
Mitochondrial Dysfunction in Vascular Wall Cells and Its Role in Atherosclerosis

 ,
, 
 ,
, 
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Risk Factors of Atherosclerosis: Oxidative Stress and LDL Modification
3. Mitochondrial Dysfunction and Inflammation
4. mtDNA Mutations in Atherosclerosis and Related Pathologies
5. The Role of Mitochondria in Different Cell Types
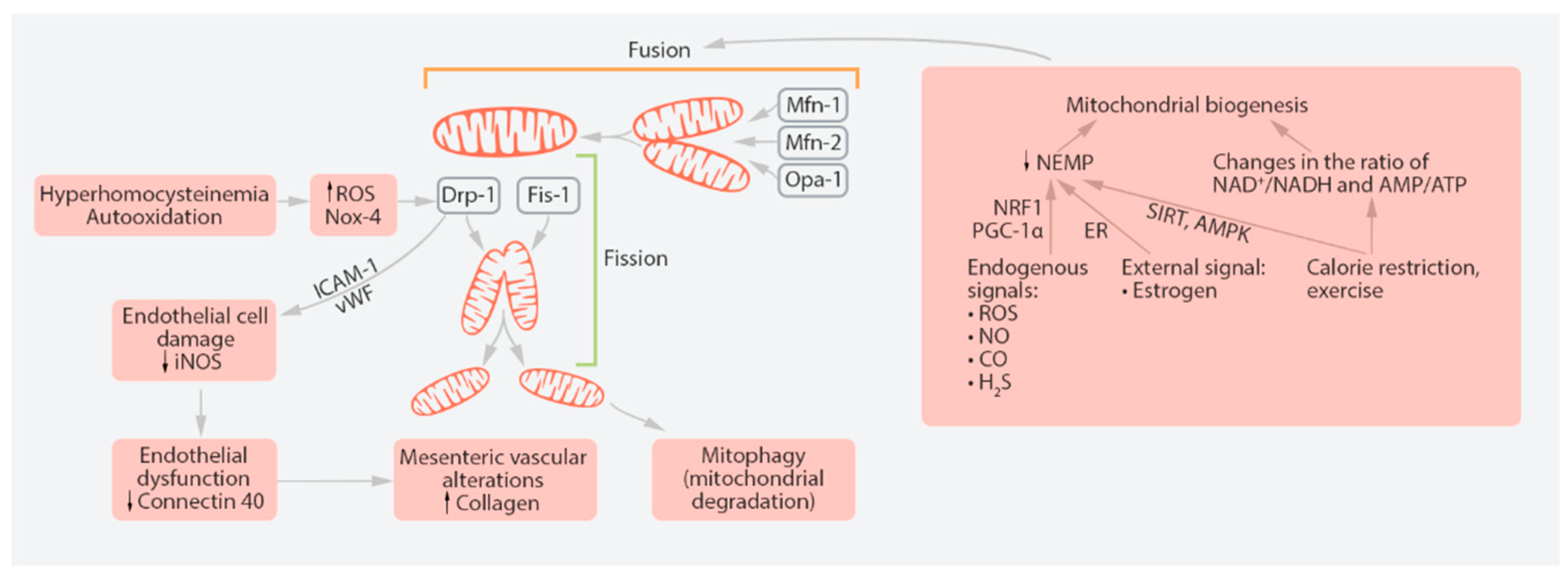
5.1. Endothelial Cells
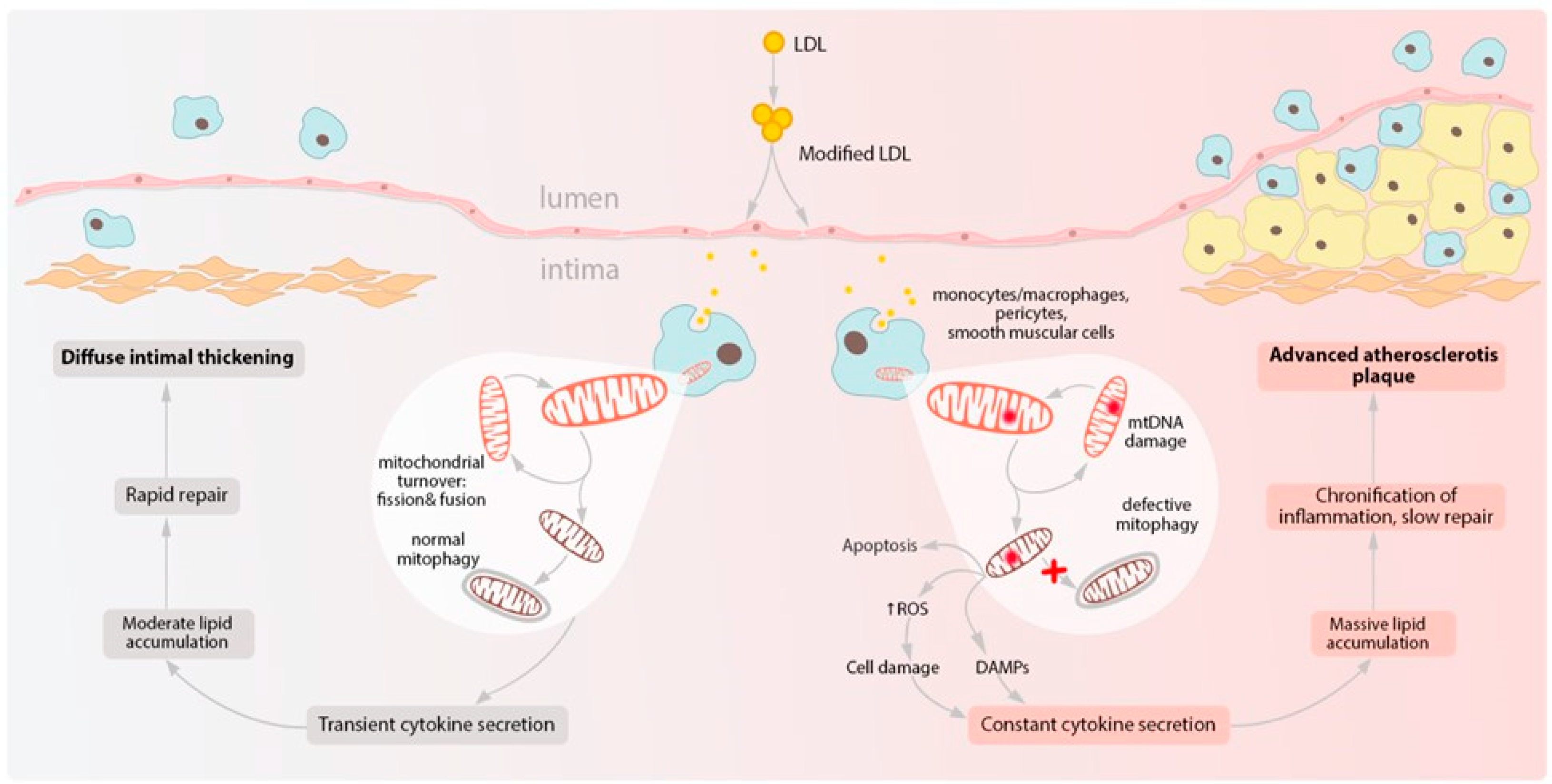
5.2. Macrophages
5.3. Pericytes and Vascular Smooth Muscle Cells
6. The Mitochondrion as a Potential Therapeutic Target
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Colín-Castelán, D.; Zaina, S. Associations between atherosclerosis and neurological diseases, beyond ischemia-induced cerebral damage. Rev. Endocr. Metab. Disord. 2019, 20, 15–25. [Google Scholar] [CrossRef]
- Skilton, M.R.; Celermajer, D.S.; Cosmi, E.; Crispi, F.; Gidding, S.S.; Raitakari, O.T.; Urbina, E.M. Natural History of Atherosclerosis and Abdominal Aortic Intima-Media Thickness: Rationale, Evidence, and Best Practice for Detection of Atherosclerosis in the Young. J. Clin. Med. 2019, 8, 1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiripurasundari, R.; Sreekumari, K.; Aravindan, K.P. Autopsy-based morphometric study of coronary atherosclerosis in young adults. Indian J. Med. Res. 2019, 150, 592–597. [Google Scholar] [PubMed]
- Ledard, N.; Liboz, A.; Blondeau, B.; Babiak, M.; Moulin, C.; Vallin, B.; Guillas, I.; Mateo, V.; Jumeau, C.; Blirando, K.; et al. Slug, a Cancer-Related Transcription Factor, is Involved in Vascular Smooth Muscle Cell Transdifferentiation Induced by Platelet-Derived Growth Factor-BB During Atherosclerosis. J. Am. Heart Assoc. 2020, 9, e014276. [Google Scholar] [CrossRef] [PubMed]
- Balzan, S.; Lubrano, V. LOX-1 receptor: A potential link in atherosclerosis and cancer. Life Sci. 2018, 198, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Sazonova, M.A.; Sinyov, V.V.; Barinova, V.A.; Ryzhkova, A.I.; Zhelankin, A.V.; Postnov, A.Y.; Sobenin, I.A.; Bobryshev, Y.V.; Orekhov, A.N. Mosaicism of Mitochondrial Genetic Variation in Atherosclerotic Lesions of the Human Aorta. Biomed. Res. Int. 2015, 2015, 825468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orekhov, A.N.; Nikiforov, N.N.; Ivanova, E.A.; Sobenin, I.A. Possible Role of Mitochondrial DNA Mutations in Chronification of Inflammation: Focus on Atherosclerosis. J. Clin. Med. 2020, 9, 978. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C. Mitochondrial DNA mutations in disease and aging. Environ. Mol. Mutagenes. 2010, 51, 440–450. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maassen, J.A.; van Essen, E.; van den Ouweland, J.M.; Lemkes, H.H. Molecular and clinical aspects of mitochondrial diabetes mellitus. Exp. Clin. Endocrinol. Diabetes 2001, 109, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.; Leu, J. The Red Queen in mitochondria: Cyto-nuclear co-evolution, hybrid breakdoxn and human disease. Front. Genet. 2015, 6, 187. [Google Scholar] [CrossRef] [Green Version]
- Ziada, A.S.; Lu, M.Y.; Ignas-Menzies, J.; Paintsil, E.; Li, M.; Ogbuagu, O.; Saberi, S.; Hsieh, A.Y.Y.; Sattha, B.; Harrigan, P.R.; et al. Mitochondrial DNA somatic mutation burden and heteroplasmy are associated with chronological age, smoking, and HIV infection. Aging Cell 2019, 18, e13018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Shen, L.; Hu, P.; Huang, R.; Cao, Y.; Deng, J.; Yuan, W.; Liu, D.; Yang, J.; Gu, H.; et al. Aging-associated mitochondrial DNA mutations alter oxidative phosphorylation machinery and cause mitochondrial dysfunctions. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2266–2273. [Google Scholar] [CrossRef] [PubMed]
