
Genetic Variants and Tumor Immune Microenvironment: Clues for Targeted Therapies in Inflammatory Breast Cancer (IBC)


 ,
, 
Abstract
:1. Introduction
2. Results
2.1. Patient Characteristics and Samples Studied by Next Generation Sequencing (NGS)
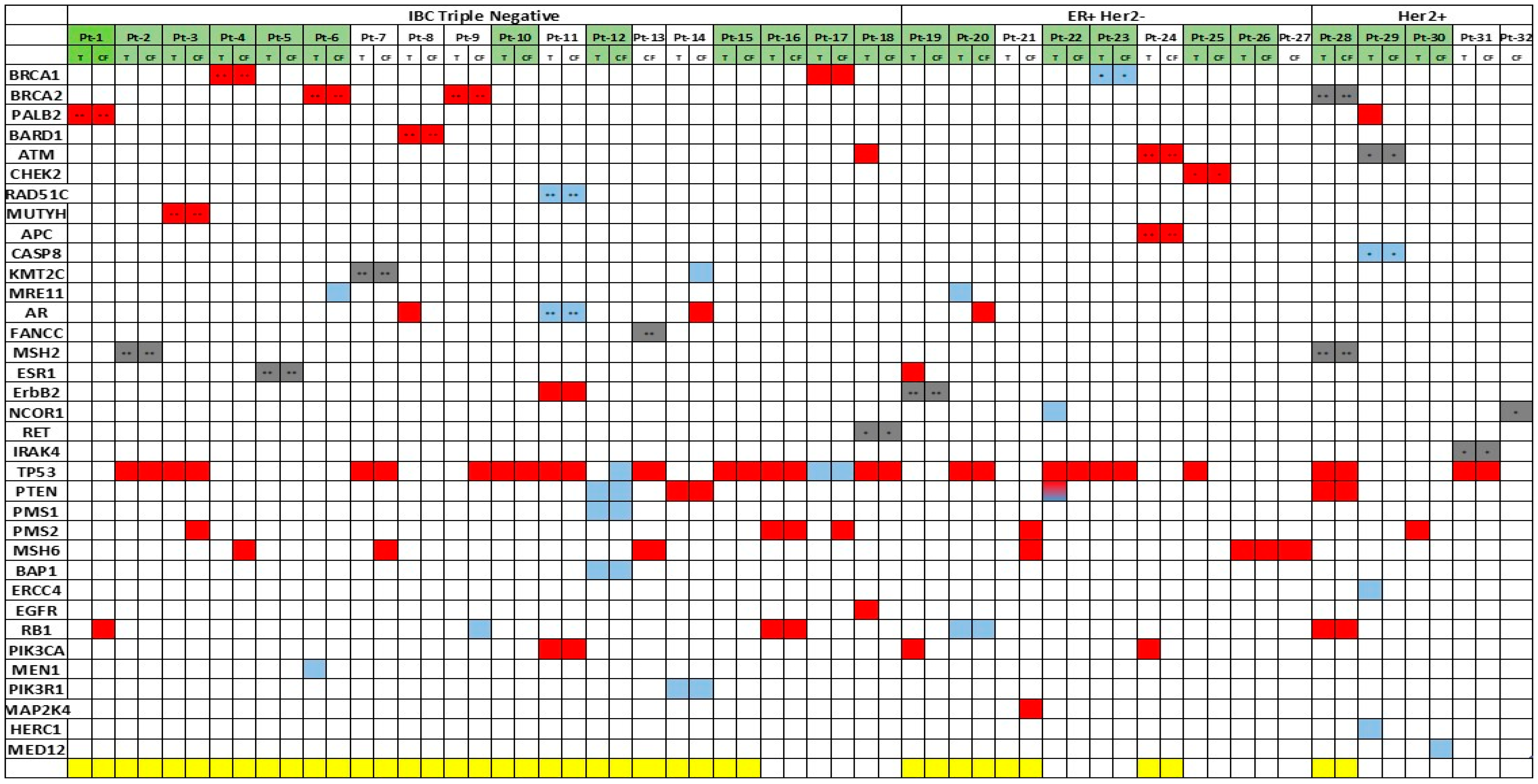
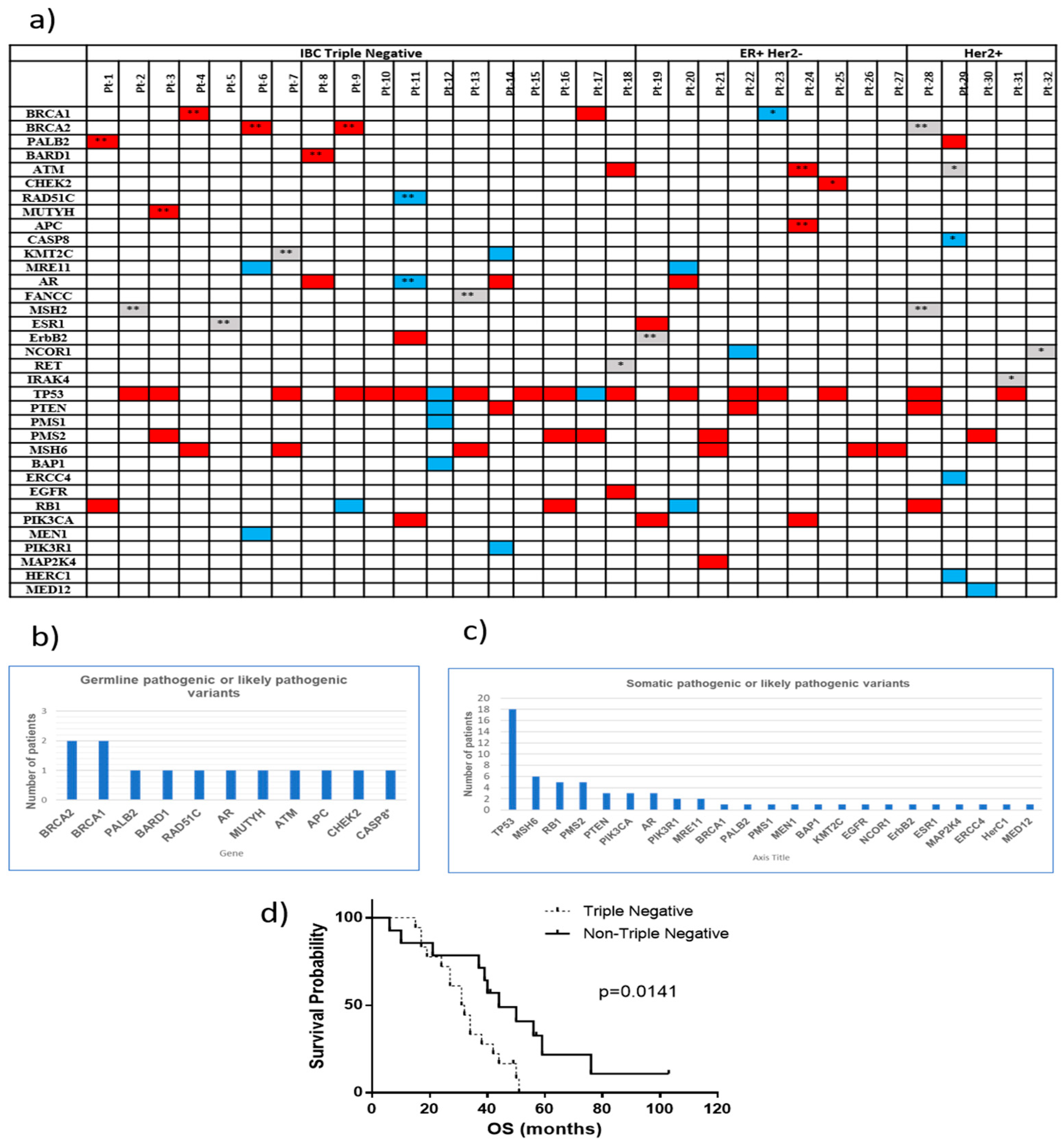
2.2. Clinical Relevant Variants in IBC Patients
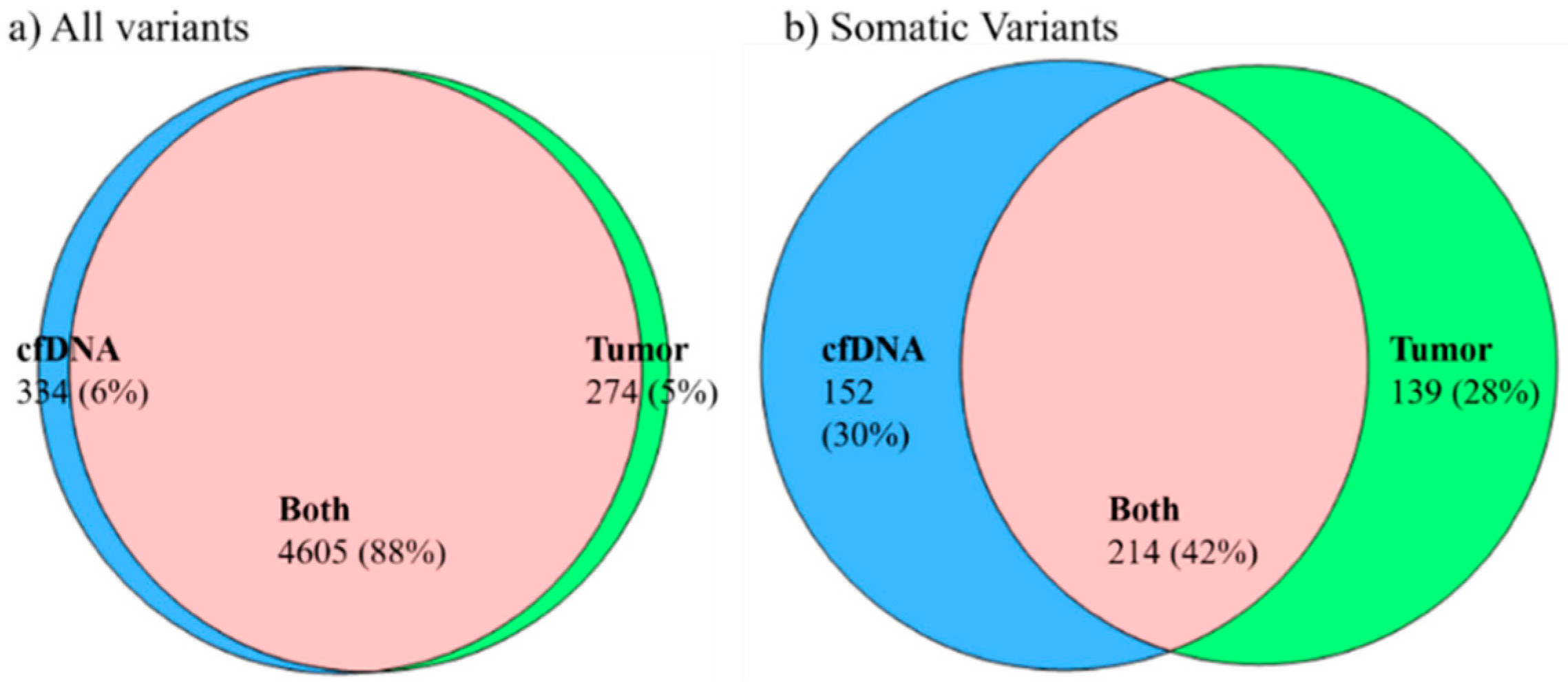
2.3. Concordance of Variants in Tumor Tissue and Paired cfDNA
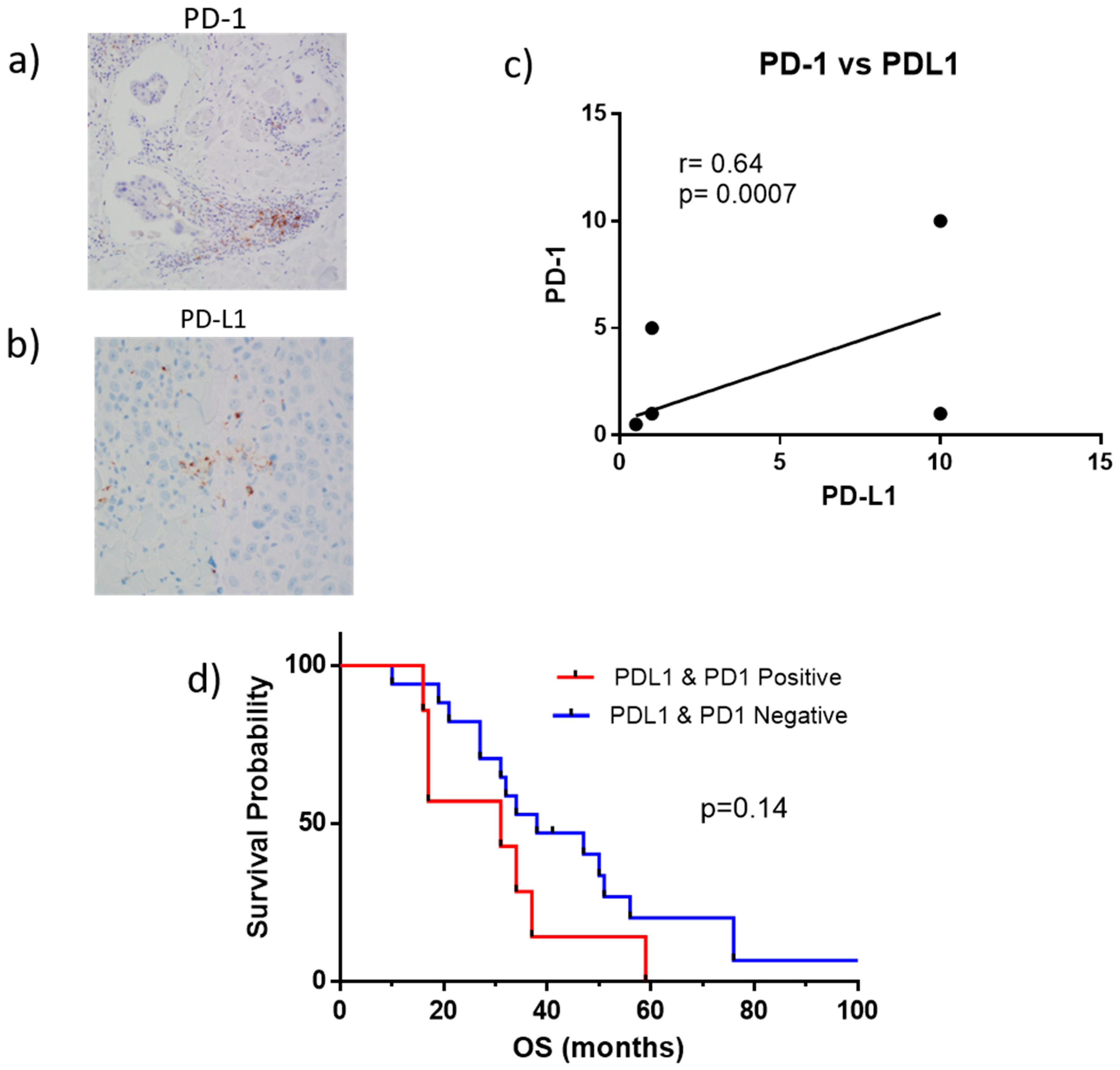
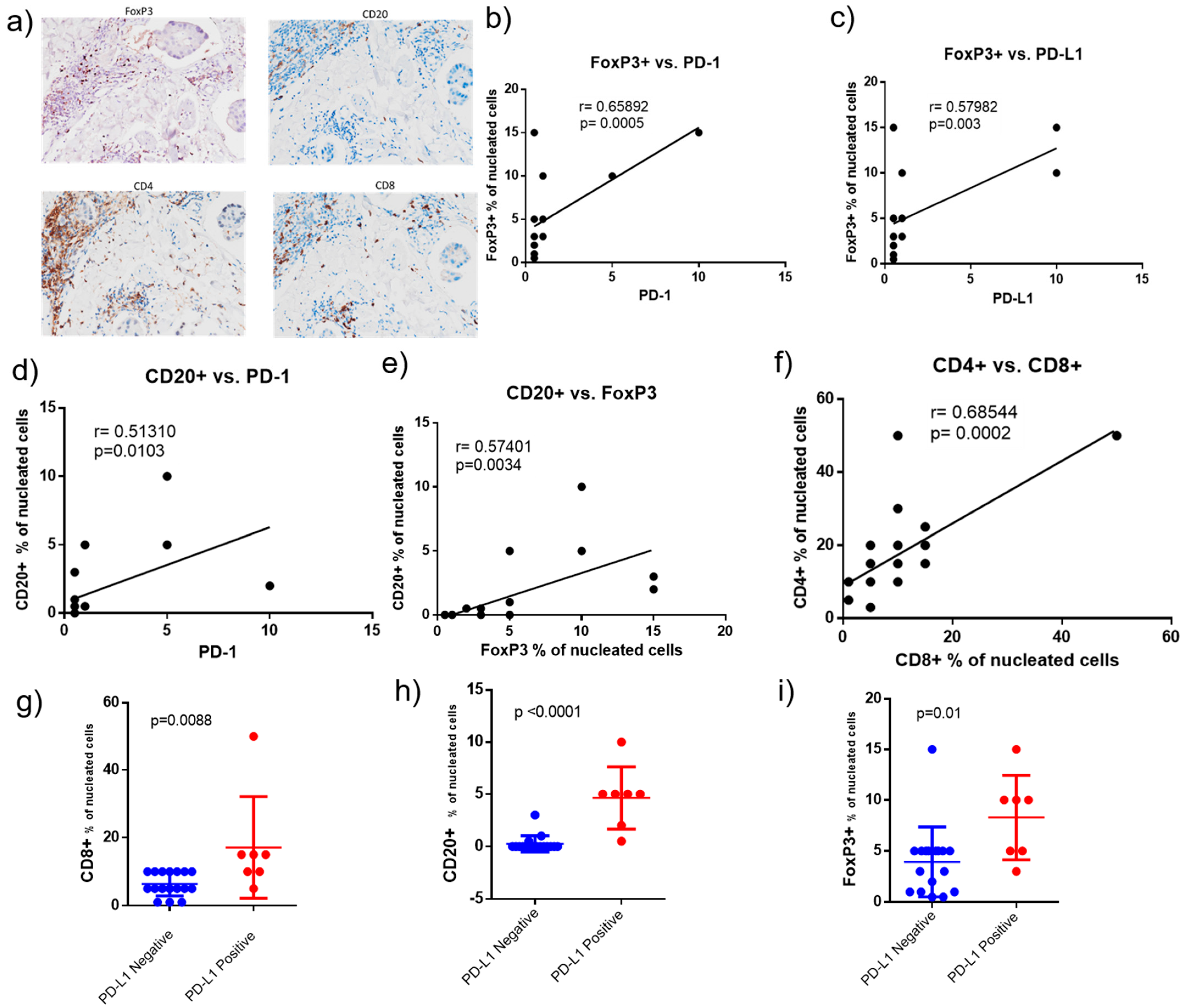
2.4. PD-1 and PD-L1 Expression in Tumor Biopsies
2.5. Correlation of Tumor Infiltrated Lymphocytes (TILs), PD-1 and PD-L1 in Tumor Biopsies
3. Discussion
4. Materials and Methods
4.1. Patient Cohort
4.2. Sample Collection for NGS
4.3. cfDNA and Genomic DNA Isolation
4.4. Library Preparation for NGS
4.5. NGS and Data Analysis
4.6. Variant Classification
4.7. Concordance of Genetic Variants in Tumor Tissue and Paired cfDNA from Plasma/Serum
4.8. Measuring Tumor-Infiltrating Lymphocytes (TILs) in Tumor Biopsies
4.9. PD-1 and PD-L1 Expression in Tumor Biopsies
4.10. Statistical Analysis
4.11. Data Availability
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AF | allele fraction |
| AR | androgen receptor |
| cfDNA | cell-free DNA |
| ER | estrogen receptor |
| FFPE | formalin fixed paraffin embedded |
| Her2 (or ErbB2) | human epidermal growth factor receptor-2 |
| HRR | homologous recombination repair |
| IBC | inflammatory breast cancer |
| IHC | immunohistochemistry |
| ICI | immune check point inhibitors |
| Indels | small insertions and deletions |
| mAb | monoclonal antibody |
| NGS | next generation sequencing |
| OCT | optimal cutting temperature compound |
| OS | overall survival |
| PARP | poly (ADP-ribose) polymerase |
| PD-1 | programmed cell death protein 1 |
| PD-L1 | programmed cell death-ligand 1 |
| PR | progesterone receptor |
| QCI | Qiagen Clinical Insight Interpret |
| RBC | red blood cells |
| SNVs | single nucleotide variants |
| TILs | tumor infiltrating lymphocytes |
| TN | triple negative |
| UMIs | unique molecular indices |
| VCF | variant call format |
References
- Cristofanilli, M.; Valero, V.; Buzdar, A.U.; Kau, S.W.; Broglio, K.R.; Gonzalez-Angulo, A.M.; Sneige, N.; Islam, R.; Ueno, N.T.; Buchholz, T.A.; et al. Inflammatory breast cancer (IBC) and patterns of recurrence: Understanding the biology of a unique disease. Cancer 2007, 110, 1436–1444. [Google Scholar] [CrossRef]
