
Prediction of Drug-Induced Liver Toxicity Using SVM and Optimal Descriptor Sets




Abstract
:1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. Molecular Descriptors
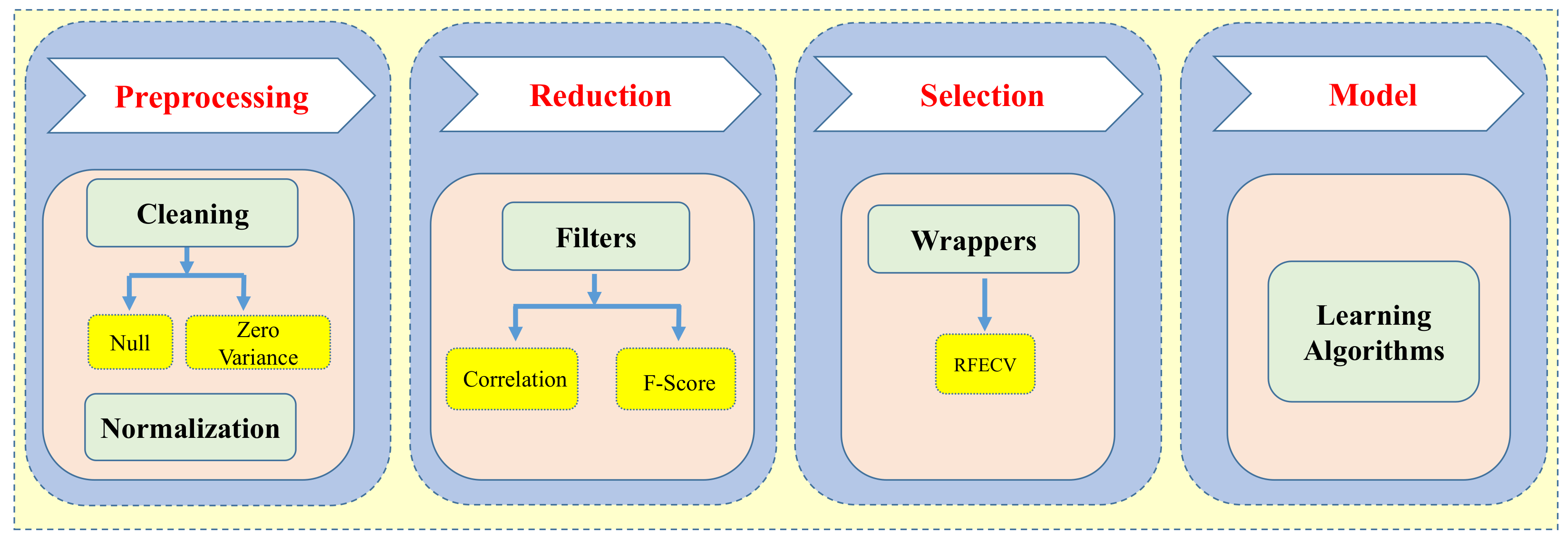
2.3. Data Preprocessing and Feature Selection
2.4. Model Building and Optimization
Support Vector Machine
2.5. Model Training and Validation
2.6. Performance Evaluation Metrics
3. Results and Discussion
3.1. Data Analysis
3.2. Performance of Models Using Cross-Validation
3.2.1. Experiments with Individual Descriptor Sets
3.2.2. Experiments with Combined Descriptor Sets
3.2.3. Comparison of SVM with Other Classifiers
3.3. Performance Comparison with Previous Work
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Almazroo, O.A.; Miah, M.K.; Venkataramanan, R. Drug metabolism in the liver. Clin. Liver Dis. 2017, 21, 1–20. [Google Scholar] [CrossRef]
- Real, M.; Barnhill, M.S.; Higley, C.; Rosenberg, J.; Lewis, J.H. Drug-induced liver injury: Highlights of the recent literature. Drug Saf. 2019, 42, 365–387. [Google Scholar] [CrossRef]
- Albrecht, W.; Kappenberg, F.; Brecklinghaus, T.; Stoeber, R.; Marchan, R.; Zhang, M.; Ebbert, K.; Kirschner, H.; Grinberg, M.; Leist, M.; et al. Prediction of human drug-induced liver injury (DILI) in relation to oral doses and blood concentrations. Arch. Toxicol. 2019, 93, 1609–1637. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.M. Drug-induced hepatotoxicity. N. Engl. J. Med. 2003, 349, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Onakpoya, I.J.; Heneghan, C.J.; Aronson, J.K. Post-marketing withdrawal of 462 medicinal products because of adverse drug reactions: A systematic review of the world literature. BMC Med. 2016, 14, 10. [Google Scholar] [CrossRef] [Green Version]
- Njoku, D.B. Drug-induced hepatotoxicity: Metabolic, genetic and immunological basis. Int. J. Mol. Sci. 2014, 15, 6990–7003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Cortes, M.; Robles-Diaz, M.; Stephens, C.; Ortega-Alonso, A.; Lucena, M.I.; Andrade, R.J. Drug induced liver injury: An update. Arch. Toxicol. 2020, 94, 3381–3407. [Google Scholar] [CrossRef] [PubMed]
- Assis, D.N.; Navarro, V.J. Human drug hepatotoxicity: A contemporary clinical perspective. Expert Opin. Drug Metab. Toxicol. 2009, 5, 463–473. [Google Scholar] [CrossRef]
- Chen, M.; Vijay, V.; Shi, Q.; Liu, Z.; Fang, H.; Tong, W. FDA-approved drug labeling for the study of drug-induced liver injury. Drug Discov. Today 2011, 16, 697–703. [Google Scholar] [CrossRef]
- Issa, A.M.; Phillips, K.A.; Van Bebber, S.; Nidamarthy, H.G.; Lasser, K.E.; Haas, J.S.; Alldredge, B.K.; Wachter, R.M.; Bates, D.W. Drug withdrawals in the United States: A systematic review of the evidence and analysis of trends. Curr. Drug Saf. 2007, 2, 177–185. [Google Scholar] [CrossRef]
- Suh, J.I. Drug-induced liver injury. Yeungnam Univ. J. Med. 2020, 37, 2. [Google Scholar] [CrossRef]
- Olson, H.; Betton, G.; Robinson, D.; Thomas, K.; Monro, A.; Kolaja, G.; Lilly, P.; Sanders, J.; Sipes, G.; Bracken, W.; et al. Concordance of the toxicity of pharmaceuticals in humans and in animals. Regul. Toxicol. Pharmacol. 2000, 32, 56–67. [Google Scholar] [CrossRef]
