
Cancer-Associated Fibroblasts in Conversation with Tumor Cells in Endometrial Cancers: A Partner in Crime


Abstract
:1. Definition of CAF
2. CAF as an Evolving Component of Tumor Microenvironment
3. CAF & CAF Conundrum
4. CAF in Endometrial Cancers
5. Characteristics of CAF-Tumor Cross-Talk
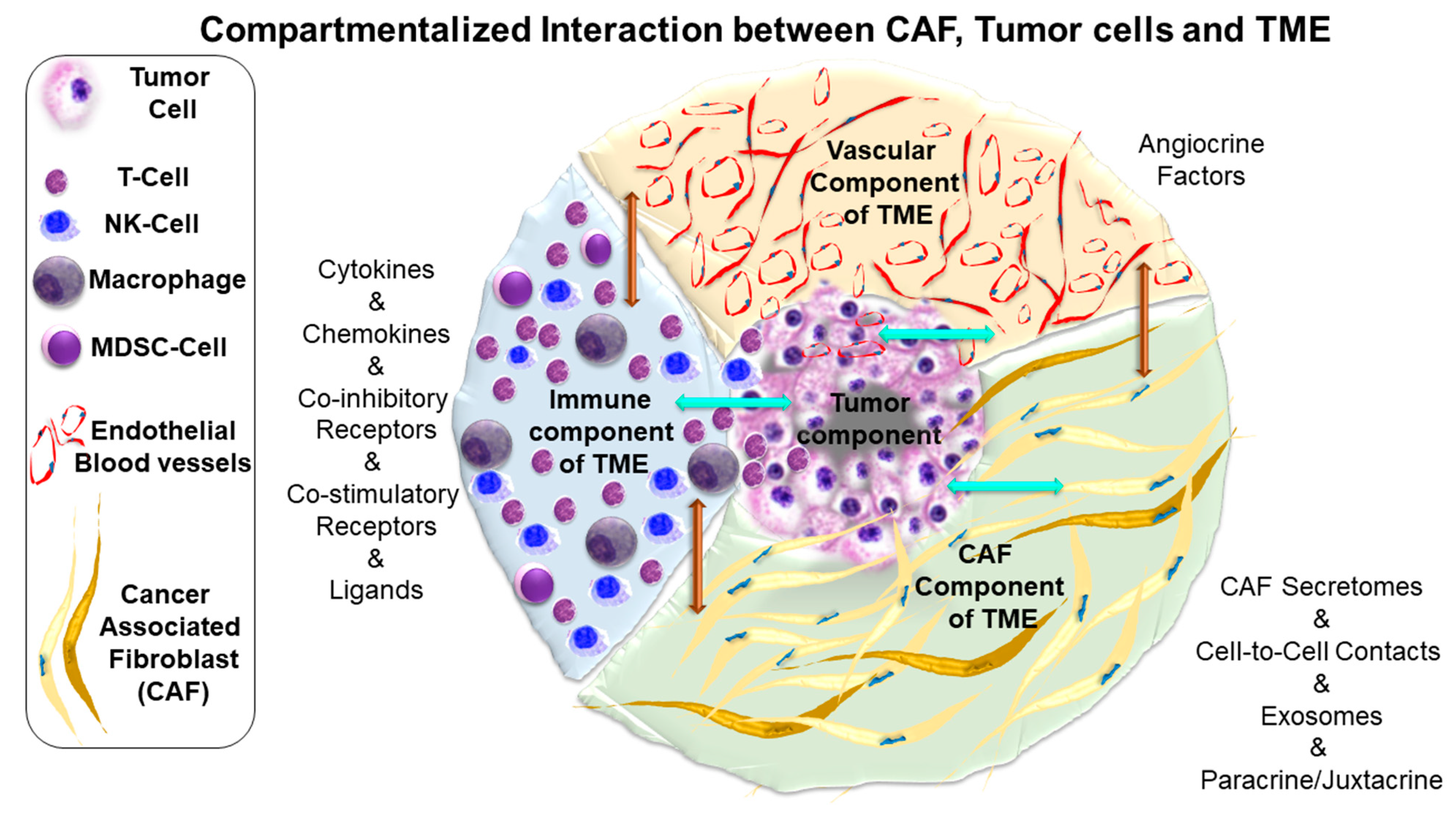
6. Language and Topic of Cross-Talk between CAF and Tumor Cells in Endometrial Cancers
7. CAF Influencing Proliferation and Growth of Tumor Cells in Endometrial Cancers
7.1. Steroid-Driven Proliferation of Endometrial Tumor Cells
7.2. Non-Steroidal Proliferation of Endometrial Tumor Cells
8. CAF Influencing Metastasis-Associated Phenotypes of Tumor Cells in Endometrial Cancers
8.1. Matrix Organization & Stromal Architecture
8.2. EMT
8.3. Migration, Invasion, and Metastatic Progression
9. CAF Influencing Immune-Defence of the Host and Immune-Surveillance of Tumor Cells by the Host in Endometrial Cancers
10. Epilogue
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviation
| CAF | Cancer-Associated Fibroblast |
| TGFA/B | Transforming Growth FactorA/B |
| TK | Tyrosine Kinase |
| STAT3 | Signal transducer and activator of transcription 3 |
| SPARC | Secreted Protein Acidic And Cysteine Rich |
| SATB2 | SATB Homeobox 2 |
| HIF-1alpha | Hypoxia Inducible Factor 1 Subunit Alpha |
| NK Cells | Natural Killer Cells |
| DNMT1 | DNA Methyltransferase 1 |
| SDF1 | Stromal Cell-derived factor 1 |
| CXCR4 | C-X-C Motif Chemokine Receptor 4 |
| PCNA | Proliferating Cell Nuclear Antigen |
| EMT | Epithelial-Mesenchymal Transition |
| CAF | Cancer-Associated Fibroblast |
| TGFA/B | Transforming Growth FactorA/B |
| VEGFC | Vascular endothelial growth factorC |
| TK | Tyrosine Kinase |
| STAT3 | Signal transducer and activator of transcription 3 |
| SPARC | Secreted Protein Acidic And Cysteine Rich |
| SATB2 | SATB Homeobox 2 |
| HIF-1alpha | Hypoxia Inducible Factor 1 Subunit Alpha |
| NK Cells | Natural Killer Cells |
| DNMT1 | DNA Methyltransferase 1 |
| SDF1 | Stromal Cell-derived factor 1 |
| CXCR4 | C-X-C Motif Chemokine Receptor 4 |
| PCNA | Proliferating Cell Nuclear Antigen |
| EMT | Epithelial-Mesenchymal Transition |
| CAF | Cancer-Associated Fibroblast |
| NK Cells | Natural Killer Cells |
| MDSC | Myeloid deriver stromal cells |
| TME | Tumor microenvironment |
References
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lövrot, J.; Larsson, C.; Sommarin, M.; Madsen, C.D.; Lindgren, D.; Pekar, G.; Karlsson, G.; et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 2018, 9, 5150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganguly, D.; Chandra, R.; Karalis, J.; Teke, M.; Aguilera, T.; Maddipati, R.; Wachsmann, M.B.; Ghersi, D.; Siravegna, G.; Zeh, H.J.; et al. Cancer-Associated Fibroblasts: Versatile Players in the Tumor Microenvironment. Cancers 2020, 12, 2652. [Google Scholar] [CrossRef] [PubMed]
- Öhlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523. [Google Scholar] [CrossRef] [PubMed]
- Ishii, G.; Ochiai, A.; Neri, S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv. Drug Deliv. Rev. 2016, 99 Pt B, 186–196. [Google Scholar] [CrossRef]