- Lim, B.; Woodward, W.A.; Wang, X.; Reuben, J.M.; Ueno, N.T. Inflammatory breast cancer biology: The tumour microenvironment is key. Nat. Rev. Cancer 2018, 18, 485–499. [Google Scholar] [CrossRef]
- Menta, A.; Fouad, T.M.; Lucci, A.; Le-Petross, H.; Stauder, M.C.; Woodward, W.A.; Ueno, N.T.; Lim, B. Inflammatory Breast Cancer: What to Know About This Unique, Aggressive Breast Cancer. Surg. Clin. N. Am. 2018, 98, 787–800. [Google Scholar] [CrossRef]
- Robertson, F.M.; Bondy, M.; Yang, W.; Yamauchi, H.; Wiggins, S.; Kamrudin, S.; Krishnamurthy, S.; Le-Petross, H.; Bidaut, L.; Player, A.N.; et al. Inflammatory breast cancer: The disease, the biology, the treatment. CA Cancer J. Clin. 2010, 60, 351–375. [Google Scholar] [CrossRef]
- Rowan, K. Inflammatory breast cancer: New hopes and many hurdles. J. Natl. Cancer Inst. 2009, 101, 1302–1304. [Google Scholar] [CrossRef] [Green Version]
- Levine, P.H.; Steinhorn, S.C.; Ries, L.G.; Aron, J.L. Inflammatory breast cancer: The experience of the surveillance, epidemiology, and end results (SEER) program. J. Natl. Cancer Inst. 1985, 74, 291–297. [Google Scholar]
- Hance, K.W.; Anderson, W.F.; Devesa, S.S.; Young, H.A.; Levine, P.H. Trends in inflammatory breast carcinoma incidence and survival: The surveillance, epidemiology, and end results program at the National Cancer Institute. J. Natl. Cancer Inst. 2005, 97, 966–975. [Google Scholar] [CrossRef]
- Anderson, W.F.; Schairer, C.; Chen, B.E.; Hance, K.W.; Levine, P.H. Epidemiology of inflammatory breast cancer (IBC). Breast Dis. 2005, 22, 9–23. [Google Scholar] [CrossRef] [Green Version]
- Wingo, P.A.; Jamison, P.M.; Young, J.L.; Gargiullo, P. Population-based statistics for women diagnosed with inflammatory breast cancer (United States). Cancer Causes Control 2004, 15, 321–328. [Google Scholar] [CrossRef] [Green Version]
- Bertucci, F.; Finetti, P.; Birnbaum, D.; Viens, P. Gene expression profiling of inflammatory breast cancer. Cancer 2010, 116, 2783–2793. [Google Scholar] [CrossRef]
- Cakar, B.; Surmeli, Z.; Oner, P.G.; Yelim, E.S.; Karabulut, B.; Uslu, R. The Impact of Subtype Distribution in Inflammatory Breast Cancer Outcome. Eur. J. Breast Health 2018, 14, 211–217. [Google Scholar] [CrossRef]
- Bertucci, F.; Finetti, P.; Vermeulen, P.; Van Dam, P.; Dirix, L.; Birnbaum, D.; Viens, P.; Van Laere, S. Genomic profiling of inflammatory breast cancer: A review. Breast 2014, 23, 538–545. [Google Scholar] [CrossRef]
- Bertucci, F.; Rypens, C.; Finetti, P.; Guille, A.; Adelaide, J.; Monneur, A.; Carbuccia, N.; Garnier, S.; Dirix, P.; Goncalves, A.; et al. NOTCH and DNA repair pathways are more frequently targeted by genomic alterations in inflammatory than in non-inflammatory breast cancers. Mol. Oncol. 2020, 14, 504–519. [Google Scholar] [CrossRef] [Green Version]