- Orekhov, A.N.; Poznyak, A.V.; Sobenin, I.A.; Nikifirov, N.N.; Ivanova, E.A. Mitochondrion as a selective target for treatment of atherosclerosis: Role of mitochondrial DNA mutations and defective mitophagy in the pathogenesis of atherosclerosis and chronic inflammation. Curr. Neuropharmacol. 2020, 18, 1064–1075. [Google Scholar] [CrossRef] [PubMed]
- Marchio, P.; Guerra-Ojeda, S.; Vila, J.M.; Aldasoro, M.; Victor, V.M.; Mauricio, M.D. Targeting Early Atherosclerosis: A Focus on Oxidative Stress and Inflammation. Oxid. Med. Cell. Longev. 2019, 2019, 8563845. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.P.; Bennett, M.R. Mitochondrial DNA damage and atherosclerosis. Trends Endocrinol. Metab. 2014, 25, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Sentman, M.L.; Brännström, T.; Westerlund, S.; Laukkanen, M.O.; Ylä-Herttuala, S.; Basu, S.; Marklund, S.L. Extracellular superoxide dismutase deficiency and atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1477–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Yu, S.; Park, H. Rice bran oil ameliorates inflammatory responses by enhancing mito-chondrial respiration in murine macrophages. PLoS ONE 2019, 14, e0222857. [Google Scholar]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar]
- Schrepfer, E.; Scorrano, L. Mitofusins, from Mitochondria to Metabolism. Mol. Cell 2016, 61, 683–694. [Google Scholar] [CrossRef] [Green Version]
- Bertholet, A.; Delerue, T.; Millet, A. Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neuobiol Dis. 2016, 90, 3–19. [Google Scholar] [CrossRef]
- Yamashita, S.; Kanki, T. How autophagy eats large mitochondria: Autophagosome formation coupled with mitochondrial fragmentation. Autophagy 2017, 13, 980–981. [Google Scholar] [CrossRef] [Green Version]
- Heo, J.M.; Ordureau, A.; Swarup, S.; Paulo, J.A.; Shen, K.; Sabatini, D.M.; Harper, J.W. RAB7A phosphorylation by TBK1 promotes mitophagy via the PINK-PARKIN pathway. Sci. Adv. 2018, 4, eaav0443. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Wang, C.; Jin, Y.; Ma, X.; Xie, T.; Wang, J.; Liu, K.; Sun, H. Disocin prevents postmeno-pausal atherosclerosis in ovariectomized LDLR−/− mice through a PGC-1α/ERα pathway leading to promotion of autophagy and inhibition of oxidative stress, inflammation and apoptosis. Pharmacol. Res. 2019, 148, 104414. [Google Scholar] [CrossRef]
- Peng, X.; Chen, H.; Li, Y.; Huang, D.; Huang, B.; Sun, D. Effects of NIX-mediated mitophagy on ox-LDL-induced macrophage pyroptosis in atherosclerosis. Cell Biol. Int. 2020, 44, 1481–1490. [Google Scholar] [CrossRef]
- Forte, M.; Schirone, L.; Ameri, P.; Basso, C.; Catalucci, D.; Modica, J.; Chimenti, C.; Crotti, L.; Fra-ti, G.; Rubattu, S.; et al. The role of mitochondrial dynamics in cardiovascular diseases. Br. J. Pharmacol. 2021, 178, 2060–2076. [Google Scholar] [CrossRef] [PubMed]
- Gkikas, I.; Palikaras, K.; Tavernarakis, N. The role of mitophagy in innate immunity. Front. Immunol. 2018, 9, 1283. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg effect: Essential part of metabolic reprogramming and central contributor to cancer progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef] [PubMed]
