
Exploring the Binding Mechanism of PF-07321332 SARS-CoV-2 Protease Inhibitor through Molecular Dynamics and Binding Free Energy Simulations


Abstract
:1. Introduction
2. Results
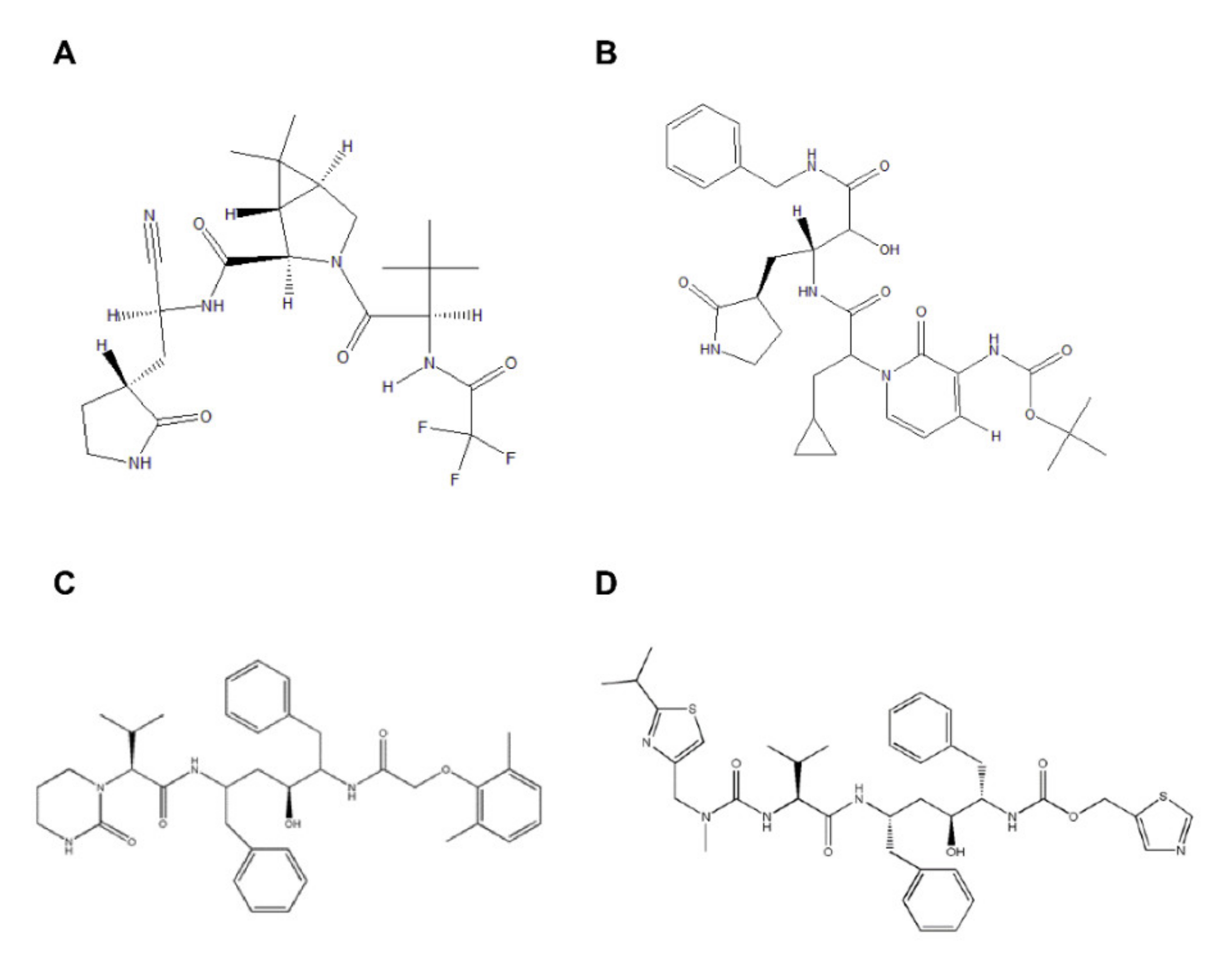
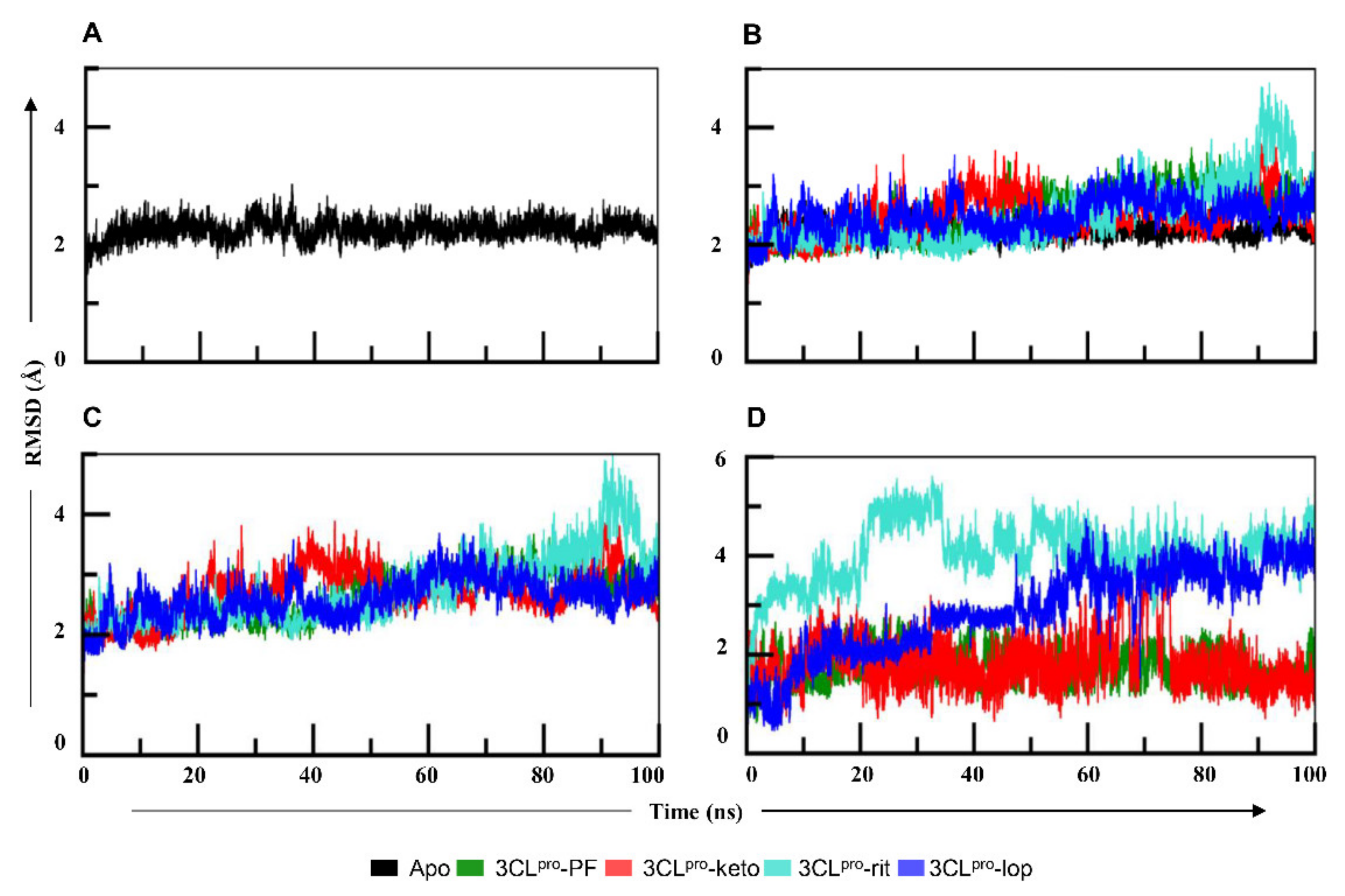
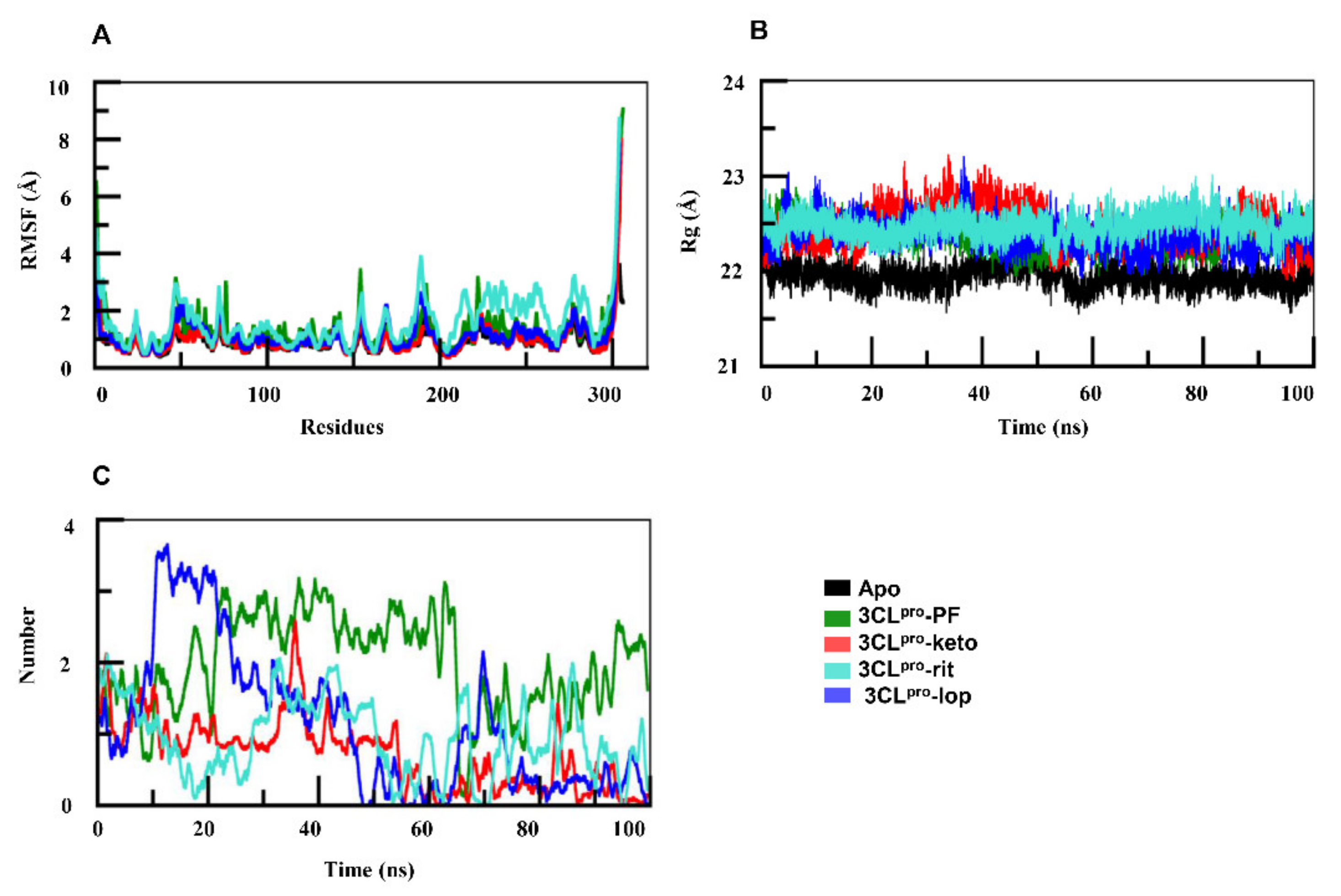