- Sertkaya, A.; Wong, H.H.; Jessup, A.; Beleche, T. Key cost drivers of pharmaceutical clinical trials in the United States. Clin. Trials 2016, 13, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Sistare, F.D.; Mattes, W.B.; LeCluyse, E.L. The promise of new technologies to reduce, refine, or replace animal use while reducing risks of drug induced liver injury in pharmaceutical development. ILAR J. 2017, 57, 186–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segall, M.D.; Barber, C. Addressing toxicity risk when designing and selecting compounds in early drug discovery. Drug Discov. Today 2014, 19, 688–693. [Google Scholar] [CrossRef]
- Przybylak, K.R.; Cronin, M.T. In silico models for drug-induced liver injury–current status. Expert Opin. Drug Metab. Toxicol. 2012, 8, 201–217. [Google Scholar] [CrossRef]
- Saini, N.; Bakshi, S.; Sharma, S. In-silico approach for drug induced liver injury prediction: Recent advances. Toxicol. Lett. 2018, 295, 288–295. [Google Scholar] [CrossRef]
- Thakkar, S.; Chen, M.; Fang, H.; Liu, Z.; Roberts, R.; Tong, W. The Liver Toxicity Knowledge Base (LKTB) and drug-induced liver injury (DILI) classification for assessment of human liver injury. Expert Rev. Gastroenterol. Hepatol. 2018, 12, 31–38. [Google Scholar] [CrossRef]
- Chierici, M.; Francescatto, M.; Bussola, N.; Jurman, G.; Furlanello, C. Predictability of drug-induced liver injury by machine learning. Biol. Direct 2020, 15, 1–10. [Google Scholar] [CrossRef]
- Xu, Y.; Dai, Z.; Chen, F.; Gao, S.; Pei, J.; Lai, L. Deep learning for drug-induced liver injury. J. Chem. Inf. Modeling 2015, 55, 2085–2093. [Google Scholar] [CrossRef] [PubMed]
- Kuna, L.; Bozic, I.; Kizivat, T.; Bojanic, K.; Mrso, M.; Kralj, E.; Smolic, R.; Wu, G.Y.; Smolic, M. Models of drug induced liver injury (DILI)–current issues and future perspectives. Curr. Drug Metab. 2018, 19, 830–838. [Google Scholar] [CrossRef]
- Marchant, C.A.; Fisk, L.; Note, R.R.; Patel, M.L.; Suárez, D. An expert system approach to the assessment of hepatotoxic potential. Chem. Biodivers. 2009, 6, 2107–2114. [Google Scholar] [CrossRef]
- Greene, N.; Fisk, L.; Naven, R.T.; Note, R.R.; Patel, M.L.; Pelletier, D.J. Developing structure- activity relationships for the prediction of hepatotoxicity. Chem. Res. Toxicol. 2010, 23, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, F.; Lombardo, A.; Manganaro, A.; Benfenati, E. A new structure-activity relationship (SAR) model for predicting drug-induced liver injury, based on statistical and expert-based structural alerts. Front. Pharmacol. 2016, 7, 442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekins, S.; Williams, A.J.; Xu, J.J. A predictive ligand-based Bayesian model for human drug-induced liver injury. Drug Metab. Dispos. 2010, 38, 2302–2308. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Ding, L.; Zou, Y.; Hu, S.Q.; Huang, H.G.; Kong, W.B.; Zhang, J. Predicting drug-induced liver injury in human with Naïve Bayes classifier approach. J. Comput. Aided Mol. Des. 2016, 30, 889–898. [Google Scholar] [CrossRef]
- Kotsampasakou, E.; Montanari, F.; Ecker, G.F. Predicting drug-induced liver injury: The importance of data curation. Toxicology 2017, 389, 139–145. [Google Scholar] [CrossRef]