- Bertucci, F.; Ueno, N.T.; Finetti, P.; Vermeulen, P.; Lucci, A.; Robertson, F.M.; Marsan, M.; Iwamoto, T.; Krishnamurthy, S.; Masuda, H.; et al. Gene expression profiles of inflammatory breast cancer: Correlation with response to neoadjuvant chemotherapy and metastasis-free survival. Ann. Oncol. 2014, 25, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Masuda, H.; Baggerly, K.A.; Wang, Y.; Iwamoto, T.; Brewer, T.; Pusztai, L.; Kai, K.; Kogawa, T.; Finetti, P.; Birnbaum, D.; et al. Comparison of molecular subtype distribution in triple-negative inflammatory and non-inflammatory breast cancers. Breast Cancer Res. 2013, 15, R112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faldoni, F.L.C.; Villacis, R.A.R.; Canto, L.M.; Fonseca-Alves, C.E.; Cury, S.S.; Larsen, S.J.; Aagaard, M.M.; Souza, C.P.; Scapulatempo-Neto, C.; Osorio, C.; et al. Inflammatory Breast Cancer: Clinical Implications of Genomic Alterations and Mutational Profiling. Cancers 2020, 12, 2816. [Google Scholar] [CrossRef] [PubMed]
- Moslehi, R.; Freedman, E.; Zeinomar, N.; Veneroso, C.; Levine, P.H. Importance of hereditary and selected environmental risk factors in the etiology of inflammatory breast cancer: A case-comparison study. BMC Cancer 2016, 16, 334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawood, S.; Merajver, S.D.; Viens, P.; Vermeulen, P.B.; Swain, S.M.; Buchholz, T.A.; Dirix, L.Y.; Levine, P.H.; Lucci, A.; Krishnamurthy, S.; et al. International expert panel on inflammatory breast cancer: Consensus statement for standardized diagnosis and treatment. Ann. Oncol. 2011, 22, 515–523. [Google Scholar] [CrossRef]
- Fouad, T.M.; Barrera, A.M.G.; Reuben, J.M.; Lucci, A.; Woodward, W.A.; Stauder, M.C.; Lim, B.; DeSnyder, S.M.; Arun, B.; Gildy, B.; et al. Inflammatory breast cancer: A proposed conceptual shift in the UICC-AJCC TNM staging system. Lancet Oncol. 2017, 18, e228–e232. [Google Scholar] [CrossRef]
- Fouad, T.M.; Kogawa, T.; Liu, D.D.; Shen, Y.; Masuda, H.; El-Zein, R.; Woodward, W.A.; Chavez-MacGregor, M.; Alvarez, R.H.; Arun, B.; et al. Overall survival differences between patients with inflammatory and noninflammatory breast cancer presenting with distant metastasis at diagnosis. Breast Cancer Res. Treat. 2015, 152, 407–416. [Google Scholar] [CrossRef] [Green Version]