2.1. Comparative Analysis of PF-07321332, α-ketoamide, Lopinavir, and Ritonavir Binding to 3CLpro
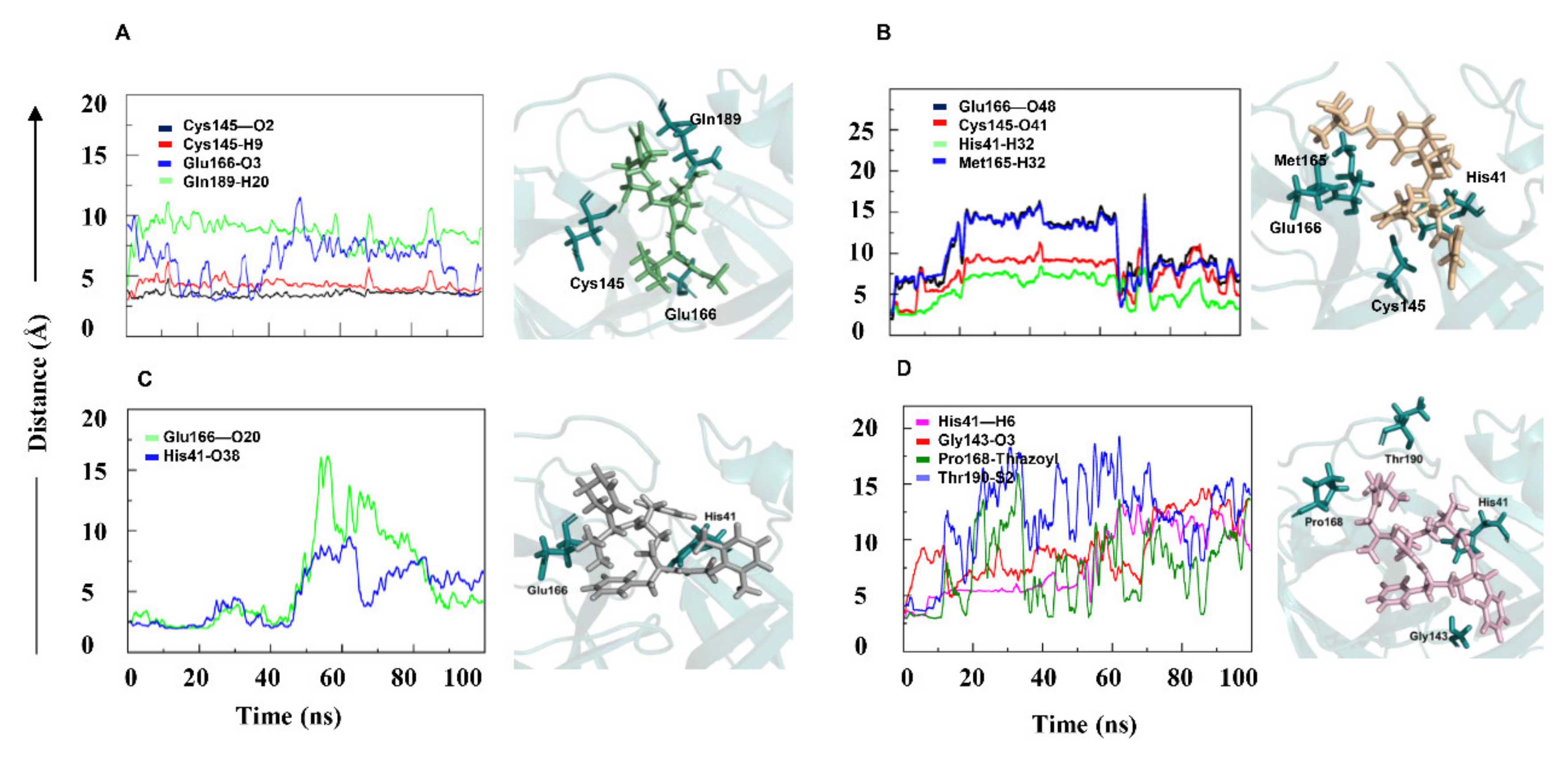
2.2. Interaction Analysis of PF-07321332, α-ketoamide, Lopinavir, and Ritonavir with 3CLpro
2.3. Binding Free Energy Calculation
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. System Preparation for Molecular Docking and MD Simulations
5.2. Molecular Docking of 3CLpro to Lopinavir and Ritonavir
5.3. Building the Ligand Topology
5.4. MD Simulations of Complexes
5.5. Binding-Free-Energy Calculation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Velavan, T.P.; Meyer, C.G. The COVID-19 epidemic. Trop. Med. Int. Health 2020, 25, 278–280. [Google Scholar] [CrossRef] [Green Version]
- Johns Hopkins Coronavirus Resource Center. COVID-19 Map. Available online: https://coronavirus.jhu.edu/map.html (accessed on 10 August 2020).