- De Preter, G.; Neveu, M.; Danhier, P.; Brisson, L.; Payen, V.L.; Porporato, P.E.; Jordan, B.F.; Sonveaux, P.; Gallez, B. Inhibition of the pentose phosphate pathway by dichloroacetate unravels a missing link between aerobic glycolysis and cancer cell proliferation. Oncotarget 2016, 7, 2910–2920. [Google Scholar] [CrossRef] [Green Version]
- Movahed, Z.G.; Yarani, R.; Mohammadi, P.; Mansouri, K. Sustained oxidative stress instigates differentiation of cancer stem cells into tumor endothelial cells: Pentose phosphate pathway, reactive oxygen species and autophagy crosstalk. Biomed. Pharmacother. 2021, 139, 111643. [Google Scholar] [CrossRef] [PubMed]
- Kaczanowski, S.; Klim, J.; Zielenkiewicz, U. An Apoptotic and Endosymbiotic Explanation of the Warburg and the Inverse Warburg Hypotheses. Int. J. Mol. Sci. 2018, 19, 3100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S. Calcium, ATP, and ROS: A mito-chondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Szabadkai, G.; Duchen, M.R. Mitochondria: The hub of cellular Ca2+ signaling. Physiology (Bethesda) 2008, 23, 84–94. [Google Scholar] [CrossRef] [Green Version]
- Orekhov, A.N.; Sobenin, I.A. Modified and Dysfunctional Lipoproteins in Atherosclerosis: Effectors or Biomarkers? Curr. Med. Chem. 2019, 26, 1512–1524. [Google Scholar] [CrossRef] [PubMed]
- Tertov, V.V.; Sobenin, I.A.; Kaplun, V.V.; Orekhov, A.N. Antioxidant content in low density lip-oprotein and lipoprotein oxidation in vivo and in vitro. Free Radic. Res. 1998, 29, 165–173. [Google Scholar] [CrossRef]
- Lopes-Virella, M.F.; Virella, G. Pathogenic role of modified LDL antibodies and immune complexes in atherosclerosis. J. Atherscler. Thromb. 2013, 20, 743–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Li, Y.; Peng, X.; Huang, D.; Gui, L.; Huang, B. Resistance of mitochondrial DNA-depleted cells against oxidized low-density lipoprotein-induced macrophage pyroptosis. Mol. Med. Rep. 2016, 13, 4393–4399. [Google Scholar] [CrossRef] [Green Version]
- Maguire, E.M.; Pearce, S.W.A.; Xiao, Q. Foam cell formation: A new target for fighting athero-sclerosis and cardiovascular disease. Vascul. Pharmacol. 2019, 112, 54–71. [Google Scholar] [CrossRef]
- Ouimet, M.; Barrett, T.J.; Fisher, E.A. HDL and Reverse Cholesterol Transport. Circ. Res. 2019, 124, 1505–1518. [Google Scholar] [CrossRef] [PubMed]
- Gaudreault, N.; Kumar, N.; Posada, J.M.; Stephens, K.B.; de Mochel, N.S.R.; Eberlé, D.; Olivas, V.R.; Kim, R.Y.; Harms, M.J.; Johnson, S.; et al. ApoE suppresses atherosclerosis by reducing lipid accumulation in circulating monocytes and the expression of inflammatory molecules on monocytes and vascular endothelium. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 264–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gistera, A.; Ketelhuth, D.F. Lipid-driven immunometabolic responses in atherosclerosis. Curr. Opin. Lipidol. 2018, 29, 375–380. [Google Scholar] [CrossRef]
- Gencer, S.; Evans, B.R.; van der Vorst, E.P.C.; Doring, Y.; Weber, C. Inflammatory Chemokines in Atherosclerosis. Cells 2021, 10, 226. [Google Scholar] [CrossRef]