- Zhang, C.; Cheng, F.; Li, W.; Liu, G.; Lee, P.W.; Tang, Y. In silico prediction of drug induced liver toxicity using substructure pattern recognition method. Mol. Inform. 2016, 35, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Nam, H. Prediction models for drug-induced hepatotoxicity by using weighted molecular fingerprints. BMC Bioinform. 2017, 18, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulliner, D.; Schmidt, F.; Stolte, M.; Spirkl, H.P.; Czich, A.; Amberg, A. Computational models for human and animal hepatotoxicity with a global application scope. Chem. Res. Toxicol. 2016, 29, 757–767. [Google Scholar] [CrossRef] [Green Version]
- Ai, H.; Chen, W.; Zhang, L.; Huang, L.; Yin, Z.; Hu, H.; Zhao, Q.; Zhao, J.; Liu, H. Predicting drug-induced liver injury using ensemble learning methods and molecular fingerprints. Toxicol. Sci. 2018, 165, 100–107. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Ye, T.; Wang, R.; Zhang, C.; Zhang, X.; Sun, G.; Sun, X. An in silico model for predicting drug-induced hepatotoxicity. Int. J. Mol. Sci. 2019, 20, 1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Suzuki, A.; Thakkar, S.; Yu, K.; Hu, C.; Tong, W. DILIrank: The largest reference drug list ranked by the risk for developing drug-induced liver injury in humans. Drug Discov. Today 2016, 21, 648–653. [Google Scholar] [CrossRef]
- Hoofnagle, J.H.; Serrano, J.; Knoben, J.E.; Navarro, V.J. LiverTox: A website on Drug-Induced Liver Injury; Wiley Online Library: Hoboken, NJ, USA, 2013. [Google Scholar]
- Chen, M.; Zhang, J.; Wang, Y.; Liu, Z.; Kelly, R.; Zhou, G.; Fang, H.; Borlak, J.; Tong, W. The liver toxicity knowledge base: A systems approach to a complex end point. Clin. Pharmacol. Ther. 2013, 93, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Liew, C.Y.; Lim, Y.C.; Yap, C.W. Mixed learning algorithms and features ensemble in hepatotoxicity prediction. J. Comput. Aided Mol. Des. 2011, 25, 855–871. [Google Scholar] [CrossRef]
- Huang, S.H.; Tung, C.W.; Fülöp, F.; Li, J.H. Developing a QSAR model for hepatotoxicity screening of the active compounds in traditional Chinese medicines. Food Chem. Toxicol. 2015, 78, 71–77. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.; Thiessen, P.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, S.; Semin, M.; Lagunin, A.; Filimonov, D.; Poroikov, V. In Silico Identification of Proteins Associated with Drug-Induced Liver Injury Based on the Prediction of Drug-Target Interactions. Mol. Inform. 2017, 36, 1600142. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V. Molecular Descriptors for Chemoinformatics: Volume I: Alphabetical Listing/Volume II: Appendices, References; John Wiley & Sons: Hoboken, NJ, USA, 2009; Volume 41. [Google Scholar]
- Steinbeck, C.; Hoppe, C.; Kuhn, S.; Floris, M.; Guha, R.; Willighagen, E.L. Recent developments of the chemistry development kit (CDK)-an open-source java library for chemo-and bioinformatics. Curr. Pharm. Des. 2006, 12, 2111–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, D.S.; Xu, Q.S.; Hu, Q.N.; Liang, Y.Z. ChemoPy: Freely available python package for computational biology and chemoinformatics. Bioinformatics 2013, 29, 1092–1094. [Google Scholar] [CrossRef] [PubMed]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef]
- Landrum, G. Rdkit documentation. Release 2013, 1, 4. [Google Scholar]