- Dai, W.; Zhang, B.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Liu, F.; Li, C.; Li, Y.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Durai, P.; Batool, M.; Shah, M.; Choi, S. Middle East respiratory syndrome coronavirus: Transmission, virology and therapeutic targeting to aid in outbreak control. Exp. Mol. Med. 2015, 47, e181. [Google Scholar] [CrossRef] [PubMed]
- Batool, M.; Shah, M.; Patra, M.C.; Yesudhas, D.; Choi, S. Structural insights into the Middle East respiratory syndrome coronavirus 4a protein and its dsRNA binding mechanism. Sci. Rep. 2017, 7, 11362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [Green Version]
- Van Boheemen, S.; de Graaf, M.; Lauber, C.; Bestebroer, T.M.; Raj, V.S.; Zaki, A.M.; Osterhaus, A.D.; Haagmans, B.L.; Gorbalenya, A.E.; Snijder, E.J.; et al. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. mBio 2012, 3, e00473-12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muramatsu, T.; Takemoto, C.; Kim, Y.T.; Wang, H.; Nishii, W.; Terada, T.; Shirouzu, M.; Yokoyama, S. SARS-CoV 3CL protease cleaves its C-terminal autoprocessing site by novel subsite cooperativity. Proc. Natl. Acad. Sci. USA 2016, 113, 12997–13002. [Google Scholar] [CrossRef] [Green Version]
- Thiel, V.; Ivanov, K.A.; Putics, A.; Hertzig, T.; Schelle, B.; Bayer, S.; Weissbrich, B.; Snijder, E.J.; Rabenau, H.; Doerr, H.W.; et al. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 2003, 84, 2305–2315. [Google Scholar] [CrossRef]
- Ren, Z.; Yan, L.; Zhang, N.; Guo, Y.; Yang, C.; Lou, Z.; Rao, Z. The newly emerged SARS-like coronavirus HCoV-EMC also has an “Achilles’ heel”: Current effective inhibitor targeting a 3C-like protease. Protein Cell 2013, 4, 248–250. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, H.; Mathieu, E.; Rodés-Guirao, L.; Appel, C.; Giattino, C.; Ortiz-Ospina, E.; Hasell, J.; Macdonald, B.; Beltekian, D.; Roser, M. Coronavirus pandemic (COVID-19). 2020. Available online: https://ourworldindata.org/covid-vaccinations (accessed on 31 July 2021).
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Kalil, A.C. Treating COVID-19-Off-Label Drug Use, Compassionate Use, and Randomized Clinical Trials During Pandemics. JAMA 2020, 323, 1897–1898. [Google Scholar] [CrossRef]
- Arabi, Y.M.; Alothman, A.; Balkhy, H.H.; Al-Dawood, A.; Al Johani, S.; Al Harbi, S.; Kojan, S.; Al Jeraisy, M.; Deeb, A.M.; Assiri, A.M.; et al. Treatment of Middle East Respiratory Syndrome with a combination of lopinavir-ritonavir and interferon-beta1b (MIRACLE trial): Study protocol for a randomized controlled trial. Trials 2018, 19, 81. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.M.; Cheng, V.C.; Hung, I.F.; Wong, M.M.; Chan, K.H.; Chan, K.S.; Kao, R.Y.; Poon, L.L.; Wong, C.L.; Guan, Y.; et al. Role of lopinavir/ritonavir in the treatment of SARS: Initial virological and clinical findings. Thorax 2004, 59, 252–256. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Chan, K.H.; Jiang, Y.; Kao, R.Y.; Lu, H.T.; Fan, K.W.; Cheng, V.C.; Tsui, W.H.; Hung, I.F.; Lee, T.S.; et al. In vitro susceptibility of 10 clinical isolates of SARS coronavirus to selected antiviral compounds. J. Clin. Virol. 2004, 31, 69–75. [Google Scholar] [CrossRef]
- Wu, C.Y.; Jan, J.T.; Ma, S.H.; Kuo, C.J.; Juan, H.F.; Cheng, Y.S.; Hsu, H.H.; Huang, H.C.; Wu, D.; Brik, A.; et al. Small molecules targeting severe acute respiratory syndrome human coronavirus. Proc. Natl. Acad. Sci. USA 2004, 101, 10012–10017. [Google Scholar] [CrossRef] [Green Version]
- Sevrioukova, I.F.; Poulos, T.L. Structure and mechanism of the complex between cytochrome P4503A4 and ritonavir. Proc. Natl. Acad. Sci. USA 2010, 107, 18422–18427. [Google Scholar] [CrossRef] [Green Version]
- Chandwani, A.; Shuter, J. Lopinavir/ritonavir in the treatment of HIV-1 infection: A review. Ther. Clin. Risk Manag. 2008, 4, 1023–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batool, M.; Ahmad, B.; Choi, S. A Structure-Based Drug Discovery Paradigm. Int. J. Mol. Sci. 2019, 20, 2783. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.C.; Chang, G.G.; Chou, C.Y. Mutation of Glu-166 blocks the substrate-induced dimerization of SARS coronavirus main protease. Biophys J. 2010, 98, 1327–1336. [Google Scholar] [CrossRef] [Green Version]