- Ueno, N.T.; Espinosa Fernandez, J.R.; Cristofanilli, M.; Overmoyer, B.; Rea, D.; Berdichevski, F.; El-Shinawi, M.; Bellon, J.; Le-Petross, H.T.; Lucci, A.; et al. International Consensus on the Clinical Management of Inflammatory Breast Cancer from the Morgan Welch Inflammatory Breast Cancer Research Program 10th Anniversary Conference. J. Cancer 2018, 9, 1437–1447. [Google Scholar] [CrossRef]
- Arias-Pulido, H.; Cimino-Mathews, A.; Chaher, N.; Qualls, C.; Joste, N.; Colpaert, C.; Marotti, J.D.; Foisey, M.; Prossnitz, E.R.; Emens, L.A.; et al. The combined presence of CD20+ B cells and PD-L1+ tumor-infiltrating lymphocytes in inflammatory breast cancer is prognostic of improved patient outcome. Breast Cancer Res. Treat. 2018, 171, 273–282. [Google Scholar] [CrossRef]
- Bertucci, F.; Finetti, P.; Colpaert, C.; Mamessier, E.; Parizel, M.; Dirix, L.; Viens, P.; Birnbaum, D.; van Laere, S. PDL1 expression in inflammatory breast cancer is frequent and predicts for the pathological response to chemotherapy. Oncotarget 2015, 6, 13506–13519. [Google Scholar] [CrossRef] [Green Version]
- Hamm, C.A.; Moran, D.; Rao, K.; Trusk, P.B.; Pry, K.; Sausen, M.; Jones, S.; Velculescu, V.E.; Cristofanilli, M.; Bacus, S. Genomic and Immunological Tumor Profiling Identifies Targetable Pathways and Extensive CD8+/PDL1+ Immune Infiltration in Inflammatory Breast Cancer Tumors. Mol. Cancer Ther. 2016, 15, 1746–1756. [Google Scholar] [CrossRef] [Green Version]
- Van Berckelaer, C.; Rypens, C.; van Dam, P.; Pouillon, L.; Parizel, M.; Schats, K.A.; Kockx, M.; Tjalma, W.A.A.; Vermeulen, P.; van Laere, S.; et al. Infiltrating stromal immune cells in inflammatory breast cancer are associated with an improved outcome and increased PD-L1 expression. Breast Cancer Res. 2019, 21, 28. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, S.V.; MacFarlane, A.W.T.; Jillab, M.; Arisi, M.F.; Yearley, J.; Annamalai, L.; Gong, Y.; Cai, K.Q.; Alpaugh, R.K.; Cristofanilli, M.; et al. Immune phenotype of patients with stage IV metastatic inflammatory breast cancer. Breast Cancer Res. 2020, 22, 134. [Google Scholar] [CrossRef] [PubMed]
- Winn, J.S.; Hasse, Z.; Slifker, M.; Pei, J.; Arisi-Fernandez, S.M.; Talarchek, J.N.; Obeid, E.; Baldwin, D.A.; Gong, Y.; Ross, E.; et al. Genetic Variants Detected Using Cell-Free DNA from Blood and Tumor Samples in Patients with Inflammatory Breast Cancer. Int. J. Mol. Sci. 2020, 21, 1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breast Cancer Association Consortium. Breast Cancer Risk Genes—Association Analysis in More than 113,000 Women. N. Engl. J. Med. 2021, 384, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Win, A.K.; Dowty, J.G.; Cleary, S.P.; Kim, H.; Buchanan, D.D.; Young, J.P.; Clendenning, M.; Rosty, C.; MacInnis, R.J.; Giles, G.G.; et al. Risk of colorectal cancer for carriers of mutations in MUTYH, with and without a family history of cancer. Gastroenterology 2014, 146, 1208–1211.e5. [Google Scholar] [CrossRef] [Green Version]
