
Molecular Dynamics-Derived Pharmacophore Model Explaining the Nonselective Aspect of KV10.1 Pore Blockers





Abstract
:1. Introduction
2. Materials and Methods
2.1. Software
2.2. Homology Modeling
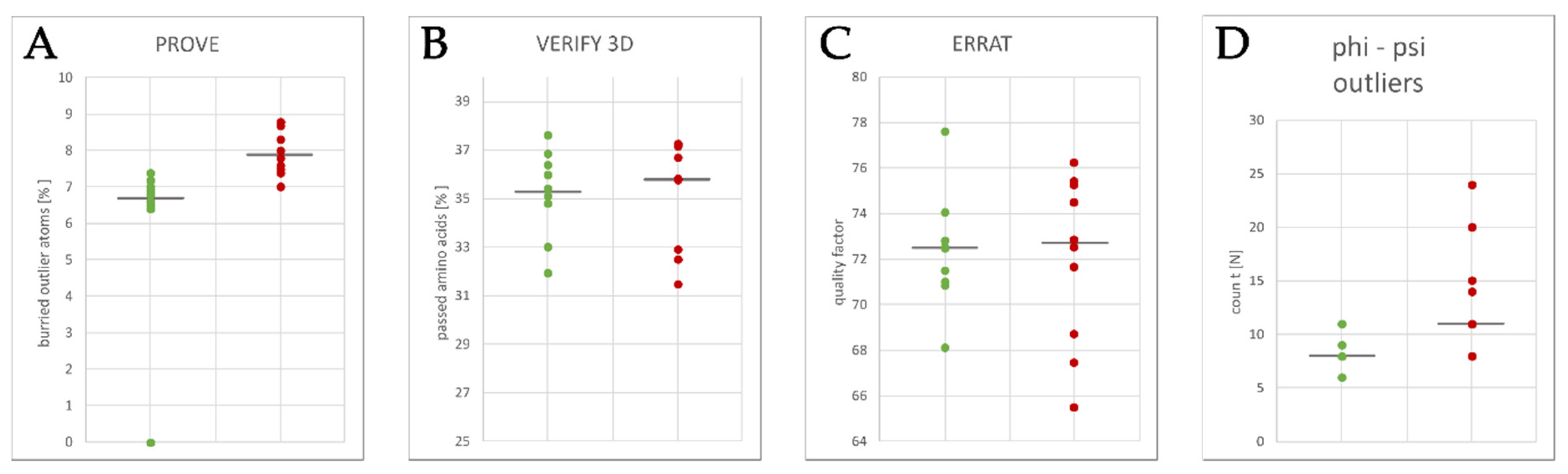
2.3. Homology Model Evaluation
2.4. Docking of Compounds
2.5. Molecular Dynamics Preparation and Simulation
2.6. Analysis of Molecular Dynamics Simulation
2.7. Pharmacophore Modeling
2.8. Virtual Library Preparation
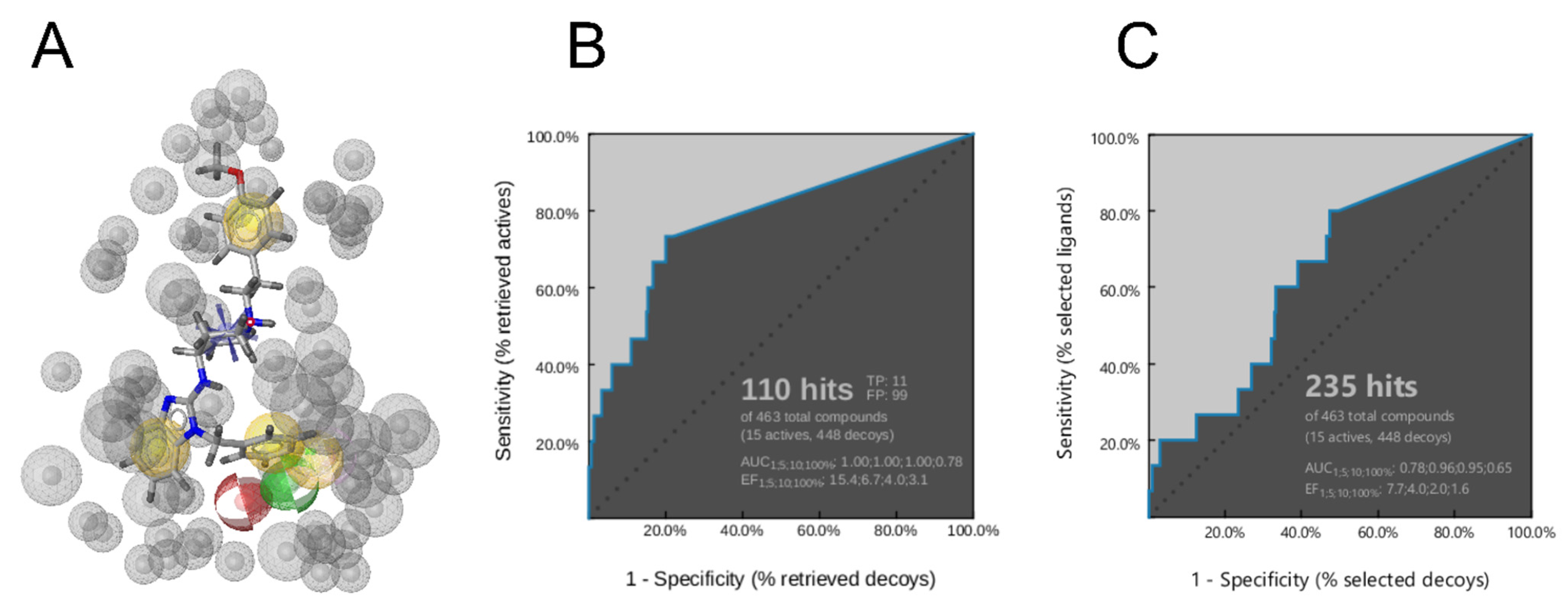
2.9. Virtual Screening
3. Results and Discussion