- Suarez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Alvarez-Cordoba, M.; Vil-lalon-Garcia, I.; Talaveron-Rey, M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Sanchez-Alcazar, J.A. From Mitochondria to Atherosclerosis: The Inflammation Path. Biomedicines 2021, 9, 258. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria for NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy pro-teins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misawa, T.; Takahama, M.; Kozaki, T.; Lee, H.; Zou, J.; Saitoh, T.; Akira, S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 2013, 14, 454–460. [Google Scholar] [CrossRef]
- Su, Y.; Wang, P.; Weng, S. The Role of Mitochondria in Immune-Cell-Mediated Tissue Regeneration and Ageing. Int. J. Mol. Sci. 2021, 22, 2668. [Google Scholar] [CrossRef] [PubMed]
- Coppi, L.; Ligorio, S.; Mitro, N.; Caruso, D.; De Fabiani, E.; Crestani, M. PGC-1s and Beyond: Disentangling the Complex Regulation of Mitochondrial and Cellular Metabolism. Int. J. Mol. Sci. 2021, 22, 6913. [Google Scholar] [CrossRef] [PubMed]
- Kadlec, A.O.; Chabowski, D.S.; Ait-Aissa, K.; Gutterman, D.D. Role of PGC-1α in Vascular Regulation: Implications for Atherosclerosis. Atherioscler. Thromb. Vasc. Biol. 2016, 36, 1467–1474. [Google Scholar] [CrossRef] [Green Version]
- Mishra, S.R.; Mahapatra, K.K.; Behera, B.P.; Patra, S.; Bhol, C.S.; Panigrahi, D.P.; Praharaj, P.P.; Singh, A.; Patil, S.; Dhiman, R.; et al. Mitochondrial dysfunction as a driver of NLRP3 inflammasome activation and its modulation through mitophagy for potential therapeutics. Int. J. Biochem. Cell Biol. 2021, 136, 106013. [Google Scholar] [CrossRef] [PubMed]
- Swiader, A.; Nahapetyan, H.; Faccini, J.; D’Angelo, R.; Mucher, E.; Elbaz, M.; Boya, P.; Vindis, C. Mitophagy acts as a safeguard mechanism against human vascular smooth muscle cell apoptosis induced by atherogenic lipids. Oncotarget 2016, 7, 28821–28835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nahapetyan, H.; Moulis, M.; Grousset, E.; Faccini, J.; Grazide, M.; Mucher, E.; Elbaz, M.; Martinet, W.; Vindis, C. Altered mitochondrial quality control in Atg7-deficient VSMCs promotes enhanced apoptosis and is linked to unstable atherosclerotic plaque phenotype. Cell Death Dis. 2019, 10, 119. [Google Scholar] [CrossRef] [Green Version]
- Markin, A.M.; Sobenin, I.A.; Grechko, A.V.; Zhang, D.; Orekhov, A.N. Cellular Mechanisms of Human Atherogenesis: Focus on Chronification of Inflammation and Mitochondrial Mutations. Front. Pharmacol. 2020, 11, 642. [Google Scholar] [CrossRef] [PubMed]
- Dabravolski, S.A.; Orekhova, V.A.; Baig, M.S.; Bezsonov, E.E.; Starodubova, A.V.; Popkova, T.V.; Orekhov, A.N. The Role of Mitochondrial Murtations and Chronic Inflammation in Diabetes. Int. J. Mol. Sci. 2021, 22, 6733. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.F. Why chloroplasts and mitochondria retain their own genomes and genetic systems: Colocation for redox regulation of gene expression. Proc. Natl. Acad. Sci. USA 2015, 112, 10231–10238. [Google Scholar] [CrossRef] [Green Version]
- Jia, Z.; Zhang, Y.; Li, Q.; Ye, Z.; Liu, Y.; Fu, C.; Cang, X.; Wang, M.; Guan, M.X. A coronary artery disease-associated tRNAThr mutation altered mitochondrial function, apoptosis and angiogenesis. Nucleic Acids Res. 2019, 47, 2056–2074. [Google Scholar] [CrossRef] [PubMed]
- Fukui, H.; Moraes, C.T. Mechanisms of formation and accumulation of mitochondrial DNA deletions in aging neurons. Hum. Mol. Genet. 2009, 18, 1028–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermulst, M.; Wanagat, J.; Kujoth, G.C.; Bielas, J.H.; Rabinovitch, P.S.; Prolla, T.A.; Loeb, L.A. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat. Genet. 2008, 40, 392–394. [Google Scholar] [CrossRef]