- Raz, Y.; Cohen, N.; Shani, O.; Bell, R.E.; Novitskiy, S.V.; Abramovitz, L.; Levy, C.; Milyavsky, M.; Leider-Trejo, L.; Moses, H.L.; et al. Bone marrow-derived fibroblasts are a functionally distinct stromal cell population in breast cancer. J. Exp. Med. 2018, 215, 3075–3093. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Daquinag, A.C.; Amaya-Manzanares, F.; Sirin, O.; Tseng, C.; Kolonin, M.G. Stromal progenitor cells from endogenous adipose tissue contribute to pericytes and adipocytes that populate the tumor microenvironment. Cancer Res. 2012, 72, 5198–5208. [Google Scholar] [CrossRef] [Green Version]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurmik, M.; Ullmann, P.; Rodriguez, F.; Haan, S.; Letellier, E. In search of definitions: Cancer-associated fibroblasts and their markers. Int. J. Cancer 2020, 146, 895–905. [Google Scholar] [CrossRef] [Green Version]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Beacham, D.A.; Cukierman, E. Stromagenesis: The changing face of fibroblastic microenvironments during tumor progression. Semin. Cancer Biol. 2005, 15, 329–341. [Google Scholar] [CrossRef]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.; et al. Stromal Elements Act to Restrain, Rather Than Support, Pancreatic Ductal Adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [Green Version]
- OÖzdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.; Zheng, X.; Wu, C.-C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biffi, G.; Tuveson, D.A. Diversity and Biology of Cancer—Associated Fibroblasts. Physiol. Rev. 2021, 101, 147–176. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Taketyama, H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 1–15. [Google Scholar] [CrossRef]
- Karagiannis, G.S.; Poutahidis, T.; Etrdman, S.E.; Kirsch, R.; Riddell, R.H.; Dialmandis, E. Cancer-Associated Fibroblasts Drive the Progression of Metastasis through both Paracrine and Mechanical Pressure on Cancer Tissue. Mol. Cancer Res. 2012, 10, 1403–1418. [Google Scholar] [CrossRef] [Green Version]
- Bissell, M.J.; Radisky, D. Putting tumours in context. Nat. Rev. Cancer 2001, 1, 46–54. [Google Scholar] [CrossRef] [Green Version]
- Liao, Z.; Tan, Z.W.; Zhu, P.; Taln, N.S. Cancer-associated fibroblasts in tumor microenvironment—Accomplices in tumor malignancy. Cell. Immunol. 2019, 343, 103729. [Google Scholar] [CrossRef]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Dzobo, K.; Dandara, C. Architecture of Cancer-Associated Fibroblasts in Tumor Microenvironment: Mapping Their Origins, Heterogeneity, and Role in Cancer Therapy Resistance. OMICS 2020, 24, 314–339. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef]
- Walter, S.G.; Scheidt, S.; Nißletr, R.; Gaisendrees, C.; Zarghooni, K.; Schildberg, F. In-Depth Characterization of Stromal Cells within the Tumor Microenvironment Yields Novel Therapeutic Targets. Cancers 2021, 13, 1466. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, H.; Mundel, T.M.; Kieran, M.; Kalluri, R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol. Ther. 2006, 5, 1640–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuzet, S.-E.; Gaggioli, C. Fibroblast activation in cancer: When seed fertilizes soil. Cell Tissue Res. 2016, 365, 607–619. [Google Scholar] [CrossRef]