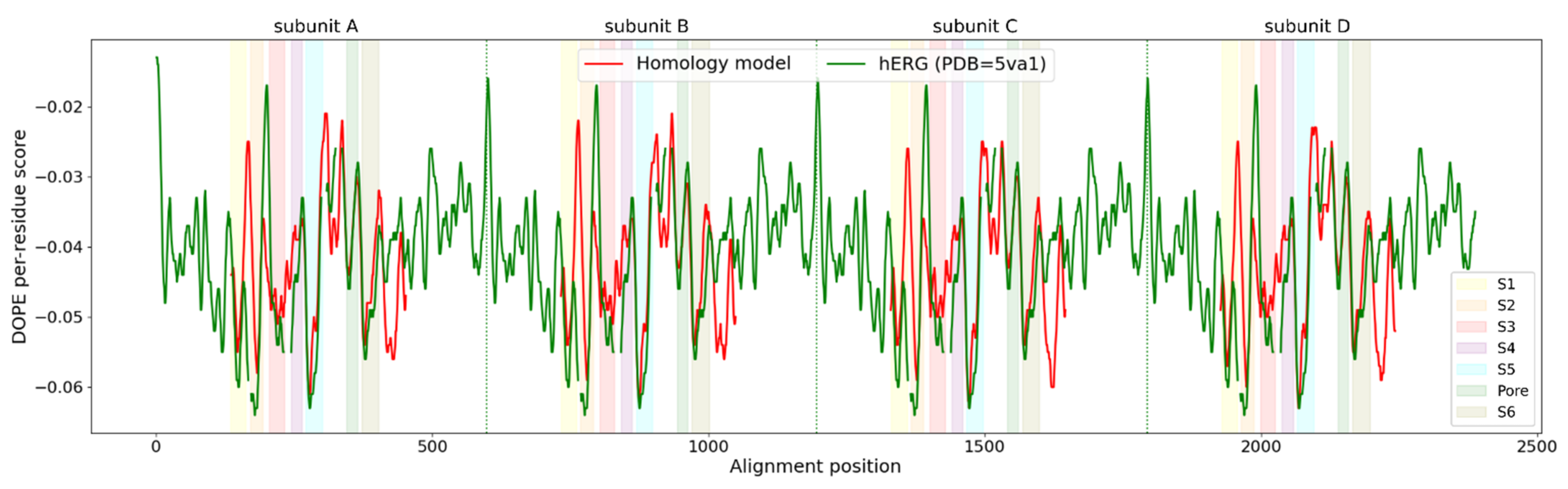
3.1. Homology Modeling of the KV10.1 Open Pore Conformation
3.2. Docking of KV10.1 Inhibitors for Binding to the Channel Pore
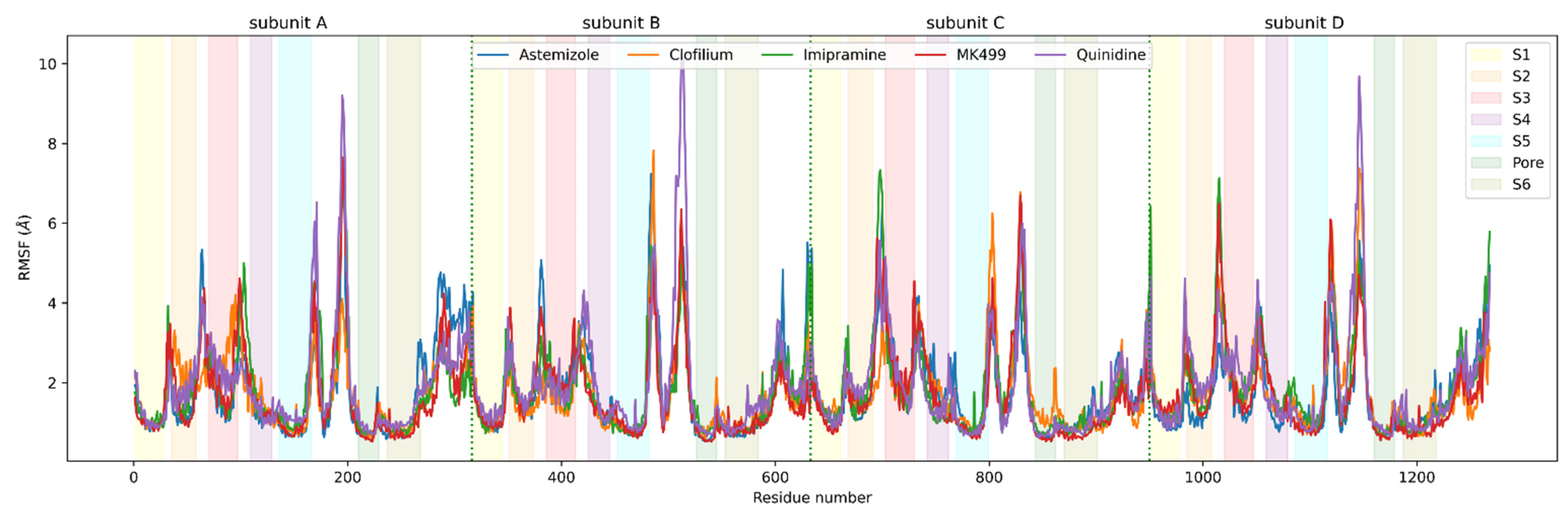
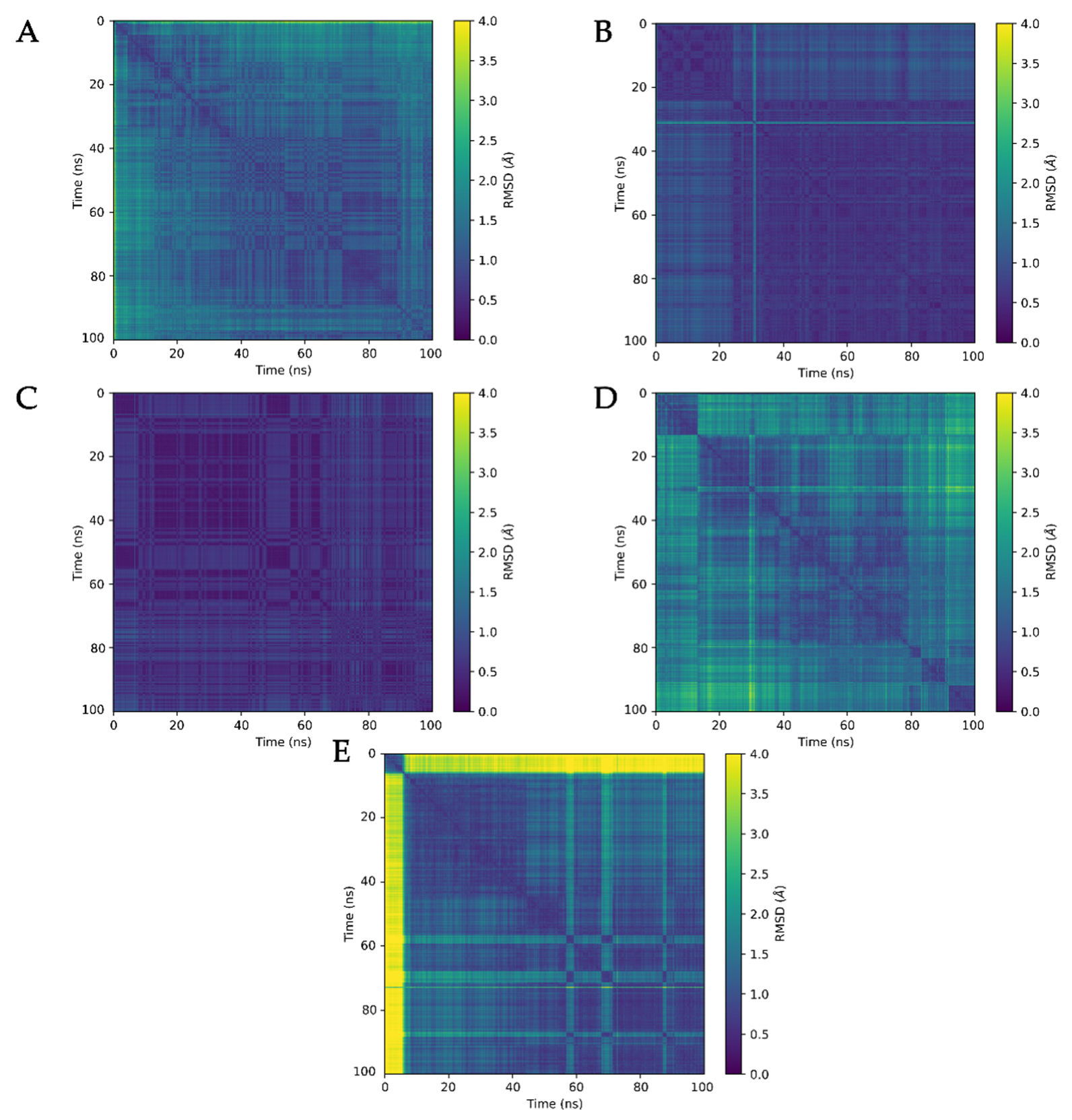
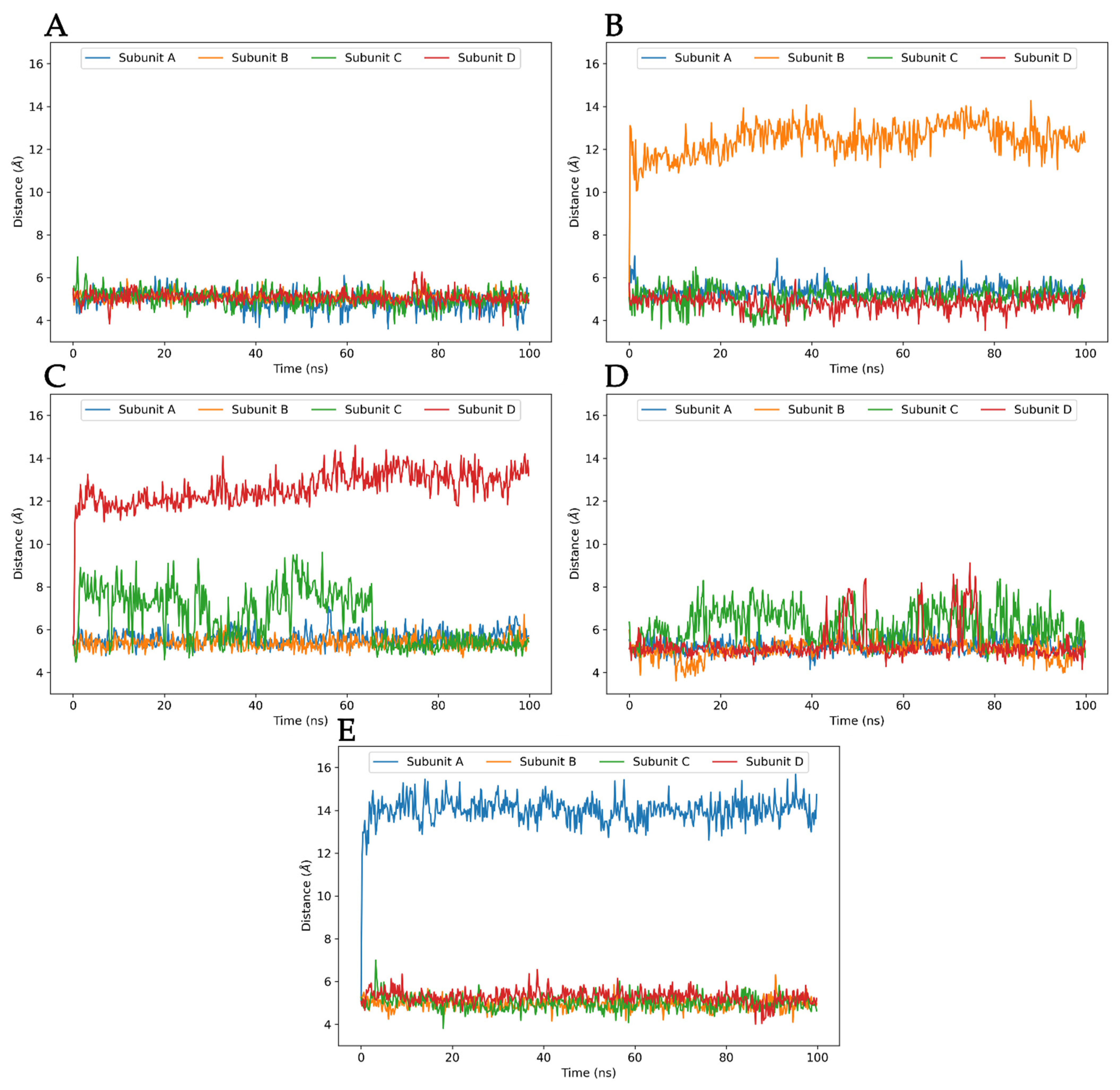
3.3. Molecular Dynamics Analysis of Ligand and Protein Stabilities
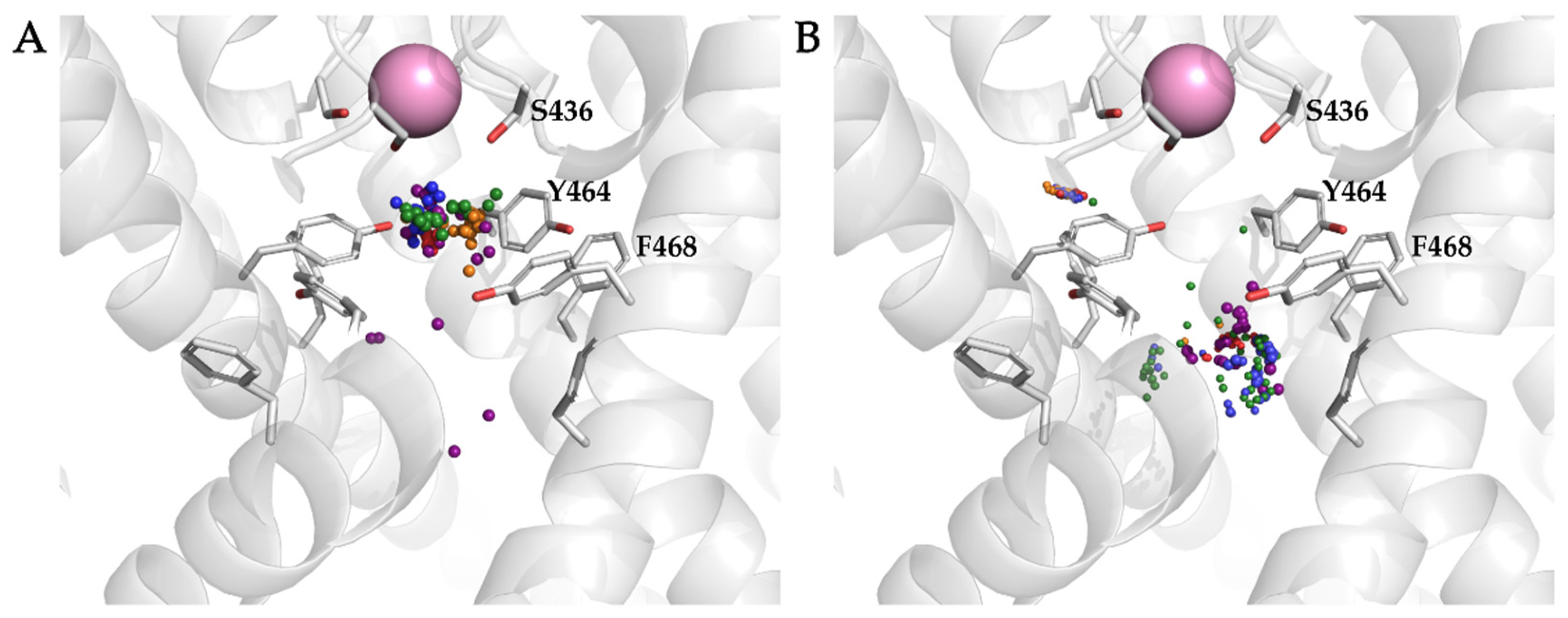
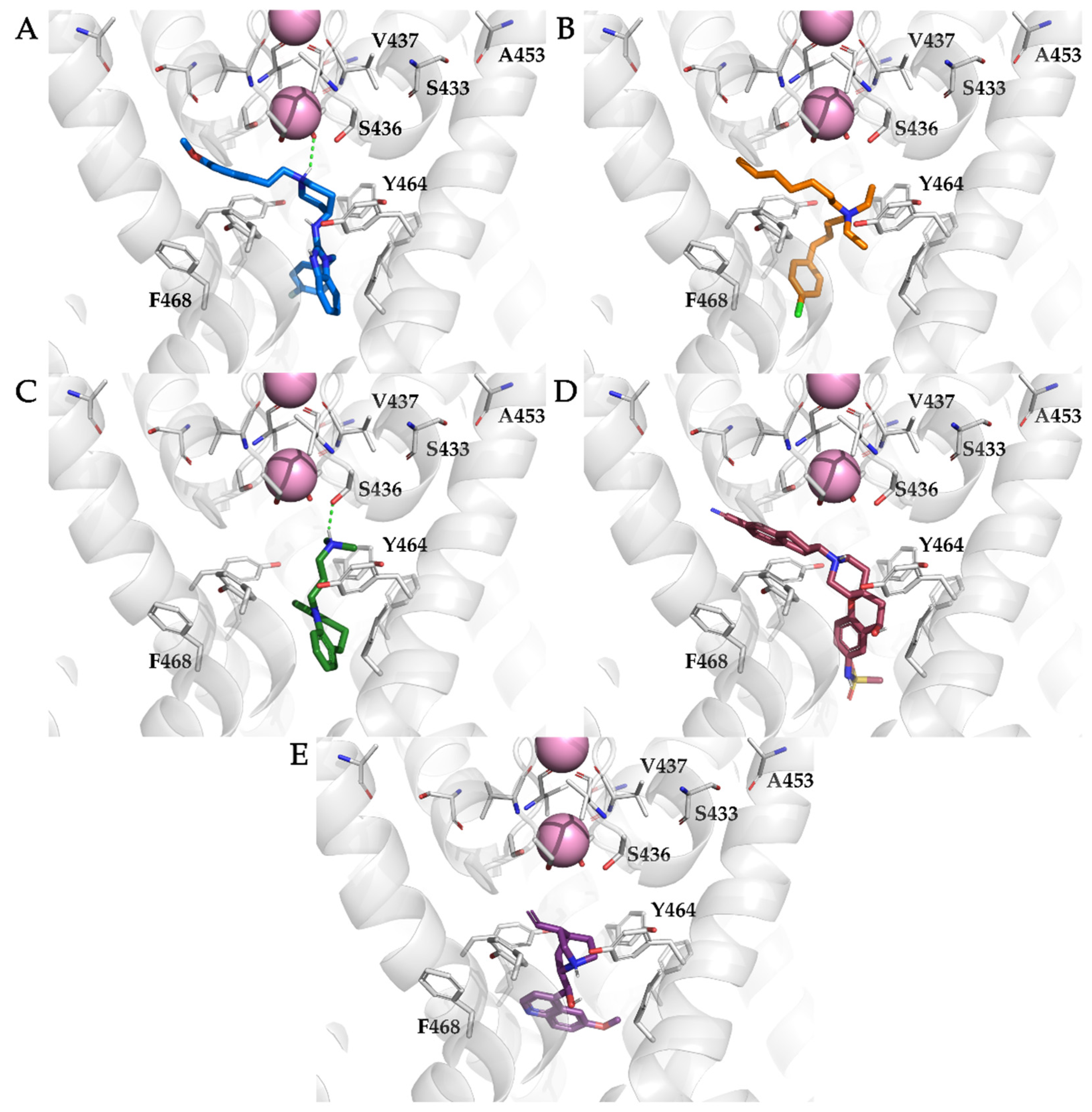
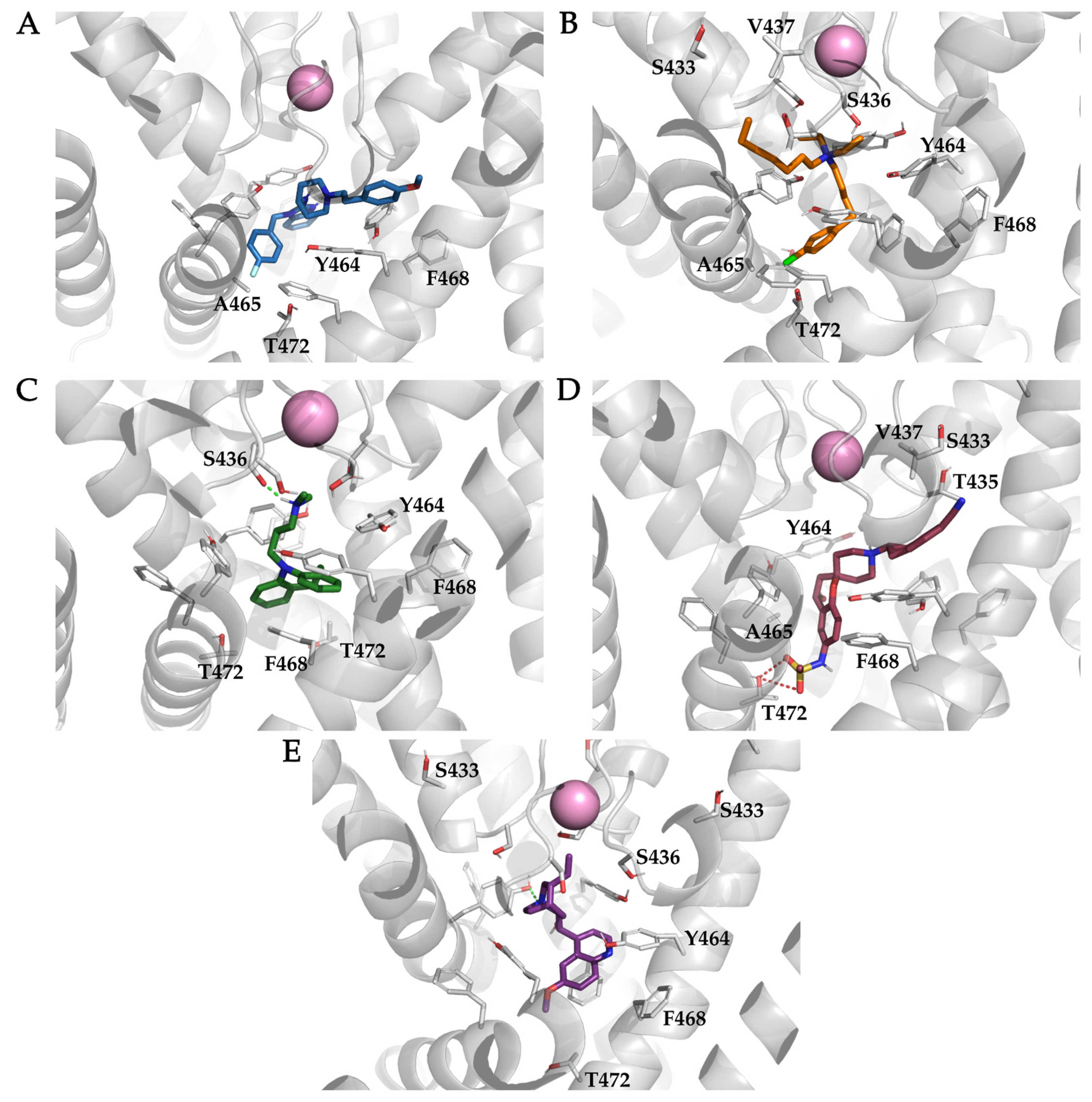
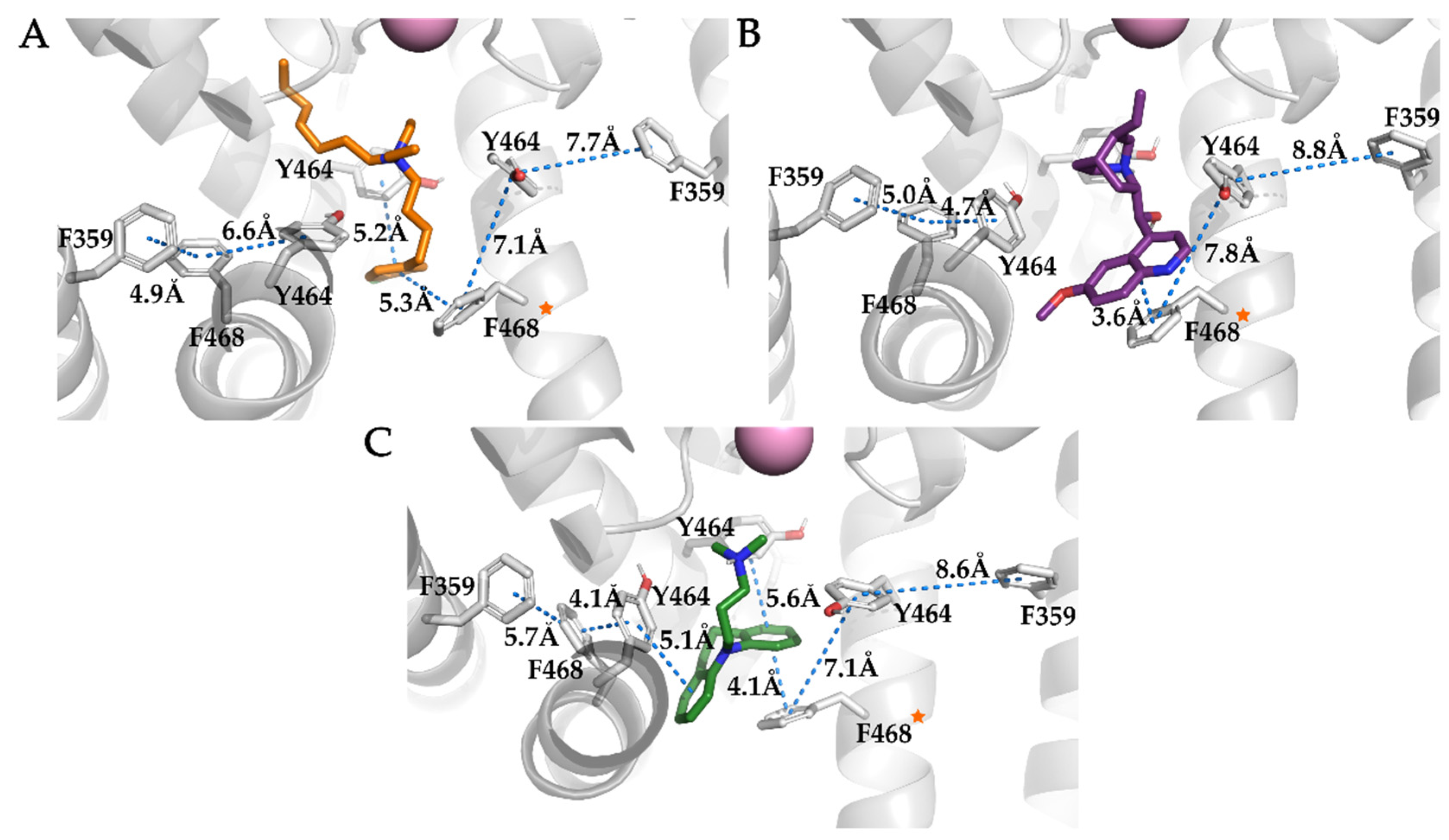
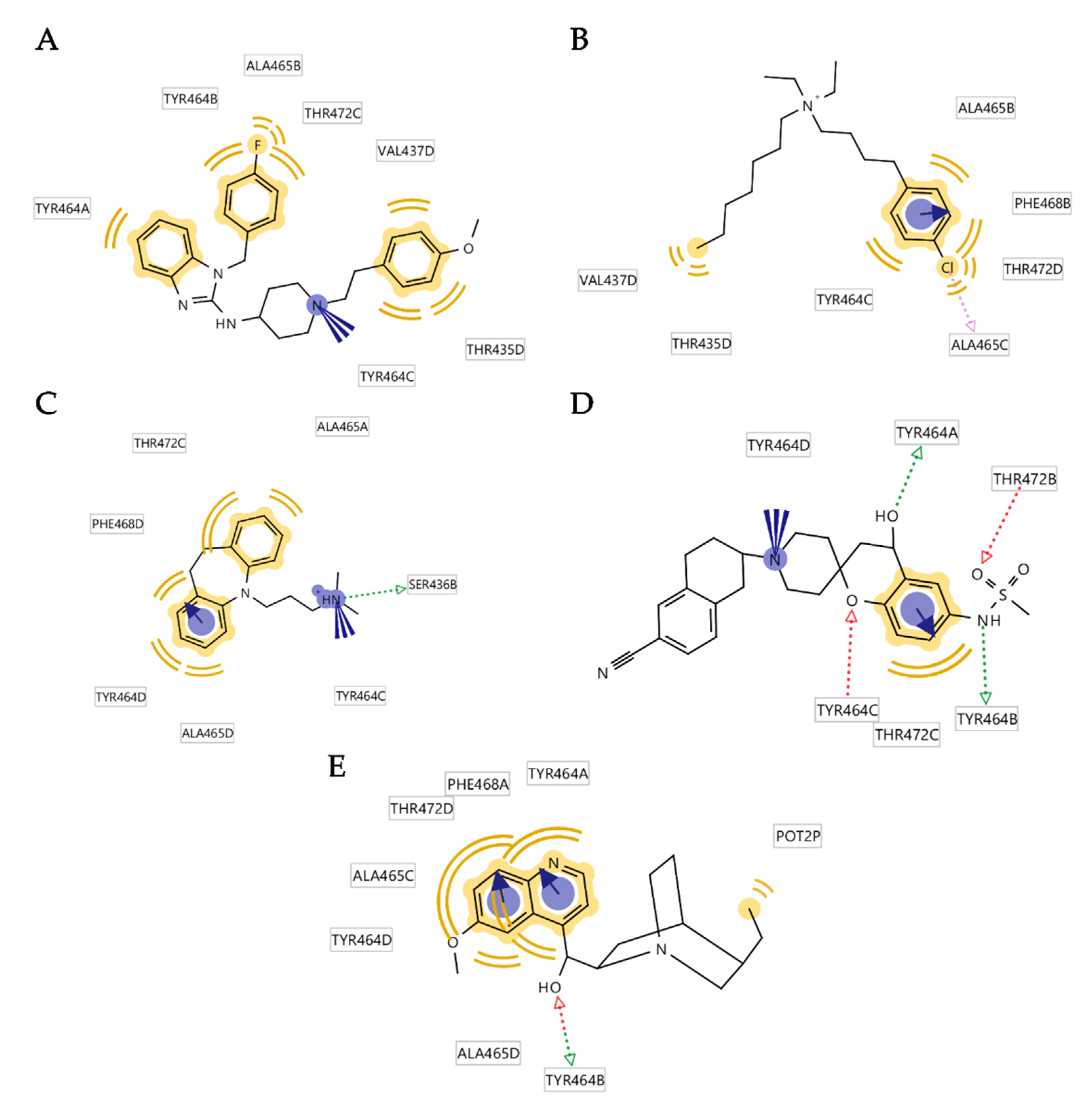