- Larsson, N.G. Somatic mitochondrial DNA mutations in mammalian aging. Annu. Rev. Biochem. 2010, 79, 683–706. [Google Scholar] [CrossRef] [PubMed]
- Orekhov, A.N.; Zhelankin, A.V.; Kolmychkova, K.I.; Mitrofanov, K.Y.; Kubekina, M.V.; Ivanova, E.A.; Sobenin, I.A. Susceptibility of monocytes to activation correlates with atherogenic mitochondrial DNA mutations. Exp. Mol. Pathol. 2015, 99, 672–676. [Google Scholar] [CrossRef]
- Orogo, A.M.; Gonzalez, E.R.; Kubli, D.A.; Baptista, I.L.; Ong, S.B.; Prolla, T.A.; Sussman, M.A.; Murphy, A.N.; Gustafsson, Å.B. Accumulation of Mitochondrial DNA Mutations Disrupts Cardi-ac Progenitor Cell Function and Reduces Survival. J. Biol. Chem. 2015, 290, 22061–22075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilkerson, R.W.; De Vries, R.L.; Lebot, P.; Wikstrom, J.D.; Torgyekes, E.; Shirihai, O.S.; Przedborski, S.; Schon, E.A. Mitochondrial autophagy in cells with mtDNA mutations results from synergistic loss of transmembrane potential and mTORC1 inhibition. Hum. Mol. Genet. 2012, 21, 978–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granatiero, V.; Giorgio, V.; Calì, T.; Patron, M.; Brini, M.; Bernardi, P.; Tiranti, V.; Zeviani, M.; Pallafacchina, G.; De Stefani, D.; et al. Reduced mitochondrial Ca(2+) transients stimulate autophagy in human fibroblasts carrying the 13514A>G mutation of the ND5 subunit of NADH dehydrogenase. Cell Death Differ. 2016, 23, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Gimbrone, M.A.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Li, Y.S.; Chien, S. Shear stress-initiated signaling and its regulation of endothelial function. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2191–2198. [Google Scholar] [CrossRef] [Green Version]
- Chistiakov, D.A.; Kashirskikh, D.A.; Khotina, V.A.; Grechko, A.V.; Orekhov, A.N. Immune-Inflammatory Responses in Atherosclerosis: The Role of Myeloid Cells. J. Clin. Med. 2019, 8, 1798. [Google Scholar] [CrossRef] [Green Version]
- Romanov, Y.A.; Balyasnikova, I.V.; Bystrevskaya, V.B.; Byzova, T.V.; Ilyinskaya, O.P.; Krushinsky, A.V.; Latsis, R.V.; Soboleva, E.; Yoon, Y.; Robotham, J.L.; et al. Endothelial heterogeneity and intimal blood-borne cells. Relation to human atherosclerosis. Ann. N. Y. Acad. Sci. 1995, 748, 12–37. [Google Scholar] [CrossRef]
- Lip, G.Y.; Blann, A. Von Willebrand factor. In Meyler’s Side Effects of Drugs; Elsevier B.V.: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Zhao, J.; Chen, H.; Liu, N.; Chen, J.; Gu, Y.; Chen, J.; Yang, K. Role of Hyperhomocysteinemia and Hyperuricemia in Pathogenesis of Atherosclerosis. J. Stroke Cerebrovasc. Dis. 2017, 26, 2695–2699. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Shi, F.; Tan, Z. Mitochondrial network structure homeostasis and cell death. Cancer Sci. 2018, 109, 3686–3694. [Google Scholar] [CrossRef]
- Rovira-Llopis, S.; Bañuls, C.; Diaz-Morales, N.; Hernandez-Mijares, A.; Rocha, M.; Victor, V.M. Mitochondrial dynamics in type 2 diabetes: Pathophysiological implications. Redox Biol. 2017, 11, 637–645. [Google Scholar] [CrossRef]
- Alevriadou, B.R.; Shanmughapriya, S.; Patel, A.; Stathopulos, P.B.; Madesh, M. Mitochondrial Ca2+ transport in the endothelium: Regulation by ions, redox signalling and mechanical forces. J. R. Soc. Interface 2017, 17, 20140672. [Google Scholar]