- Shen, T.; Li, Y.; Zhu, S.; Yu, J.; Zhang, B.; Chetn, X.; Zhalng, Z.; Ma, Y.; Niu, Y.; Shang, Z. YAP1 plays a key role of the conversion of normal fibroblasts into cancer-associated fibroblasts that contribute to prostate cancer progression. J. Exp. Clin. Cancer Res. 2020, 39, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Olumi, A.F.; Grossfeld, G.D.; Hayward, S.W.; Carroll, P.R.; Tlsty, T.D.; Cunhaet, G.R. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999, 59, 5002–5011. [Google Scholar] [PubMed]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal Fibroblasts Present in Invasive Human Breast Carcinomas Promote Tumor Growth and Angiogenesis through Elevated SDF-1/CXCL12 Secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Sneddon, J.B.; Zhen, H.H.; Montgomery, K.; van de Rijn, M.; Tward, A.D.; West, R.; Gladstone, H.; Chang, H.Y.; Morganroth, G.S.; Oro, A.E.; et al. Bone morphogenetic protein antagonist gremlin 1 is widely expressed by cancer-associated stromal cells and can promote tumor cell proliferation. Proc. Natl. Acad. Sci. USA 2006, 103, 14842–14847. [Google Scholar] [CrossRef] [Green Version]
- Polyak, K.; Haviv, I.; Campbell, I. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009, 25, 30–38. [Google Scholar] [CrossRef]
- Cardone, A.; Tolino, A.; Zarcone, R.; Caracciolo, G.B.; Tartaglia, E. Prognostic value of desmoplastic reaction and lymphocytic infiltration in the management of breast cancer. Panminerva Med. 1997, 39, 174–177. [Google Scholar] [PubMed]
- Kellermann, M.G.; Sobral, L.; da Silva, S.D.; Zecchin, K.G.; Graner, E.; Lopes, M.A.; Kowalski, L.P.; Coletta, R.D. Mutual paracrine effects of oral squamous cell carcinoma cells and normal oral fibroblasts: Induction of fibroblast to myofibroblast transdifferentiation and modulation of tumor cell proliferation. Oral Oncol. 2008, 44, 509–517. [Google Scholar] [CrossRef] [PubMed]
- De Nola, R.; Metnga, A.; Castegna, A.; Loizzi, V.; Ranieri, G.; Cicinelli, E.; Cormio, G. The Crowded Crosstalk between Cancer Cells and Stromal Microenvironment in Gynecological Malignancies: Biological Pathways and Therapeutic Implication. Int. J. Mol. Sci. 2019, 20, 2401. [Google Scholar] [CrossRef] [Green Version]
- Tao, L.; Huang, G.; Song, H.; Chetn, Y.; Chen, L. Cancer associated fibroblasts: An essential role in the tumor microenvironment. Oncol. Lett. 2017, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef] [Green Version]
- Vennin, C.; Murphy, K.J.; Morton, J.; Cox, T.R.; Pajic, M.; Timpson, P. Reshaping the Tumor Stroma for Treatment of Pancreatic Cancer. Gastroenterology 2018, 154, 820–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vennin, C.; Apgi, A.P.G.I.; Mélénec, P.; Rouet, R.; Nobis, M.; Cazet, A.S.; Murphy, K.J.; Herrmann, D.; Reed, D.A.; Lucas, M.C.; et al. CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nat. Commun. 2019, 10, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdogan, B.; Webb, D.J. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem. Soc. Trans. 2017, 45, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Winuthayanon, W.; Lierz, S.; Delarosa, K.C.; Sampels, S.R.; Donoghue, L.J.; Hewitt, S.C.; Korach, K.S. Juxtacrine Activity of Estrogen Receptor alpha in Uterine Stromal Cells is Necessary for Estrogen-Induced Epithelial Cell Proliferation. Sci. Rep. 2017, 7, 8377. [Google Scholar] [CrossRef]
- Imai, A.; Matsunami, K.; Iida, K.; Tamaya, T. Inhibitory action of estradiol on growth promoting activity in extract from uterine cancers. Biosci. Rep. 1990, 10, 47–53. [Google Scholar] [CrossRef]