3.4. Analysis of Binding Interactions of KV10.1 Inhibitors in the Molecular Dynamics Simulations
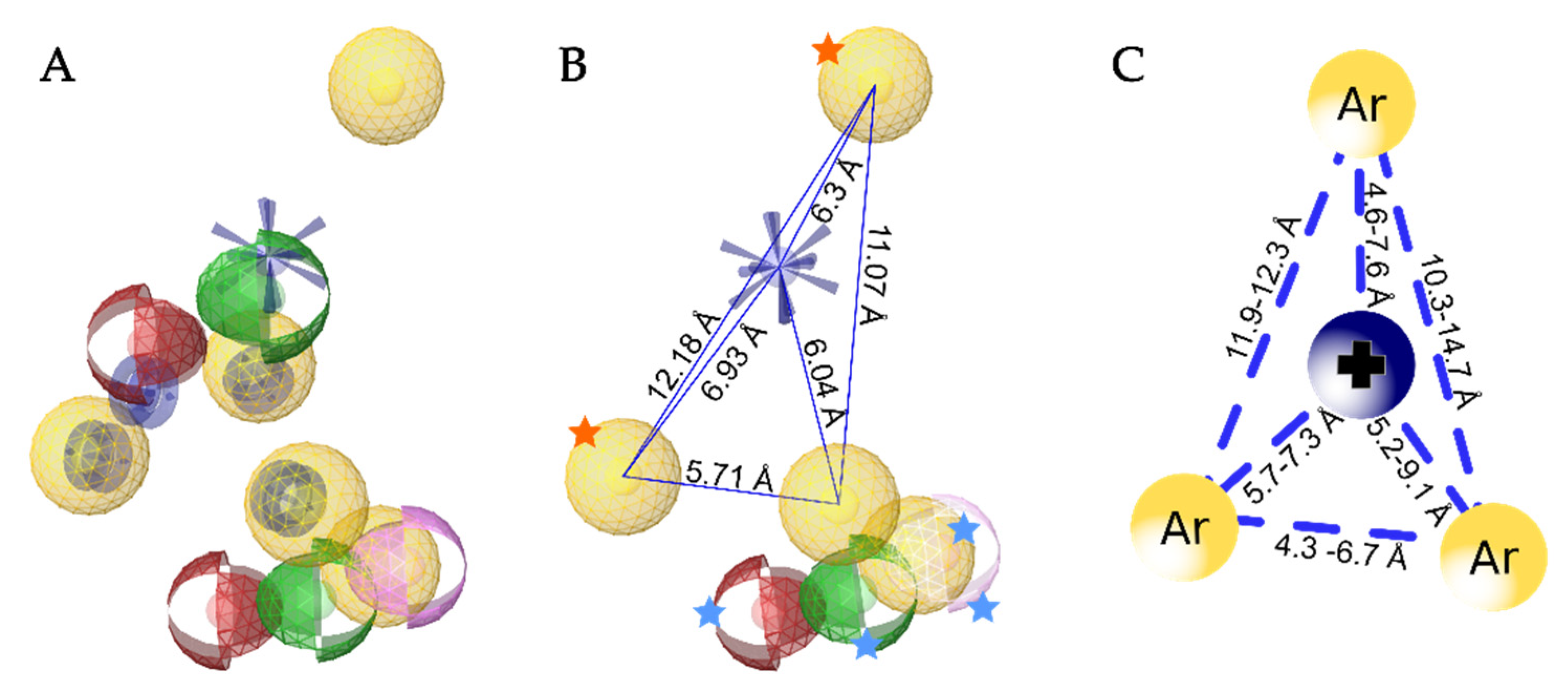
3.5. Creation of the Merged Structure-Based Pharmacophore Model
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Warmke, J.W.; Ganetzky, B. A family of potassium channel genes related to eag in Drosophila and mammals. Proc. Natl. Acad. Sci. USA 1994, 91, 3438–3442. [Google Scholar] [CrossRef] [Green Version]