- Li, X.; Wu, G.; Han, F.; Wang, K.; Bai, X.; Jia, Y.; Li, Z.; Cai, W.; Zhang, W.; Su, L.; et al. SIRT1 activation promotes angiogenesis in diabetic wounds by protecting endothelial cells against oxidative stress. Arch. Biochem. Biophys. 2019, 661, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.; Zhang, J.; Ren, Y.; Du, Z.; Li, Q.; Wang, Y.; Wei, S.; Yang, L.; Zhang, J.; Liu, C.; et al. Irisin alleviates liver ischemia-reperfusion injury by inhibiting excessive mitochondrial fission, promoting mitochondrial biogenesis and decreasing oxidative stress. Redox Biol. 2019, 20, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Akira, S. Toll-Like Receptor Signaling and Its Inducible Proteins. Microbiol. Spectr. 2016, 4, 4–6. [Google Scholar] [CrossRef]
- Dela Cruz, C.; Kang, M. Mitochondrial dysfunction and damage associated molecular patterns (DAMPs) in chronic inflammatory diseases. Mitochondrion 2018, 41, 37–44. [Google Scholar] [CrossRef]
- Walsh, M.C.; Lee, J.; Choi, Y. Tumor necrosis factor receptor- associated factor 6 (TRAF6) regulation of development, function, and homeostasis of the immune system. Immunol. Rev. 2015, 266, 72–92. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Hoyt, L.; Randall, M.; Ather, J. Mitochondrial ROS induced by chronic ethanol exposure promote hyper-activation of the NLRP3 inflammasome. Redox Biol. 2017, 12, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Liang, H.; Zen, K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef] [Green Version]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.C.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Gruning, N.; Kruger, A.; Alam, M.T.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 2015, 90, 927–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koelwyn, G.J.; Corr, E.M.; Erbay, E.; Moore, K.J. Regulation of macrophage immunometabolism in atherosclerosis. Nat. Immunol. 2018, 19, 526–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberale, L.; Dallegri, F.; Montecucco, F.; Carbone, F. Pathophysiological relevance of macrophage subsets in atherogenesis. Thromb. Haemost. 2017, 117, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Orekhov, A.N.; Bobryshev, Y.V.; Chistiakov, D.A. The complexity of cell composition of the intima of large arteries: Focus on pericyte-like cells. Cardiovasc. Res. 2014, 103, 438–451. [Google Scholar] [CrossRef]
- Juchem, G.; Weiss, D.R.; Gansera, B.; Kemkes, B.M.; Mueller-Hoecker, J.; Nees, S. Pericytes in the macrovascular intima: Possible physiological and pathogenetic impact.love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar]
- Summerhill, V.; Orekhov, A. Pericytes in Atherosclerosis. Adv. Exp. Med. Biol. 2019, 1147, 279–297. [Google Scholar]
- Ivanova, E.A.; Bobryshev, Y.V.; Orekhov, A.N. Intimal pericytes as the second line of immune defence in atherosclerosis. World J. Cardiol. 2015, 7, 583–593. [Google Scholar] [CrossRef]
- Cao, Y.L.; Liu, D.J.; Zhang, H.G. MiR-7 regulates the PI3K/AKT/VEGF pathway of retinal capillary endothelial cell and retinal pericytes in diabetic rat model through IRS-1 and inhibits cell proliferation. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 4427–4430. [Google Scholar]
- Zheng, L.S.; Ishii, Y.; Zhao, Q.L.; Kondo, T.; Sasahara, M. PDGF suppresses oxidative stress induced Ca2+ overload and calpain activation in neurons. Oxid. Med. Cell. Longev. 2013, 2013, 367206. [Google Scholar] [CrossRef] [Green Version]