- Senol, S.; Sayar, I.; Ceyran, A.B.; Ibiloglu, I.; Akalin, I.; Firat, U.; Kosemetin, D.; Zerk, R.E.; Aydin, A. Stromal Clues in Endometrial Carcinoma: Loss of Expression of beta-Catenin, Epithelial-Mesenchymal Transition Regulators, and Estrogen-Progesterone Receptor. Int. J. Gynecol. Pathol. 2016, 35, 238–248. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, J.; Hori, M.; Ichigo, S.; Morishita, S.; Tamaya, T. Estrogen induces expression of c-fos and c-jun via activation of protein kinase C in an endometrial cancer cell line and fibroblasts derived from human uterine endometrium. Gynecol. Endocrinol. 1996, 10, 109–118. [Google Scholar] [CrossRef]
- Fujimoto, J.; Ichigo, S.; Hori, M.; Morishita, S.; Tamaya, T. Estrogen induces c-Ha-ras expression via activation of tyrosine kinase in uterine endometrial fibroblasts and cancer cells. J. Steroid Biochem. Mol. Biol. 1995, 55, 25–33. [Google Scholar] [CrossRef]
- Fujimoto, J.; Ichigo, S.; Hori, M.; Morishita, S.; Tamaya, T. Oestrogen Induces C-Ha-Ras Expression in the Fibroblasts Derived from Human Uterine Endometrium. Ann. Clin. Biochem. Int. J. Lab. Med. 1995, 32, 487–492. [Google Scholar] [CrossRef]
- Pineda, M.J.; Lu, Z.; Cao, D.; Kim, J.J. Influence of Cancer-Associated Endometrial Stromal Cells on Hormone-Driven Endometrial Tumor Growth. Horm. Cancer 2015, 6, 131–141. [Google Scholar] [CrossRef] [Green Version]
- Subramaniam, K.S.; Tham, S.T.; Mohamed, Z.; Woo, Y.L.; Adenan, N.A.M.; Chung, I. Cancer-Associated Fibroblasts Promote Proliferation of Endometrial Cancer Cells. PLoS ONE 2013, 8, e68923. [Google Scholar] [CrossRef]
- Subramaniam, K.S.; Omar, I.S.; Kwong, S.C.; Mohamed, Z.; Woo, Y.L.; Adenan, N.A.M.; Chung, I. Cancer-associated fibroblasts promote endometrial cancer growth via activation of interleukin-6/STAT-3/c-Myc pathway. Am. J. Cancer Res. 2016, 6, 200–213. [Google Scholar]
- Rinehart, C.A.; Watson, J.M.; Torti, V.R.; Palmieri, D. The Role of Interleukin-1 in Interactive Senescence and Age-Related Human Endometrial Cancer. Exp. Cell Res. 1999, 248, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, D.; Watson, J.M.; Rinehart, C.A. Age-related expression of PEDF/EPC-1 in human endometrial stromal fibroblasts: Implications for interactive senescence. Exp. Cell Res. 1999, 247, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Pankova, D.; Chen, Y.; Terajima, M.; Schliekelman, M.J.; Baird, B.N.; Fahrenholtz, M.; Sun, L.; Gill, B.J.; Vadakkan, T.J.; Kim, M.P.; et al. Cancer-Associated Fibroblasts Induce a Collagen Cross-link Switch in Tumor Stroma. Mol. Cancer Res. 2015, 14, 287–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, S.; Asanoma, K.; Yagi, H.; Onoyama, I.; Hori, E.; Matsumura, Y.; Okugawa, K.; Yahata, H.; Kato, K. Fibronectin mediates activation of stromal fibroblasts by SPARC in endometrial cancer cells. BMC Cancer 2021, 21, 1–12. [Google Scholar] [CrossRef]
- Wang, X.; Sun, X.; Mu, L.; Chen, W. Cancer-Associated Fibroblasts Induce Epithelial-Mesenchymal Transition in Endometrial Cancer Cells by Regulating Pituitary Tumor Transforming Gene. Cancer Investig. 2019, 37, 134–143. [Google Scholar] [CrossRef]