- Recanatini, M.; Poluzzi, E.; Masetti, M.; Cavalli, A.; Ponti, F.D. QT prolongation through hERG K+ channel blockade: Current knowledge and strategies for the early prediction during drug development. Med. Res. Rev. 2005, 25, 133–166. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Barakat, K.H. Development of Safe Drugs: The hERG Challenge. Med. Res. Rev. 2017, 38, 525–555. [Google Scholar] [CrossRef]
- Pardo, L.A.; Stühmer, W. The roles of K+ channels in cancer. Nat. Rev. Cancer 2014, 14, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Movsisyan, N.; Pardo, L.A. Kv10.1 Regulates Microtubule Dynamics during Mitosis. Cancers 2020, 12, 2409. [Google Scholar] [CrossRef] [PubMed]
- Pardo, L.A.; Gomez-Varela, D.; Major, F.; Sansuk, K.; Leurs, R.; Downie, B.R.; Tietze, L.F.; Stuhmer, W. Approaches Targeting KV10.1 Open a Novel Window for Cancer Diagnosis and Therapy. Curr. Med. Chem. 2012, 19, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Whicher, J.R.; MacKinnon, R. Structure of the voltage-gated K+ channel Eag1 reveals an alternative voltage sensing mechanism. Science 2016, 353, 664–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; MacKinnon, R. Cryo-EM Structure of the Open Human Ether-à-go-go -Related K + Channel hERG. Cell 2017, 169, 422–430.e10. [Google Scholar] [CrossRef] [Green Version]
- Barros, F.; de la Peña, P.; Domínguez, P.; Sierra, L.M.; Pardo, L.A. The EAG Voltage-Dependent K+ Channel Subfamily: Similarities and Differences in Structural Organization and Gating. Front. Pharmacol. 2020, 11, 411. [Google Scholar] [CrossRef] [Green Version]
- Ident and Sim. Available online: https://www.bioinformatics.org/sms2/ident_sim.html (accessed on 13 May 2021).
