Vis. Sci. 2018, 59, 6075–6088. [Google Scholar] [CrossRef] [Green Version]
- Rapp, B.M.; Saadatzedeh, M.R.; Ofstein, R.H.; Bhavsar, J.R.; Tempel, Z.S.; Moreno, O.; Morone, P.; Booth, D.A.; Traktuev, D.O.; Dalsing, M.C.; et al. Resident Endothelial Progenitor Cells from Human Placenta have Greater Vasculogenic Potential than Circulating Endothelial Progenitor Cells from Umbilical Cord Blood. Cell Med. 2011, 2, 85–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smadja, D.M.; Melero-Martin, J.M.; Eikenboom, J.; Bowman, M.; Sabatier, F.; Randi, A.M. Standardization of methods to quantify and culture endothelial colony-forming cells derived from peripheral blood: Position paper from the International Society on Thrombosis and Haemostasis SSC. J. Thromb. Haemost. 2019, 17, 1190–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-L, A.; Sanz-Rodriguez, F.; Zarrabeitia, R.; Pérez-Molino, A.; Hebbel, R.P.; Nguyen, J.; Bernabéu, C.; Botella, L.M. Blood outgrowth endothelial cells from Hereditary Haemorrhagic Telangiectasia patients reveal abnormalities compatible with vascular lesions. Cardiovasc. Res. 2005, 68, 235–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, K. Signaling mechanisms and functional roles of cofilin phosphorylation and dephosphorylation. Cell Signal. 2013, 25, 457–469. [Google Scholar] [CrossRef]
- Kauskot, A.; Poirault-Chassac, S.; Adam, F.; Muczynski, V.; Aymé, G.; Casari, C.; Bordet, J.-C.; Soukaseum, C.; Rothschild, C.; Proulle, V.; et al. LIM kinase/cofilin dysregulation promotes macrothrombocytopenia in severe von Willebrand disease-type 2B. JCI Insight 2016, 1, 1–17. [Google Scholar] [CrossRef]
- Dalal, P.J.; Muller, W.A.; Sullivan, D.P. Endothelial Cell Calcium Signaling during Barrier Function and Inflammation. Am. J. Pathol. 2020, 190, 535–542. [Google Scholar] [CrossRef] [Green Version]
- Cerutti, C.; Ridley, A.J. Endothelial cell-cell adhesion and signaling. Exp. Cell Res. 2017, 358, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Rodriguez, F.; Guerrero-Esteo, M.; Botella, L.M.; Banville, D.; Vary, C.P.H.; Bernabéu, C. Endoglin regulates cytoskeletal organization through binding to ZRP-1, a member of the LIM family of proteins. J. Biol. Chem. 2004, 279, 32858–32868. [Google Scholar] [CrossRef] [Green Version]
- Rossi, E.; Bernabeu, C.; Smadja, D.M. Endoglin as an adhesion molecule in mature and progenitor endothelial cells: A function beyond TGF-β. Front. Med. 2019, 6, 1–8. [Google Scholar] [CrossRef]
- Rossi, E.; Lopez-Novoa, J.M.; Bernabeu, C. Endoglin involvement 1 in integrin-mediated cell adhesion as a putative pathogenic mechanism in Hereditary Hemorrhagic Telangectasia type 1 (HHT1). Front. Genet. 2014, 5, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Bernabeu, C.; Bayrak-Toydemir, P.; Mcdonald, J.; Letarte, M. Potential Second-Hits in Hereditary Hemorrhagic Telangiectasia. J. Clin. Med. 2020, 9, 3571. [Google Scholar] [CrossRef] [PubMed]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Le Bras, A.; Sacharidou, A.; Itagaki, K.; Zhan, Y.; Kondo, M.; Carman, C.V.; Davis, G.E.; Aird, W.C.; Oettgen, P. ETS-related gene (ERG) controls endothelial cell permeability via transcriptional regulation of the claudin 5 (CLDN5) gene. J. Biol. Chem. 2012, 287, 6582–6591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torsney, E.; Charlton, R.; Parums, D.; Collis, M.; Arthur, H.M. Inducible expression of human endoglin during inflammation and wound healing in vivo. Inflamm. Res. 2002, 51, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Oberleithner, H.; Riethmüller, C.; Schillers, H.; MacGregor, G.A.; De Wardener, H.E.; Hausberg, M. Plasma sodium stiffens vascular endothelium and reduces nitric oxide release. Proc. Natl. Acad. Sci. USA 2007, 104, 16281–16286. [Google Scholar] [CrossRef] [Green Version]
- Smadja, D.M.; Chocron, R.; Rossi, E.; Poitier, B.; Pya, Y.; Bekbossynova, M.; Peronino, C.; Rancic, J.; Roussel, J.C.; Kindo, M.; et al. Autoregulation of Pulsatile Bioprosthetic Total Artificial Heart is Involved in Endothelial Homeostasis Preservation. Thromb. Haemost. 2020, 120, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Rossi, E.; Goyard, C.; Cras, A.; Dizier, B.; Bacha, N.; Lokajczyk, A.; Guerin, C.L.; Gendron, N.; Planquette, B.; Mignon, V.; et al. Co-injection of mesenchymal stem cells with endothelial progenitor cells accelerates muscle recovery in hind limb ischemia through an endoglin-dependent mechanism. Thromb. Haemost. 2017, 117, 1908–1918. [Google Scholar] [CrossRef] [PubMed]