- Wasielewski, M.; Out, A.A.; Vermeulen, J.; Nielsen, M.; van den Ouweland, A.; Tops, C.M.; Wijnen, J.T.; Vasen, H.F.; Weiss, M.M.; Klijn, J.G.; et al. Increased MUTYH mutation frequency among Dutch families with breast cancer and colorectal cancer. Breast Cancer Res. Treat. 2010, 124, 635–641. [Google Scholar] [CrossRef]
- Rennert, G.; Lejbkowicz, F.; Cohen, I.; Pinchev, M.; Rennert, H.S.; Barnett-Griness, O. MutYH mutation carriers have increased breast cancer risk. Cancer 2012, 118, 1989–1993. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Goncalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Rana, H.Q.; Sacca, R.; Drogan, C.; Gutierrez, S.; Schlosnagle, E.; Regan, M.M.; Speare, V.; LaDuca, H.; Dolinsky, J.; Garber, J.E.; et al. Prevalence of germline variants in inflammatory breast cancer. Cancer 2019, 125, 2194–2202. [Google Scholar] [CrossRef]
- Daly, M.B.; Pilarski, R.; Berry, M.; Buys, S.S.; Farmer, M.; Friedman, S.; Garber, J.E.; Kauff, N.D.; Khan, S.; Klein, C.; et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast and Ovarian, Version 2.2017. J. Natl. Compr. Cancer Netw. 2017, 15, 9–20. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef] [Green Version]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Bialkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar] [CrossRef] [Green Version]
- Buisson, R.; Dion-Cote, A.M.; Coulombe, Y.; Launay, H.; Cai, H.; Stasiak, A.Z.; Stasiak, A.; Xia, B.; Masson, J.Y. Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat. Struct. Mol. Biol. 2010, 17, 1247–1254. [Google Scholar] [CrossRef] [Green Version]
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.N.H.; Harrell, M.I.; Kuiper, M.J.; Ho, G.Y.; Barker, H.; Jasin, M.; et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2017, 7, 984–998. [Google Scholar] [CrossRef] [Green Version]
- Calderaro, J.; Petitprez, F.; Becht, E.; Laurent, A.; Hirsch, T.Z.; Rousseau, B.; Luciani, A.; Amaddeo, G.; Derman, J.; Charpy, C.; et al. Intra-tumoral tertiary lymphoid structures are associated with a low risk of early recurrence of hepatocellular carcinoma. J. Hepatol. 2019, 70, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Zitvogel, L.; Sautes-Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef]