- Gómez-Varela, D.; Contreras-Jurado, C.; Furini, S.; García-Ferreiro, R.; Stühmer, W.; Pardo, L.A. Different relevance of inactivation and F468 residue in the mechanisms of hEag1 channel blockage by astemizole, imipramine and dofetilide. FEBS Lett. 2006, 580, 5059–5066. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Seebohm, G.; Sanguinetti, M.C. Position of aromatic residues in the S6 domain, not inactivation, dictates cisapride sensitivity of HERG and eag potassium channels. Proc. Natl. Acad. Sci. USA 2002, 99, 12461–12466. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, D.; Ghanta, A.; Kauffman, G.W.; Sanguinetti, M.C. Physicochemical Features of the hERG Channel Drug Binding Site. J. Biol. Chem. 2004, 279, 10120–10127. [Google Scholar] [CrossRef] [Green Version]
- Ekins, S. Three-Dimensional Quantitative Structure-Activity Relationship for Inhibition of Human Ether-a-Go-Go-Related Gene Potassium Channel. J. Pharmacol. Exp. Ther. 2002, 301, 427–434. [Google Scholar] [CrossRef] [Green Version]
- Cavalli, A.; Poluzzi, E.; De Ponti, F.; Recanatini, M. Toward a Pharmacophore for Drugs Inducing the Long QT Syndrome: Insights from a CoMFA Study of HERG K+ Channel Blockers. J. Med. Chem. 2002, 45, 3844–3853. [Google Scholar] [CrossRef] [PubMed]
- Pearlstein, R.A.; Vaz, R.J.; Kang, J.; Chen, X.-L.; Preobrazhenskaya, M.; Shchekotikhin, A.E.; Korolev, A.M.; Lysenkova, L.N.; Miroshnikova, O.V.; Hendrix, J.; et al. Characterization of HERG potassium channel inhibition using CoMSiA 3D QSAR and homology modeling approaches. Bioorg. Med. Chem. Lett. 2003, 13, 1829–1835. [Google Scholar] [CrossRef]
- Toplak, Ž.; Hendrickx, L.A.; Abdelaziz, R.; Shi, X.; Peigneur, S.; Tomašič, T.; Tytgat, J.; Peterlin-Mašič, L.; Pardo, L.A. Overcoming challenges of HERG potassium channel liability through rational design: Eag1 inhibitors for cancer treatment. Med. Res. Rev. 2021. [Google Scholar] [CrossRef]
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-coffee: A novel method for fast and accurate multiple sequence alignment 1 1Edited by J. Thornton. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lüthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pontius, J.; Richelle, J.; Wodak, S.J. Deviations from Standard Atomic Volumes as a Quality Measure for Protein Crystal Structures. J. Mol. Biol. 1996, 264, 121–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. Procheck: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Brooks, B.R.; Brooks, C.L.; MacKerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- LigandScout v.4.4. Inte:Ligand. Available online: https://www.inteligand.com/ligandscout (accessed on 10 May 2021).
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef] [PubMed]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Sieb, C.; Thiel, K.; Wiswedel, B. KNIME: The Konstanz Information Miner. In Studies in Classification, Data Analysis, and Knowledge Organization (GfKL 2007); Springer: Berlin/Heidelberg, Germany, 2007. [Google Scholar]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Landrum, G. RDKit: Open-Source Cheminformatics Software. 2016. Available online: https://www.rdkit.org/ (accessed on 10 May 2021).
- Inte:Ligand Expert KNIME Extensions. Inte:Ligand GmbH. Available online: http://www.inteligand.com/knime-nodes (accessed on 10 January 2021).
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 497, D480–D490. [Google Scholar]
- García-Ferreiro, R.E.; Kerschensteiner, D.; Major, F.; Monje, F.; Stühmer, W.; Pardo, L.A. Mechanism of Block of hEag1 K + Channels by Imipramine and Astemizole. J. Gen. Physiol. 2004, 124, 301–317. [Google Scholar] [CrossRef] [Green Version]
- Gessner, G.; Heinemann, S.H. Inhibition of hEAG1 and hERG1 potassium channels by clofilium and its tertiary analogue LY97241. Br. J. Pharmacol. 2003, 138, 161–171. [Google Scholar] [CrossRef] [Green Version]
- Schönherr, R.; Gessner, G.; Löber, K.; Heinemann, S.H. Functional distinction of human EAG1 and EAG2 potassium channels. FEBS Lett. 2002, 514, 204–208. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, J.; Terlau, H.; Wunder, F.; Brüggemann, A.; Pardo, L.A.; Marquardt, A.; Stühmer, W.; Pongs, O. Functional expression of a rat homologue of the voltage gated either á go-go potassium channel reveals differences in selectivity and activation kinetics between the Drosophila channel and its mammalian counterpart. EMBO J. 1994, 13, 4451–4458. [Google Scholar] [CrossRef]
- Gessner, G.; Zacharias, M.; Bechstedt, S.; Schönherr, R.; Heinemann, S.H. Molecular determinants for high-affinity block of human EAG potassium channels by antiarrhythmic agents. Mol. Pharmacol. 2004, 65, 1120–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016; Available online: https://gaussian.com/ (accessed on 11 May 2021).
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CGenFF Interface at Paramchem.org. Available online: https://cgenff.umaryland.edu/ (accessed on 11 May 2021).
- Mironenko, A.; Zachariae, U.; de Groot, B.L.; Kopec, W. The persistent question of potassium channel permeation mechanisms. J. Mol. Biol. 2021, 167002. [Google Scholar] [CrossRef] [PubMed]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Gowers, R.; Linke, M.; Barnoud, J.; Reddy, T.; Melo, M.; Seyler, S.; Domański, J.; Dotson, D.; Buchoux, S.; Kenney, I.; et al. MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations. In Proceedings of the 15th Python in Science Conference, Austin, TX, USA, 11–17 July 2016; pp. 98–105. [Google Scholar]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Toplak, Ž.; Hendrickx, L.A.; Gubič, Š.; Možina, Š.; Žegura, B.; Štern, A.; Novak, M.; Shi, X.; Peigneur, S.; Tytgat, J.; et al. 3D Pharmacophore-Based Discovery of Novel KV10.1 Inhibitors with Antiproliferative Activity. Cancers 2021, 13, 1244. [Google Scholar] [CrossRef]
- Mitcheson, J.S.; Chen, J.; Lin, M.; Culberson, C.; Sanguinetti, M.C. A structural basis for drug-induced long QT syndrome. Proc. Natl. Acad. Sci. USA 2000, 97, 12329–12333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Șterbuleac, D.; Maniu, C.L. Computer Simulations Reveal a Novel Blocking Mode of the hERG Ion Channel by the Antiarrhythmic Agent Clofilium. Mol. Inform. 2018, 37, 1700142. [Google Scholar] [CrossRef] [PubMed]