- Basatemur, G.L.; Jørgensen, H.F.; Clarke, M.C.H.; Bennett, M.R.; Mallat, Z. Vascular smooth muscle cells in atherosclerosis. Nat. Rev. Cardiol. 2019, 16, 727–744. [Google Scholar] [CrossRef]
- Martínez, M.S.; García, A.; Luzardo, E.; Chávez-Castillo, M.; Olivar, L.C.; Salazar, J.; Velasco, M.; Rojas Quintero, J.J.; Bermúdez, V. Energetic metabolism in cardiomyocytes: Molecular basis of heart ischemia and arrhythmogenesis. Vessel Plus 2017, 1, 130–141. [Google Scholar] [CrossRef]
- Fu, D.; Wu, M.; Zhang, J.; Du, M.; Yang, S.; Hammad, S.M.; Wilson, K.; Chen, J.; Lyons, T.J. Mechanisms of modified LDL-induced pericyte loss and retinal injury in diabetic retinopathy. Diabetologia 2012, 55, 3128–3140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollazadeh, H.; Tavana, E.; Fanni, G.; Bo, S.; Banach, M.; Pirro, M.; von Haehling, S.; Jamialahmadi, T.; Sahebkar, A. Effects of statins on mitochondrial pathways. J. Cahexia Sarcopenia Muscle 2021, 12, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Avram, V.F.; Chamkha, I.; Asander-Frostner, E.; Ehinger, J.; Timar, R.Z.; Hansson, M.J.; Muntean, D.M.; Elmer, E. Cell-Permeable Succinate Rescues Mitochondrial Respiration in Cellular Models of Statin Toxicity. Int. J. Mol. Sci. 2021, 22, 424. [Google Scholar] [CrossRef] [PubMed]
- Bharath, L.P.; Nikolajczyk, B.S. The intersection of metformin and inflammation. Am. J. Physiol. Cell Physiol. 2021, 320, C873–C879. [Google Scholar] [CrossRef]
- Wu, S.; Zou, M. AMPK, Mitochondrial Function, and Cardiovascular Disease. Int. J. Mol. Sci. 2020, 21, 4987. [Google Scholar] [CrossRef]
- Guan, Y.; Drake, J.C.; Yan, Z. Exercise-Induced Mitophagy in Skeletal Muscke and Heart. Exerc. Sport Sci. Rev. 2019, 47, 151–156. [Google Scholar] [CrossRef]
- Gureev, A.P.; Shaforostova, E.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1α Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef] [Green Version]
- Montaigne, D.; Butruille, L.; Dtaels, B. PPAR control of metabolism and cardiovascular functions. Nat. Rev. Cardiol. 2021, in press. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Sukhorukov, V.N.; Nikiforov, N.G.; Kubekina, M.V.; Sobenin, I.A.; Foxx, K.K.; Pintus, S.; Stegmaier, P.; Stelmashenko, D.; Kel, A.; et al. Signaling Pathways Potentially Responsible for Foam Cell Formation: Cholesterol Accumulation or Inflammatory Response-What is First? Int. J. Mol. Sci. 2020, 21, 2716. [Google Scholar] [CrossRef] [Green Version]
- Oyewole, A.O.; Birch-Machin, M.A. Mitochondria-targeted antioxidants. FASEB J. 2015, 29, 4766–4771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teixeira, J.; Deus, C.M.; Borges, F.; Oliveira, P.J. Mitochondria: Targeting mitochondrial reactive oxygen species with mitochondriotropic polyphenolic-based antioxidants. Int. J. Biochem. Cell Biol. 2018, 97, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Ben-Meir, A.; Burstein, E.; Borrego-Alvarez, A.; Chong, J.; Wong, E.; Yavorska, T.; Naranian, T.; Chi, M.; Wang, Y.; Bentov, Y.; et al. Coenzyme Q10 restores oocyte mitochondrial function and fertility during reproductive aging. Aging Cell 2015, 14, 887–895. [Google Scholar] [CrossRef]
- Blanco, L.P.; Pedersen, H.L.; Wang, X.; Lightfoot, Y.L.; Seto, N.; Carmona-Rivera, C.; Yu, Z.X.; Hoffmann, V.; Yuen, P.S.T.; Kaplan, M.J. Improved Mitochondrial Metabolism and Reduced Inflammation Following Attenuation of Murine Lupus with Coenzyme Q10 Analog Idebenone. Arthritis Rheumatol. 2020, 72, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Magwere, T.; West, M.; Riyahi, K.; Murphy, M.P.; Smith, R.A.; Partridge, L. The effects of exogenous antioxidants on lifespan and oxidative stress resistance in Drosophila melanogaster. Mech. Ageing Dev. 2006, 127, 356–370. [Google Scholar] [CrossRef]