- Borst, J.; Ahrends, T.; Babala, N.; Melief, C.J.M.; Kastenmuller, W. CD4+ T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2018, 18, 635–647. [Google Scholar] [CrossRef]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef]
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spranger, S.; Spaapen, R.M.; Zha, Y.; Williams, J.; Meng, Y.; Ha, T.T.; Gajewski, T.F. Up-regulation of PD-L1, IDO, and Tregs in the melanoma tumor microenvironment is driven by CD8+ T cells. Sci. Transl. Med. 2013, 5, 200ra116. [Google Scholar] [CrossRef] [Green Version]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [Green Version]
- Veneris, J.T.; Matulonis, U.A.; Liu, J.F.; Konstantinopoulos, P.A. Choosing wisely: Selecting PARP inhibitor combinations to promote anti-tumor immune responses beyond BRCA mutations. Gynecol. Oncol. 2020, 156, 488–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domchek, S.M.; Postel-Vinay, S.; Im, S.A.; Park, Y.H.; Delord, J.P.; Italiano, A.; Alexandre, J.; You, B.; Bastian, S.; Krebs, M.G.; et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): An open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020, 21, 1155–1164. [Google Scholar] [CrossRef]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- R Foundation for Statistical Computing. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org/ (accessed on 1 August 2020).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | IBC Subtype | Age | Race | OS (months) | Gene | Variant Description | Protein Variant | Function |
|---|---|---|---|---|---|---|---|---|
| TN IBC | ||||||||
| Pt-1 | TN | 47 | W | 27 | PALB2 | c.1317delG | F440fs*12 | Loss |
| Pt-2 | TN | 55 | W | 38 | MSH2 | c.1724A>G | D575G | Unknown |
| Pt-3 | TN | 46 | W | 17 | MUTYH | c.1145G>A | G382D | Loss |
| Pt-4 | TN | 43 | W (AJ) | 42 | BRCA1 | c.68_69delAG | E23fs*17 | Loss |
| Pt-5 | TN | 61 | W | 31 | ESR1 | c.805C>T | R269C | Loss |
| Pt-6 | TN | 44 | W | 19 | BRCA2 | c.7976G>A | R2659K | Loss |
| Pt-7 | TN | 48 | W | 27 | KMT2C | c.10432C>G | Q3478E | Loss |
| Pt-8 | TN | 32 | W | 49 (2) | BARD1 | c.2300_2301delTG | V767fs*4 | Loss |
| Pt-9 | TN | 43 | W | 51 | BRCA2 | c.3975_3978dupTGCT | A1327fs*4 | Loss |
| Pt-10 | TN | 45 | AA | 15 | None | |||
| Pt-11 | TN | 57 | W | 31 | RAD51C | c.428A>G | Q143R | Loss |
| AR | c.2395C>G | Q799E | Loss | |||||
| Pt-12 | TN (Her2+ at month 26) | 44 | W | 34 | None | |||
| Pt-13 | TN | 42 | W | 24 | FANCC | c.823T>C | F275L | Loss |
| Pt-14 | TN | 50 | AA | 34 | None | |||
| Pt-15 | TN | 43 | W | 17 | None | |||