- Xie, R.; Schlumbrecht, M.P.; Shipley, G.L.; Xie, S.; Bassett, R.L., Jr.; Broaddus, R.R. S100A4 mediates endometrial cancer invasion and is a target of TGF-beta1 signaling. Lab. Investig. 2009, 89, 937–947. [Google Scholar] [CrossRef] [Green Version]
- Maehara, Y.; Kakeji, Y.; Kabashima, A.; Emi, Y.; Watanabe, A.; Akazawa, K.; Baba, H.; Kohnoe, S.; Sugimachi, K. Role of transforming growth factor-beta 1 in invasion and metastasis in gastric carcinoma. J. Clin. Oncol. 1999, 17, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Kasai, H.; Allen, J.T.; Mason, R.M.; Kamimura, T.; Zhang, Z. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir. Res. 2005, 6, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zavadil, J.; Bottinger, E.P. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene 2005, 24, 5764–5774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, H.; Danoff, T.M.; Kalluri, R.; Neilson, E.G. Early role of Fsp1 in epithelial-mesenchymal transformation. Am. J. Physiol. Physiol. 1997, 273, F563–F574. [Google Scholar] [CrossRef]
- Lawson, W.E.; Polosukhin, V.V.; Zoia, O.; Stathopoulos, G.T.; Han, W.; Plieth, D.; Loyd, J.E.; Neilson, E.G.; Blackwell, T.S. Characterization of Fibroblast-specific Protein 1 in Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2005, 171, 899–907. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of Endothelial to Mesenchymal Transition as a Source for Carcinoma-Associated Fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhang, W.; Sun, X.; Lin, Y.; Chen, W. Cancer-associated fibroblasts induce epithelial-mesenchymal transition through secreted cytokines in endometrial cancer cells. Oncol. Lett. 2018, 15, 5694–5702. [Google Scholar] [CrossRef] [Green Version]
- Di Leva, G.; Croce, C.M. Roles of small RNAs in tumor formation. Trends Mol. Med. 2010, 16, 257–267. [Google Scholar] [CrossRef] [Green Version]
- Aprelikova, O.; Yu, X.; Palla, J.; Wei, B.-R.; John, S.; Yi, M.; Stephens, R.; Simpson, R.M.; Risinger, J.I.; Jazaeri, A.; et al. The role of miR-31 and its target gene SATB2 in cancer-associated fibroblasts. Cell Cycle 2010, 9, 4387–4398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shurin, M.R. MicroRNAs are invading the tumor microenvironment: Fibroblast microRNAs regulate tumor cell motility and invasiveness. Cell Cycle 2010, 9, 4430–4431. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.-T.; Zhou, Z.-Y.; Luo, Y.-L.; Luo, Q.; Chen, S.-B.; Zhao, J.-C.; Chen, Q.-R. Exosomal lncRNA NEAT1 from cancer-associated fibroblasts facilitates endometrial cancer progression via miR-26a/b-5p-mediated STAT3/YKL-40 signaling pathway. Neoplasia 2021, 23, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Wang, Y.; Liu, H.; Shen, W. Extracellular vesicle encapsulated microRNA-320a inhibits endometrial cancer by suppression of the HIF1α/VEGFA axis. Exp. Cell Res. 2020, 394, 112113. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Bueno, G.; Hardisson, D.; Sánchez, C.; Sarrió, D.; Cassia, R.; García-Rostán, G.; Prat, J.; Guo, M.; Herman, J.G.; Matías-Guiu, X.; et al. Abnormalities of the APC/beta-catenin pathway in endometrial cancer. Oncogene 2002, 21, 7981–7990. [Google Scholar] [CrossRef] [Green Version]
- Aprelikova, O.; Palla, J.; Hibler, B.; Yu, X.; Greer, Y.E.; Yi, M.; Stephens, R.; Maxwell, G.L.; Jazaeri, A.; Risinger, J.I.; et al. Silencing of miR-148a in cancer-associated fibroblasts results in WNT10B-mediated stimulation of tumor cell motility. Oncogene 2012, 32, 3246–3253. [Google Scholar] [CrossRef] [Green Version]