- Conley, B.A.; Koleva, R.; Smith, J.D.; Kacer, D.; Zhang, D.; Bernabéu, C.; Vary, C.P.H. Endoglin controls cell migration and composition of focal adhesions: Function of the cytosolic domain. J. Biol. Chem. 2004, 279, 27440–27449. [Google Scholar] [CrossRef] [Green Version]
- Nevo, N.; Lecourt, S.; Bièche, I.; Kucia, M.; Cras, A.; Blandinieres, A.; Vacher, S.; Gendron, N.; Guerin, C.L.; Ratajczak, M.Z.; et al. Valproic Acid Decreases Endothelial Colony Forming Cells Differentiation and Induces Endothelial-to-Mesenchymal Transition-like Process. Stem Cell Rev. Rep. 2020, 16, 357–368. [Google Scholar] [CrossRef]
- Jerkic, M.; Rivas-Elena, J.V.; Prieto, M.; Carrón, R.; Sanz-Rodríguez, F.; Pérez-Barriocanal, F.; Rodríguez-Barbero, A.; Bernabéu, C.; López-Novoa, J.M. Endoglin regulates nitric oxide-dependent vasodilatation. FASEB J. 2004, 18, 609–611. [Google Scholar] [CrossRef]
- Bolte, S.; Cordelières, F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006, 224, 213–232. [Google Scholar] [CrossRef] [PubMed]
- Blandinières, A.; Gendron, N.; Bacha, N.; Bièche, I.; Chocron, R.; Nunes, H.; Nevo, N.; Rossi, E.; Crestani, B.; Lecourt, S.; et al. Interleukin-8 release by endothelial colony-forming cells isolated from idiopathic pulmonary fibrosis patients might contribute to their pathogenicity. Angiogenesis 2019, 22, 325–339. [Google Scholar] [CrossRef]
- Şener, L.T.; Albeniz, G.; Dinç, B.; Albeniz, I. iCELLigence real-time cell analysis system for examining the cytotoxicity of drugs to cancer cell lines. Exp. Ther. Med. 2017, 14, 1866–1870. [Google Scholar] [CrossRef] [PubMed]
- Elaïb, Z.; Adam, F.; Berrou, E.; Bordet, J.C.; Prévost, N.; Bobe, R.; Bryckaert, M.; Rosa, J.P. Full activation of mouse platelets requires ADP secretion regulated by SERCA3 ATPase-dependent calcium stores. Blood 2016, 128, 1129–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Fiser, A.; Sali, A. Modeller: Generation and refinement of homology-based protein structure models. Methods Enzymol. 2003, 374, 461–491. [Google Scholar] [CrossRef]
- Guerrero-Esteo, M.; Sanchez-Elsner, T.; Letamendia, A.; Bernabeu, C. Extracellular and cytoplasmic domains of endoglin interact with the transforming growth factor-beta receptors I and II. J. Biol. Chem. 2002, 277, 29197–29209. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maupetit, J.; Derreumaux, P.; Tuffery, P. PEP-FOLD: An online resource for de novo peptide structure prediction. Nucleic Acids Res. 2009, 37, 498–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knecht, V.; Marrink, S.J. Molecular dynamics simulations of lipid vesicle fusion in atomic detail. Biophys. J. 2007, 92, 4254–4261. [Google Scholar] [CrossRef] [Green Version]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Wassenaar, T.A.; Ingólfsson, H.I.; Prieß, M.; Marrink, S.J.; Schäfer, L.V. Mixing MARTINI: Electrostatic coupling in hybrid atomistic-coarse-grained biomolecular simulations. J. Phys. Chem. B 2013, 117, 3516–3530. [Google Scholar] [CrossRef] [PubMed]
- Martoňák, R.; Laio, A.; Parrinello, M. Predicting Crystal Structures: The Parrinello-Rahman Method Revisited. Phys. Rev. Lett. 2003, 90, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossi, E.; Kauskot, A.; Saller, F.; Frezza, E.; Poirault-Chassac, S.; Lokajczyk, A.; Bourdoncle, P.; Saubaméa, B.; Gaussem, P.; Pericacho, M.; et al. Endoglin Is an Endothelial Housekeeper against Inflammation: Insight in ECFC-Related Permeability through LIMK/Cofilin Pathway. Int. J. Mol. Sci. 2021, 22, 8837. https://doi.org/10.3390/ijms22168837
Rossi E, Kauskot A, Saller F, Frezza E, Poirault-Chassac S, Lokajczyk A, Bourdoncle P, Saubaméa B, Gaussem P, Pericacho M, et al. Endoglin Is an Endothelial Housekeeper against Inflammation: Insight in ECFC-Related Permeability through LIMK/Cofilin Pathway. International Journal of Molecular Sciences. 2021; 22(16):8837. https://doi.org/10.3390/ijms22168837
Chicago/Turabian StyleRossi, Elisa, Alexandre Kauskot, François Saller, Elisa Frezza, Sonia Poirault-Chassac, Anna Lokajczyk, Pierre Bourdoncle, Bruno Saubaméa, Pascale Gaussem, Miguel Pericacho, and et al. 2021. "Endoglin Is an Endothelial Housekeeper against Inflammation: Insight in ECFC-Related Permeability through LIMK/Cofilin Pathway" International Journal of Molecular Sciences 22, no. 16: 8837. https://doi.org/10.3390/ijms22168837
APA StyleRossi, E., Kauskot, A., Saller, F., Frezza, E., Poirault-Chassac, S., Lokajczyk, A., Bourdoncle, P., Saubaméa, B., Gaussem, P., Pericacho, M., Bobe, R., Bachelot-Loza, C., Pasquali, S., Bernabeu, C., & Smadja, D. M. (2021). Endoglin Is an Endothelial Housekeeper against Inflammation: Insight in ECFC-Related Permeability through LIMK/Cofilin Pathway. International Journal of Molecular Sciences, 22(16), 8837. https://doi.org/10.3390/ijms22168837