- Dong, J.; Cao, D.S.; Miao, H.Y.; Liu, S.; Deng, B.C.; Yun, Y.H.; Wang, N.N.; Lu, A.P.; Zeng, W.B.; Chen, A.F. ChemDes: An integrated web-based platform for molecular descriptor and fingerprint computation. J. Cheminform. 2015, 7, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeys, Y.; Inza, I.; Larranaga, P. A review of feature selection techniques in bioinformatics. Bioinformatics 2007, 23, 2507–2517. [Google Scholar] [CrossRef] [Green Version]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Freedman, D.A. Statistical Models and Causal Inference: A Dialogue with the Social Sciences; Cambridge University Press: Cambridge, UK, 2010. [Google Scholar]
- Güneş, S.; Polat, K.; Yosunkaya, Ş. Multi-class f-score feature selection approach to classification of obstructive sleep apnea syndrome. Expert Syst. Appl. 2010, 37, 998–1004. [Google Scholar] [CrossRef]
- Darst, B.F.; Malecki, K.C.; Engelman, C.D. Using recursive feature elimination in random forest to account for correlated variables in high dimensional data. BMC Genet. 2018, 19, 65. [Google Scholar] [CrossRef] [Green Version]
- Guyon, I.; Weston, J.; Barnhill, S.; Vapnik, V. Gene selection for cancer classification using support vector machines. Mach. Learn. 2002, 46, 389–422. [Google Scholar] [CrossRef]
- Abdelbaky, I.; Tayara, H.; Chong, K.T. Prediction of kinase inhibitors binding modes with machine learning and reduced descriptor sets. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef]
- Khanal, J.; Lim, D.Y.; Tayara, H.; Chong, K.T. i6ma-stack: A stacking ensemble-based computational prediction of dna n6-methyladenine (6ma) sites in the rosaceae genome. Genomics 2021, 113, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Minerali, E.; Foil, D.H.; Zorn, K.M.; Lane, T.R.; Ekins, S. Comparing Machine Learning Algorithms for Predicting Drug-Induced Liver Injury (DILI). Mol. Pharm. 2020, 17, 2628–2637. [Google Scholar] [CrossRef]
- Li, X.; Chen, Y.; Song, X.; Zhang, Y.; Li, H.; Zhao, Y. The development and application of in silico models for drug induced liver injury. RSC Adv. 2018, 8, 8101–8111. [Google Scholar] [CrossRef] [Green Version]
- Ben-Hur, A.; Weston, J. A user’s guide to support vector machines. In Data Mining Techniques for the Life Sciences; Springer: Berlin/Heidelberg, Germany, 2010; pp. 223–239. [Google Scholar]
- Chicco, D.; Jurman, G. The advantages of the Matthews correlation coefficient (MCC) over F1 score and accuracy in binary classification evaluation. BMC Genom. 2020, 21, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lučić, B.; Batista, J.; Bojović, V.; Lovrić, M.; Kržić, A.S.; Bešlo, D.; Nadramija, D.; Vikić-Topić, D. Estimation of random accuracy and its use in validation of predictive quality of classification models within predictive challenges. Croat. Chem. Acta 2019, 92, 379–391. [Google Scholar] [CrossRef] [Green Version]
- Butina, D. Unsupervised data base clustering based on daylight’s fingerprint and Tanimoto similarity: A fast and automated way to cluster small and large data sets. J. Chem. Inf. Comput. Sci. 1999, 39, 747–750. [Google Scholar] [CrossRef]
- Van der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Zhang, Z.; Beck, M.W.; Winkler, D.A.; Huang, B.; Sibanda, W.; Goyal, H. Opening the black box of neural networks: Methods for interpreting neural network models in clinical applications. Ann. Transl. Med. 2018, 6, 216. [Google Scholar] [CrossRef]