- Olson, P.; Lu, J.; Zhang, H.; Shai, A.; Chun, M.G.; Wang, Y.; Libutti, S.K.; Nakakura, E.K.; Golub, T.R.; Hanahan, D. MicroRNA dynamics in the stages of tumorigenesis correlate with hallmark capabilities of cancer. Genes Dev. 2009, 23, 2152–2165. [Google Scholar] [CrossRef] [Green Version]
- Budhu, A.; Jia, H.-L.; Forgues, M.; Liu, C.-G.; Goldstein, D.; Lam, A.; Zanetti, K.A.; Ye, Q.-H.; Qin, L.-X.; Croce, C.M.; et al. Identification of metastasis-related microRNAs in hepatocellular carcinoma. Hepatology 2008, 47, 897–907. [Google Scholar] [CrossRef]
- Jiang, L.; Gonda, T.A.; Gamble, M.V.; Salas, M.; Seshan, V.; Tu, S.; Twaddell, W.S.; Hegyi, P.; Lazar, G.; Steele, I.; et al. Global Hypomethylation of Genomic DNA in Cancer-Associated Myofibroblasts. Cancer Res. 2008, 68, 9900–9908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiegl, H.; Millinger, S.; Goebel, G.; Müller-Holzner, E.; Marth, C.; Laird, P.W.; Widschwendter, M. Breast Cancer DNA Methylation Profiles in Cancer Cells and Tumor Stroma: Association with HER-2/neu Status in Primary Breast Cancer. Cancer Res. 2006, 66, 29–33. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Lu, W.; Qu, J.; Ye, L.; Du, G.; Wan, X. Loss of exosomal miR-148b from cancer-associated fibroblasts promotes endometrial cancer cell invasion and cancer metastasis. J. Cell. Physiol. 2018, 234, 2943–2953. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.; Konno, Y.; Watari, H.; Hosaka, M.; Noguchi, M.; Sakuragi, N. The impact of microRNA-mediated PI3K/AKT signaling on epithelial-mesenchymal transition and cancer stemness in endometrial cancer. J. Transl. Med. 2014, 12, 231. [Google Scholar] [CrossRef] [PubMed]
- Teng, F.; Tian, W.-Y.; Wang, Y.-M.; Zhang, Y.-F.; Guo, F.; Zhao, J.; Gao, C.; Xue, F.-X. Cancer-associated fibroblasts promote the progression of endometrial cancer via the SDF-1/CXCR4 axis. J. Hematol. Oncol. 2016, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Barrett, R.L.; Puré, E. Cancer-associated fibroblasts and their influence on tumor immunity and immunotherapy. eLife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, O.; Annibali, D.; Aguzzi, C.; Tuyaerts, S.; Amant, F.; Morelli, M.B.; Santoni, G.; Amantini, C.; Maggi, F.; Nabissi, M. The Controversial Role of PD-1 and Its Ligands in Gynecological Malignancies. Front. Oncol. 2019, 9, 1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, C.; Miki, Y.; Saito, R.; Hata, S.; Abe, J.; Sato, I.; Okada, Y.; Sasano, H. PD-L1 Induction by Cancer-Associated Fibroblast-Derived Factors in Lung Adenocarcinoma Cells. Cancers 2019, 11, 1257. [Google Scholar] [CrossRef] [Green Version]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef]
- Ford, K.; Hanley, C.J.; Mellone, M.; Szyndralewiez, C.; Heitz, F.; Wiesel, P.; Wood, O.; Machado, M.; Lopez, M.-A.; Ganesan, A.-P.; et al. NOX4 Inhibition Potentiates Immunotherapy by Overcoming Cancer-Associated Fibroblast-Mediated CD8 T-cell Exclusion from Tumors. Cancer Res. 2020, 80, 1846–1860. [Google Scholar] [CrossRef] [Green Version]