| Pt-16 | TN | 43 | AA | 44 | None | |||
| Pt-17 | TN | 48 | AA | 50 | None | |||
| Pt-18 | TN | 61 | W | 32 | None | |||
| Non-TN IBC | ||||||||
| Pt-19 | ER+ Her2− | 34 | W | 76 | ErbB2 | c.2689C>T | R897W | Loss |
| Pt-20 | ER+ Her2− (TN at month 8) | 66 | W | 10 | None | |||
| Pt-21 | ER+ Her2− | 39 | W | 103 (1) | None | |||
| Pt-22 | ER+ Her2− | 44 | W | 50 | None | |||
| Pt-23 | ER+ Her2− | 53 | AA | 6 | BRCA1 | c.5123C>T | A1708V | Loss |
| Pt-24 | ER+ Her2− | 50 | W | 41 (1) | ATM | c.6095G>A | R2032K | Loss |
| APC | c.3386T>C | L1129S | Loss | |||||
| Pt-25 | ER+ Her2− | 49 | W | 40 | CHEK2 | c.573+1G>A Splicing | Loss | |
| Pt-26 | ER+ Her2− | 57 | W | 57 (1) | None | |||
| Pt-27 | ER+ Her2− | 45 | W | 39 | None | |||
| Pt-28 | ER+ Her2+ (ER+ Her2− at month 9) | 56 | W | 21 | BRCA2 | c.9976A>T | K3326* | Loss |
| MSH2 | c.965G>A | G322D | Loss | |||||
| Pt-29 | ER+ Her2+ (ER− Her2+ at month 34) | 32 | W | 44 | † ATM | c.4324T>C | Y1442H | Loss |
| †CASP8 | c.152-1_155delGATTA | Loss | ||||||
| Pt-30 | ER+ Her2+ (ER+ Her2− at month 49) | 37 | H | 56 | None | |||
| Pt-31 | ER− Her2+ | 69 | Asian | 37 | † IRAK4 | c.529A>G | T177A | Loss |
| Pt-32 | ER− Her2+ | 37 | Asian | 59 | † NCOR1 | c.6641G>A | R2214H | Loss |
| Patient ID | IBC Subtype | Tumor Tissue Sample | Total Lymphocytes | CD3 | CD4+ | CD8+ | CD20 | CD68 | FOXP3+ | PD-L1 | PD-1 | OS |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| % | % | % | % | % | % | % | (% ITC positive) | (% ITC positive) | months | |||
| Pt-1 | TN | punch biopsy | 10 | 5 | 5 | 1 | 0 | 5 | ˂1 | ˂1 | ˂1 | 27 |
| Pt-2 | TN | punch biopsy | 20 | 10 | 20 | 5 | 0 | 30 | 5 | ˂1 | ˂1 | 38 |
| Pt-3 | TN | chest wall, punch biopsy | 10 | 10 | 20 | 15 | 5 | 25 | 10 | 10 | 1 | 17 |
| Pt-5 | TN | skin, chest wall | 50 | 30 | 25 | 15 | 5 | 10 | 10 | 1 | 5 | 31 |
| Pt-6 | TN | skin, punch biopsy | 5 | 5 | 10 | 5 | 0 | 10 | 1 | ˂1 | ˂1 | 19 |
| Pt-7 | TN | skin, punch biopsy | 5 | 5 | 3 | 5 | 0 | 5 | 3 | ˂1 | ˂1 | 27 |
| Pt-9 | TN | skin, punch biopsy | 40 | 20 | 15 | 10 | 3 | 25 | 15 | ˂1 | ˂1 | 51 |
| Pt-11 | TN | skin, punch biopsy | 5 | 2 | 5 | 1 | 0 | 10 | 5 | ˂1 | ˂1 | 31 |
| Pt-12 | TN | skin, punch biopsy | 10 | 10 | 10 | 5 | 0 | 30 | 5 | ˂1 | ˂1 | 34 |
| Pt-14 | TN | breast tissue | 10 | 10 | 15 | 15 | 2 | 10 | 15 | 10 | 10 | 34 |
| Pt-15 | TN | skin, punch biopsy | 20 | 15 | 20 | 5 | ˂1 | 15 | 3 | 1 | 1 | 17 |
| Pt-17 | TN | skin, punch biopsy | 15 | 15 | 10 | 10 | 0 | 20 | 5 | ˂1 | ˂1 | 50 |
| Pt-18 | TN | skin, punch biopsy | 10 | 5 | 10 | 10 | 0 | 5 | 3 | ˂1 | ˂1 | 32 |