- Perry, M.; Stansfeld, P.J.; Leaney, J.; Wood, C.; de Groot, M.J.; Leishman, D.; Sutcliffe, M.J.; Mitcheson, J.S. Drug Binding Interactions in the Inner Cavity of hERG Channels: Molecular Insights from Structure-Activity Relationships of Clofilium and Ibutilide Analogs. Mol. Pharmacol. 2006, 69, 509–519. [Google Scholar] [CrossRef]
- Perry, M.; de Groot, M.J.; Helliwell, R.; Leishman, D.; Tristani-Firouzi, M.; Sanguinetti, M.C.; Mitcheson, J. Structural Determinants of HERG Channel Block by Clofilium and Ibutilide. Mol. Pharmacol. 2004, 66, 240–249. [Google Scholar] [CrossRef] [Green Version]
- Sănchez-Chapula, J.A.; Ferrer, T.; Navarro-Polanco, R.A.; Sanguinetti, M.C. Voltage-Dependent Profile of HumanEther-a-go-go-Related Gene Channel Block Is Influenced by a Single Residue in the S6 Transmembrane Domain. Mol. Pharmacol. 2003, 63, 1051–1058. [Google Scholar] [CrossRef] [Green Version]
- Louvel, J.; Carvalho, J.F.S.; Yu, Z.; Soethoudt, M.; Lenselink, E.B.; Klaasse, E.; Brussee, J.; IJzerman, A.P. Removal of Human Ether-à-go-go Related Gene (hERG) K+ Channel Affinity through Rigidity: A Case of Clofilium Analogues. J. Med. Chem. 2013, 56, 9427–9440. [Google Scholar] [CrossRef]
- Saxena, P.; Zangerl-Plessl, E.-M.; Linder, T.; Windisch, A.; Hohaus, A.; Timin, E.; Hering, S.; Stary-Weinzinger, A. New potential binding determinant for hERG channel inhibitors. Sci. Rep. 2016, 6, 24182. [Google Scholar] [CrossRef] [Green Version]
- Negami, T.; Araki, M.; Okuno, Y.; Terada, T. Calculation of absolute binding free energies between the hERG channel and structurally diverse drugs. Sci. Rep. 2019, 9, 16586. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Lamothe, S.M.; Hou, X.; Moon, T.C.; Kurata, H.T.; Houghton, M.; Barakat, K.H. A structure-based computational workflow to predict liability and binding modes of small molecules to hERG. Sci. Rep. 2020, 10, 16262. [Google Scholar] [CrossRef]
- Brown, S.; Sonntag, D.P.; Sanguinetti, M.C. A Highly Conserved Alanine in the S6 Domain of the hERG1 K+ Channel is Required for Normal Gating. Cell. Physiol. Biochem. 2008, 22, 601–610. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.-J.; Soohoo, S.M.; Tiwari, P.B.; Piszczek, G.; Brelidze, T.I. Chlorpromazine binding to the PAS domains uncovers the effect of ligand modulation on EAG channel activity. J. Biol. Chem. 2020, jbc.RA119.012377. [Google Scholar] [CrossRef] [PubMed]
- Barriga-Montoya, C.; Huanosta-Gutiérrez, A.; Reyes-Vaca, A.; Hernández-Cruz, A.; Picones, A.; Gómez-Lagunas, F. Correction to: Inhibition of the K+ conductance and Cole-Moore shift of the oncogenic Kv10.1 channel by amiodarone. Pflüg. Arch. Eur. J. Physiol. 2018, 470, 981–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreels, L.; Bhat, C.; Voráčová, M.; Peigneur, S.; Goovaerts, H.; Mäki-Lohiluoma, E.; Zahed, F.; Pardo, L.A.; Yli-Kauhaluoma, J.; Kiuru, P.; et al. Synthesis of novel purpurealidin analogs and evaluation of their effect on the cancer-relevant potassium channel KV10.1. PLoS ONE 2017, 12, e0188811. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | KV10.1 IC50 [μM] | Cells and Technique Used in the Assay |
|---|---|---|---|
| Astemizole |  | 0.196 | HEK-293 cells; whole-cell patch clamp [37] |
| 2.8 ± 0.1 | Xenopus oocytes; two-electrode voltage clamp [11] | ||
| Clofilium |  | 0.255 ± 0.035 | CHO-K1 cells; whole-cell patch clamp [38] |
| 0.001 ± 0.001 | Xenopus oocytes; inside-out patch clamp [38] | ||
| Imipramine |  | 40.2 ± 3.0 | Xenopus oocytes; two-electrode voltage clamp [11] |
| MK-499 |  | 43.5 ± 4.7 | Xenopus oocytes; whole-cell patch clamp [11] |
| Quinidine |  | 1.4 ± 0.1 | CHO cells; whole-cell patch clamp [39] |
| 400 ± 200 | Xenopus oocytes; two-electrode voltage clamp [40] | ||
| 2.1 ± 0.4 | Xenopus oocytes; inside-out patch clamp [41] |
| Compound | GlideScore [kcal/mol] |
|---|---|
| Astemizole | −11.459 |
| Clofilium | −9.465 |
| Imipramine | −8.995 |
| MK-499 | −9.631 |
| Quinidine | −7.287 |
| Astemizole | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A465_A | A465_B | A465_C | S461_C | T435_D | T472_A | T472_C | Y464_A | Y464_B | Y464_C | V437_D | ||||||||||||
| 50 | 98 | 87 | 51 | 92 | 62 | 98 | 33 | 88 | 39 | 24 | 95 | 93 | 19 | 93 | ||||||||
| Clofilium | ||||||||||||||||||||||
| A465_A | A465_C | Y464_C | V437_D | F468_B | F468_C | T472_D | ||||||||||||||||
| 94 | 97 | 28 | 93 | 90 | 84 | 93 | 99 | |||||||||||||||
| Imipramine | ||||||||||||||||||||||
| A465_B | A465_D | F468_D | S436_B | T472_C | Y464_A | Y464_B | Y464_D | |||||||||||||||
| 65 | 99 | 98 | 32 | 95 | 96 | 75 | 14 | 73 | 97 | 50 | ||||||||||||
| MK-499 | ||||||||||||||||||||||
| S461_A | T472_B | Y464_B | Y464_C | Y464_D | V437_D | |||||||||||||||||
| 32 | 43 | 96 | 52 | 55 | 100 | 66 | 15 | 61 | 80 | |||||||||||||
| Quinidine | ||||||||||||||||||||||
| A465_C | A465_D | F468_A | Y464_A | Y464_B | Y464_D | |||||||||||||||||
| 82 | 92 | 99 | 79 | 93 | 87 | 51 | 94 | |||||||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toplak, Ž.; Merzel, F.; Pardo, L.A.; Peterlin Mašič, L.; Tomašič, T. Molecular Dynamics-Derived Pharmacophore Model Explaining the Nonselective Aspect of KV10.1 Pore Blockers. Int. J. Mol. Sci. 2021, 22, 8999. https://doi.org/10.3390/ijms22168999
Toplak Ž, Merzel F, Pardo LA, Peterlin Mašič L, Tomašič T. Molecular Dynamics-Derived Pharmacophore Model Explaining the Nonselective Aspect of KV10.1 Pore Blockers. International Journal of Molecular Sciences. 2021; 22(16):8999. https://doi.org/10.3390/ijms22168999
Chicago/Turabian StyleToplak, Žan, Franci Merzel, Luis A. Pardo, Lucija Peterlin Mašič, and Tihomir Tomašič. 2021. "Molecular Dynamics-Derived Pharmacophore Model Explaining the Nonselective Aspect of KV10.1 Pore Blockers" International Journal of Molecular Sciences 22, no. 16: 8999. https://doi.org/10.3390/ijms22168999
APA StyleToplak, Ž., Merzel, F., Pardo, L. A., Peterlin Mašič, L., & Tomašič, T. (2021). Molecular Dynamics-Derived Pharmacophore Model Explaining the Nonselective Aspect of KV10.1 Pore Blockers. International Journal of Molecular Sciences, 22(16), 8999. https://doi.org/10.3390/ijms22168999