- Inoue, T.; Adachi, K.; Kawana, K.; Taguchi, A.; Nagamatsu, T.; Fujimoto, A.; Tomio, K.; Yamashita, A.; Eguchi, S.; Nishida, H.; et al. Cancer-associated fibroblast suppresses killing activity of natural killer cells through downregulation of poliovirus receptor (PVR/CD155), a ligand of activating NK receptor. Int. J. Oncol. 2016, 49, 1297–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, P.; Huang, X.; Zhou, X.; Lu, Z.; Liu, F.; Shi, Z.; Lu, L.; Wu, Y.; Chen, Y. Loss of CD155 expression predicts poor prognosis in hepatocellular carcinoma. Histopathology 2014, 66, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Stoker, M.G.P.; Shearer, M.; O’Neill, C. Growth Inhibition of Polyoma-Transformed Cells by Contact with Static Normal Fibroblasts. J. Cell Sci. 1966, 1, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, Q.; Tan, Y.; Tang, Y.; Yet, J.; Yualn, B.; Yu, W. Cancer-Associated Fibroblasts Suppress Cancer Development: The Other Side of the Coin. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef]
- Neesse, A.; Bauer, C.A.; Öhlund, D.; Lauth, M.; Buchholz, M.; Michl, P.; Tuveson, D.A.; Gress, T.M. Stromal biology and therapy in pancreatic cancer: Ready for clinical translation? Gut 2019, 68, 159–171. [Google Scholar] [CrossRef]
- Bissell, M.J.; Hines, W.C. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 2011, 17, 320–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizutani, Y.; Kobayashi, H.; Iida, T.; Asai, N.; Masamune, A.; Hara, A.; Esaki, N.; Ushida, K.; Mii, S.; Shiraki, Y.; et al. Meflin-Positive Cancer-Associated Fibroblasts Inhibit Pancreatic Carcinogenesis. Cancer Res. 2019, 79, 5367–5381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyai, Y.; Esaki, N.; Takahashi, M.; Enomoto, A. Cancer-associated fibroblasts that restrain cancer progression: Hypotheses and perspectives. Cancer Sci. 2020, 111, 1047–1057. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| CAF-Tumor Cell Cross-Talk | Language of Cross-Talk (Mode of Signal Transduction between CAF and Tumor Cell) | Agenda of Cross-Talk (How Cross-Talk Alter Tumor Cell Functions?) | Result of Cross-Talk (Which Tumor Phenotypes Are Affected by Cross-Talk?) | Reference (PMID) |
|---|---|---|---|---|
| Growth | ||||
| Angiogenesis focused signals | TGFA, TGFB2 and TGFBR1 and VEGFC | Steroid Hormone-driven proliferation | Growth | 25976290 |
| Estrogen increased cHa-ras expression and tyrosine kinase (TK) activity in fibroblast | Induction of c-Ha-RAS transcripts in endometrial cancers; persistent activation of TK led to overexpression of c-Ha-RAS in some endometrial cancer cells under predominant estrogen milieu | Estrogen-driven | Growth | 7577718 |
| Estrogen induces expression of c-fos and c-jun in endometrial cancer cell line and fibroblasts | via activation of protein kinase C | Estrogen-driven | Growth | 8701784 |
| Proliferation | ||||
| STAT-3 target genes | Interleukin-6/STAT-3/c-Myc pathway | Non-steroidal | Proliferation | 27186396 |
| CAFs secrete higher levels of macrophage chemoattractant protein (MCP)-1, interleukin (IL)-6, IL-8, RANTES, and vascular endothelial growth factor (VEGF) | CAF secretome induced cell proliferation | PI3K/AKT and MAPK/ERK signaling | Proliferation | 23922669 |
| Stromagenesis, MA-Phenotypes, EMT, Progression & Stemness | ||||
| CAF induces gene expression | Pituitary tumor transforming gene in tumor cells | Invasion, Migration, and EMT | MA-Phenotypes | 30961403 |