- Alam, W.; Tayara, H.; Chong, K.T. XG-ac4C: Identification of N4-acetylcytidine (ac4C) in mRNA using eXtreme gradient boosting with electron-ion interaction pseudopotentials. Sci. Rep. 2020, 10, 1–10. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Descriptor Set | Descriptors Count |
|---|---|
| PaDEL 1&2D | 1544 |
| Chemopy 1&2D | 633 |
| CDK | 275 |
| RDKit | 196 |
| Total | 2648 |
| Descriptor Set | Selected No. of Descriptors | ACC | SPE | SEN | MCC |
|---|---|---|---|---|---|
| CDK | 77 | 0.771 | 0.772 | 0.773 | 0.545 |
| Chemopy | 104 | 0.766 | 0.763 | 0.773 | 0.536 |
| PaDEL | 91 | 0.781 | 0.749 | 0.815 | 0.565 |
| RDKit | 86 | 0.752 | 0.729 | 0.777 | 0.507 |
| Best Combination Descriptor Sets | Optimal No. of Descriptors | ACC | SPE | SEN | MCC |
|---|---|---|---|---|---|
| PaDEL-RDKit | 132 | 0.796 | 0.784 | 0.809 | 0.593 |
| PaDEL-RDKit-CDK | 162 | 0.804 | 0.796 | 0.813 | 0.609 |
| PaDEL-RDKit-CDK-Chemopy | 155 | 0.811 | 0.783 | 0.840 | 0.623 |
| Descriptor Set | Descriptor Type | Total-Type | Total-Set | % of Selection |
|---|---|---|---|---|
| PaDEL 1&2D | Autocorrelation Descriptors | 46 | 83 | 54 |
| E-state Descriptors | 13 | |||
| Topological Descriptors | 11 | |||
| Constitutional Descriptors | 11 | |||
| Others | 2 | |||
| Chemopy 1&2D | MOE-type descriptors | 11 | 37 | 24 |
| E-state Descriptors | 11 | |||
| Autocorrelation Descriptors | 10 | |||
| Others | 5 | |||
| CDK | Topological Descriptors | 17 | 28 | 18 |
| Kappa Descriptors | 5 | |||
| Autocorrelation Descriptors | 3 | |||
| Others | 5 | |||
| RDKit | Constitutional descriptors | 7 | 7 | 4 |
| Model Name | No. of Compounds | Test Method | ACC | SPE | SEN |
|---|---|---|---|---|---|
| Proposed Model | 1253 | 10-fold CV | 0.811 | 0.783 | 0.840 |
| 208 | External Validation | 0.756 | 0.708 | 0.807 | |
| Ensemble Model [33] | 1254 | 10-fold CV | 0.783 | 0.748 | 0.818 |
| 204 | External Validation | 0.730 | 0.658 | 0.773 | |
| Ensemble-Top5 [32] | 1241 | 5-fold CV | 0.711 | 0.603 | 0.799 |
| SVM [27] | 978 | 5-fold CV | 0.797 | 0.585 | 0.948 |
| 88 | External Validation | 0.750 | 0.379 | 0.932 | |
| RF [28] | 996 | 10-fold CV | 0.65 | 0.62 | 0.68 |
| 966 | External Validation | 0.58 | 0.38 | 0.75 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaganathan, K.; Tayara, H.; Chong, K.T. Prediction of Drug-Induced Liver Toxicity Using SVM and Optimal Descriptor Sets. Int. J. Mol. Sci. 2021, 22, 8073. https://doi.org/10.3390/ijms22158073
Jaganathan K, Tayara H, Chong KT. Prediction of Drug-Induced Liver Toxicity Using SVM and Optimal Descriptor Sets. International Journal of Molecular Sciences. 2021; 22(15):8073. https://doi.org/10.3390/ijms22158073
Chicago/Turabian StyleJaganathan, Keerthana, Hilal Tayara, and Kil To Chong. 2021. "Prediction of Drug-Induced Liver Toxicity Using SVM and Optimal Descriptor Sets" International Journal of Molecular Sciences 22, no. 15: 8073. https://doi.org/10.3390/ijms22158073
APA StyleJaganathan, K., Tayara, H., & Chong, K. T. (2021). Prediction of Drug-Induced Liver Toxicity Using SVM and Optimal Descriptor Sets. International Journal of Molecular Sciences, 22(15), 8073. https://doi.org/10.3390/ijms22158073