- Braakhuis, A.J.; Nagulan, R.; Somerville, V. The Effect of MitoQ on Aging-Related Biomarkers: A Systematic Review and Meta-Analysis. Oxid. Med. Cell. Longev. 2018, 2018, 8575263. [Google Scholar] [CrossRef]
- Isaev, N.K.; Stelmashook, E.V.; Genrikhs, E.E.; Korshunova, G.A.; Sumbatyan, N.V.; Kapkaeva, M.R.; Skulachev, V.P. Neuroprotective properties of mitochondria-targeted antioxidants of the SkQ-type. Rev. Neurosci. 2016, 27, 849–855. [Google Scholar] [CrossRef]
- Wang, Q.; Xu, J.; Geng, R.; Cai, J.; Li, J.; Xie, C.; Tang, W.; Shen, Q.; Huang, W.; Fan, Q. High performance one-for-all phototheranostics: NIR-II fluorescence imaging guided mitochondria-targeting phototherapy with a single-dose injection and 808 nm laser irradiation. Biomaterials 2020, 231, 119671. [Google Scholar] [CrossRef]
- Su, X.; Chen, Y.; Wang, X.; Wang, Y.; Wang, P.; Li, L.; Liu, Q. PpIX induces mitochondria-related apoptosis in murine leukemia L1210 cells. Drug Chem. Toxicol. 2014, 37, 348–356. [Google Scholar] [CrossRef]
- Mahmoudi, K.; Garvey, K.L.; Bouras, A.; Cramer, G.; Stepp, H.; Jesu Raj, J.G.; Bozec, D.; Busch, T.M.; Hadjipanayis, C.G. 5-aminolevulinic acid photodynamic therapy for the treatment of high-grade gliomas. J. Neurooncol. 2019, 141, 595–607. [Google Scholar] [CrossRef]
- Palmeira, C.M.; Teodoro, J.S.; Amorim, J.A.; Steegborn, C.; Sinclair, D.A.; Rolo, A.P. Mitohormesis and metabolic health: The interplay between ROS, cAMP and sirtuins. Free Radic. Biol. Med. 2019, 141, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakovljevic, N.K.; Pavlovic, K.; Jotic, A.; Lalic, K.; Stoiljkovic, M.; Lukic, L.; Milicic, T.; Macesic, M.; Stanarcic Gajovic, J.; Lalic, N.M. Targeting Mitochondria in Diabetes. Int. J. Mol. Sci. 2021, 22, 6642. [Google Scholar] [CrossRef] [PubMed]
- Lambers, S.; Van Laethem, C.; Van Acker, K.; Calders, P. Influence of combined exercise training on indices of obesity, diabetes and cardiovascular risk in type 2 diabetes patients. Clin. Rehabil. 2008, 22, 483–492. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salnikova, D.; Orekhova, V.; Grechko, A.; Starodubova, A.; Bezsonov, E.; Popkova, T.; Orekhov, A. Mitochondrial Dysfunction in Vascular Wall Cells and Its Role in Atherosclerosis. Int. J. Mol. Sci. 2021, 22, 8990. https://doi.org/10.3390/ijms22168990
Salnikova D, Orekhova V, Grechko A, Starodubova A, Bezsonov E, Popkova T, Orekhov A. Mitochondrial Dysfunction in Vascular Wall Cells and Its Role in Atherosclerosis. International Journal of Molecular Sciences. 2021; 22(16):8990. https://doi.org/10.3390/ijms22168990
Chicago/Turabian StyleSalnikova, Diana, Varvara Orekhova, Andrey Grechko, Antonina Starodubova, Evgeny Bezsonov, Tatyana Popkova, and Alexander Orekhov. 2021. "Mitochondrial Dysfunction in Vascular Wall Cells and Its Role in Atherosclerosis" International Journal of Molecular Sciences 22, no. 16: 8990. https://doi.org/10.3390/ijms22168990
APA StyleSalnikova, D., Orekhova, V., Grechko, A., Starodubova, A., Bezsonov, E., Popkova, T., & Orekhov, A. (2021). Mitochondrial Dysfunction in Vascular Wall Cells and Its Role in Atherosclerosis. International Journal of Molecular Sciences, 22(16), 8990. https://doi.org/10.3390/ijms22168990