| Pt-19 | ER+ Her2− | skin, biopsy | 5 | 1 | 10 | 1 | 0 | 2 | ˂1 | ˂1 | ˂1 | 76 |
| Pt-20 | ER+ Her2− (TN at month 8) | skin, punch biopsy | 10 | 10 | 15 | 5 | ˂1 | 15 | 2 | ˂1 | ˂ 1 | 10 |
| Pt-24 | ER+ Her2− | breast core biopsy | 5 | 5 | 10 | 5 | 0 | 15 | 1 | ˂1 | ˂1 | 41 * |
| Pt-28 | ER+ Her2+ (Her2− at month 9) | skin, punch biopsy | 10 | 10 | 15 | 10 | 0 | 10 | 5 | ˂1 | ˂1 | 21 |
| Pt-30 | ER+ Her2+ (Her2− at month 49) | skin, punch biopsy | 5 | 5 | 15 | 5 | 0 | 5 | 1 | ˂1 | ˂1 | 56 |
| Pt-31 | ER− Her2+ | skin, punch biopsy | 20 | 10 | 10 | 10 | 5 | 25 | 5 | 1 | 1 | 37 |
| Pt-32 | ER− Her2+ | skin, punch biopsy | 50 | 20 | 50 | 50 | 5 | 5 | 5 | 1 | 1 | 59 |
| Pt-33 | ER− Her2+ | skin, punch biopsy | 30 | 15 | 20 | 10 | 10 | 20 | 10 | 1 | 5 | 16 |
| Pt-34 | ER− Her2+ | liver | 30 | 20 | 30 | 10 | 1 | 20 | 5 | ˂1 | ˂1 | 127 * |
| Pt-35 | TN/history of melanoma | skin, punch biopsy | 50 | 30 | 50 | 10 | 0 | 15 | 5 | ˂1 | ˂1 | 47 |
| Pt-36 | ER− Her2+ | skin, punch biopsy | 10 | 20 | 15 | 10 | 0 | 20 | 5 | ˂1 | ˂1 | 76 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, Y.; Nagarathinam, R.; Arisi, M.F.; Gerratana, L.; Winn, J.S.; Slifker, M.; Pei, J.; Cai, K.Q.; Hasse, Z.; Obeid, E.; et al. Genetic Variants and Tumor Immune Microenvironment: Clues for Targeted Therapies in Inflammatory Breast Cancer (IBC). Int. J. Mol. Sci. 2021, 22, 8924. https://doi.org/10.3390/ijms22168924
Gong Y, Nagarathinam R, Arisi MF, Gerratana L, Winn JS, Slifker M, Pei J, Cai KQ, Hasse Z, Obeid E, et al. Genetic Variants and Tumor Immune Microenvironment: Clues for Targeted Therapies in Inflammatory Breast Cancer (IBC). International Journal of Molecular Sciences. 2021; 22(16):8924. https://doi.org/10.3390/ijms22168924
Chicago/Turabian StyleGong, Yulan, Rajeswari Nagarathinam, Maria F. Arisi, Lorenzo Gerratana, Jennifer S. Winn, Michael Slifker, Jianming Pei, Kathy Q. Cai, Zachary Hasse, Elias Obeid, and et al. 2021. "Genetic Variants and Tumor Immune Microenvironment: Clues for Targeted Therapies in Inflammatory Breast Cancer (IBC)" International Journal of Molecular Sciences 22, no. 16: 8924. https://doi.org/10.3390/ijms22168924
APA StyleGong, Y., Nagarathinam, R., Arisi, M. F., Gerratana, L., Winn, J. S., Slifker, M., Pei, J., Cai, K. Q., Hasse, Z., Obeid, E., Noriega, J., Sebastiano, C., Ross, E., Alpaugh, K., Cristofanilli, M., & Fernandez, S. V. (2021). Genetic Variants and Tumor Immune Microenvironment: Clues for Targeted Therapies in Inflammatory Breast Cancer (IBC). International Journal of Molecular Sciences, 22(16), 8924. https://doi.org/10.3390/ijms22168924