| SPARC-expressing endometrial cancer cells | SPARC from tumor cells activated fibroblasts in the presence of fibronectin | A matricellular glycoprotein, SPARC | Stromagenesis, mobility and invasion of cancer cells | 33579227 |
| Exosome-mediated transfer of miR-148b from CAF to tumor cells | Downregulated miR-148b in CAF induced EMT of cancer cell as a result of relieving the suppression of DNMT1 | miR-148b as a tumor suppressor by binding to its downstream target gene, DNMT1 | EMT | 30146796 |
| MicroRNA and transcriptional regulators | SATB2 gene | miR-31 | Migration & Invasion | 20980827; 21088483 |
| SDF-1α is a novel independent poor prognostic factor | Paracrine- or autocrine- activation of the PI3K/AKT and MAPK/ERK signalings | SDF1alpha /CXCR4 axis | Progression | 26851944 |
| Invasive myofibroblasts adjacent to malignant epithelial cells of endometrial cancers showed frequently intensive positive staining of VEGF, IGF1, and EGF, the cognate receptors such as Fetal liver kinase-1/Kinase Insert Domain-containing receptor/VEGF receptor-2, fms-like tyrosine kinase-1/VEGF receptor-1, and EGRF, several cell cycle regulators such as cyclins and cyclin- dependent kinases, and estrogen receptor alpha | Growth factors, their cognate receptors, and HIF-1alpha | Myofibroblasts, as well as cancer epithelial cells, are positive staining for PCNA and Ki-67 | Progression | 11595701 |
| Activation of the WNT/β-catenin pathway in CAFs | miR-148a expression is suppressed in CAFs; Silencing of miR-148a in CAFs promotes the migration of endometrial cancer cells by targeting Wnt10B to activate the Wnt/β-catenin pathway | WNT10B stimulated migration of endometrial cancer cell lines; WNT10B is a direct target of miR-148a in endometrial CAFs | Cell motility & Invasion | 22890324 |
| Metabolic Reprogramming | ||||
| ROS, produced by CAFs or tumor cells; CAFs-derived exosomes | CAFs exhibit the Warburg effect and activation of the autophagic pathway | A metabolic symbiosis between epithelial cancer cells and CAFs | Metabolic Reprogramming in tumors | 26445347 |
| Immunological Reprogramming | ||||
| Suppression of NK cell activity by CAFs | Cell-to-cell interaction required CAF-induced decrease in NK cell activity | Exosome-independent | Immunological Reprogramming | 27499237 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pradip, D.; Jennifer, A.; Nandini, D. Cancer-Associated Fibroblasts in Conversation with Tumor Cells in Endometrial Cancers: A Partner in Crime. Int. J. Mol. Sci. 2021, 22, 9121. https://doi.org/10.3390/ijms22179121
Pradip D, Jennifer A, Nandini D. Cancer-Associated Fibroblasts in Conversation with Tumor Cells in Endometrial Cancers: A Partner in Crime. International Journal of Molecular Sciences. 2021; 22(17):9121. https://doi.org/10.3390/ijms22179121
Chicago/Turabian StylePradip, De, Aske Jennifer, and Dey Nandini. 2021. "Cancer-Associated Fibroblasts in Conversation with Tumor Cells in Endometrial Cancers: A Partner in Crime" International Journal of Molecular Sciences 22, no. 17: 9121. https://doi.org/10.3390/ijms22179121
APA StylePradip, D., Jennifer, A., & Nandini, D. (2021). Cancer-Associated Fibroblasts in Conversation with Tumor Cells in Endometrial Cancers: A Partner in Crime. International Journal of Molecular Sciences, 22(17), 9121. https://doi.org/10.3390/ijms22179121