- Fornasier, E.; Macchia, M.L.; Giachin, G.; Sosic, A.; Pavan, M.; Sturlese, M.; Salata, C.; Moro, S.; Gatto, B.; Bellanda, M.; et al. A novel conformational state for SARS-CoV-2 main protease. bioxRiv 2021. [Google Scholar] [CrossRef]
- Bartas, M.; Brazda, V.; Bohalova, N.; Cantara, A.; Volna, A.; Stachurova, T.; Malachova, K.; Jagelska, E.B.; Porubiakova, O.; Cerven, J.; et al. In-Depth Bioinformatic Analyses of Nidovirales Including Human SARS-CoV-2, SARS-CoV, MERS-CoV Viruses Suggest Important Roles of Non-canonical Nucleic Acid Structures in Their Lifecycles. Front. Microbiol. 2020, 11, 1583. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J.J.; Vasudevan, K.; Pragasam, A.K.; Gunasekaran, K.; Veeraraghavan, B.; Mutreja, A. Evolutionary Tracking of SARS-CoV-2 Genetic Variants Highlights an Intricate Balance of Stabilizing and Destabilizing Mutations. mBio 2021, 12, e0118821. [Google Scholar] [CrossRef]
- West, A.P., Jr.; Wertheim, J.O.; Wang, J.C.; Vasylyeva, T.I.; Havens, J.L.; Chowdhury, M.A.; Gonzalez, E.; Fang, C.E.; Di Lonardo, S.S.; Hughes, S.; et al. Detection and characterization of the SARS-CoV-2 lineage B.1.526 in New York. Nat. Commun. 2021, 12, 4886. [Google Scholar] [CrossRef]
- Kim, J.S.; Jang, J.H.; Kim, J.M.; Chung, Y.S.; Yoo, C.K.; Han, M.G. Genome-Wide Identification and Characterization of Point Mutations in the SARS-CoV-2 Genome. Osong Public Health Res. Perspect. 2020, 11, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Goswami, P.; Bartas, M.; Lexa, M.; Bohalova, N.; Volna, A.; Cerven, J.; Cervenova, V.; Pecinka, P.; Spunda, V.; Fojta, M.; et al. SARS-CoV-2 hot-spot mutations are significantly enriched within inverted repeats and CpG island loci. Brief. Bioinform. 2021, 22, 1338–1345. [Google Scholar] [CrossRef]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe Covid-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef]
- Ul Qamar, M.T.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.L. Structural basis of SARS-CoV-2 3CL(pro) and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020, 10, 313–319. [Google Scholar] [CrossRef]
- Paasche, A.; Zipper, A.; Schäfer, S.; Ziebuhr, J.; Schirmeister, T.; Engels, B. Evidence for Substrate Binding-Induced Zwitterion Formation in the Catalytic Cys-His Dyad of the SARS-CoV Main Protease. Biochemistry 2014, 53, 5930–5946. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Yu, H.; Yang, H.; Xue, F.; Wu, Z.; Shen, W.; Li, J.; Zhou, Z.; Ding, Y.; Zhao, Q.; et al. Structures of two coronavirus main proteases: Implications for substrate binding and antiviral drug design. J. Virol. 2008, 82, 2515–2527. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Shi, D.; Zhou, S.; Liu, H.; Liu, H.; Yao, X. Molecular dynamics simulations and novel drug discovery. Expert Opin. Drug Discov. 2018, 13, 23–37. [Google Scholar] [CrossRef]
- Borhani, D.W.; Shaw, D.E. The future of molecular dynamics simulations in drug discovery. J. Comput. Aided Mol. Des. 2012, 26, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Mengist, H.M.; Dilnessa, T.; Jin, T. Structural Basis of Potential Inhibitors Targeting SARS-CoV-2 Main Protease. Front. Chem. 2021, 9, 622898. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.D.; Gao, K.; Chen, J.; Wang, R.; Wei, G.W. Unveiling the molecular mechanism of SARS-CoV-2 main protease inhibition from 137 crystal structures using algebraic topology and deep learning. Chem. Sci. 2020, 11, 12036–12046. [Google Scholar] [CrossRef]
- Jukic, M.; Skrlj, B.; Tomsic, G.; Plesko, S.; Podlipnik, C.; Bren, U. Prioritisation of Compounds for 3CL(pro) Inhibitor Development on SARS-CoV-2 Variants. Molecules 2021, 26, 3003. [Google Scholar] [CrossRef]
- Prajapati, J.; Patel, R.; Goswami, D.; Saraf, M.; Rawal, R.M. Sterenin M as a potential inhibitor of SARS-CoV-2 main protease identified from MeFSAT database using molecular docking, molecular dynamics simulation and binding free energy calculation. Comput. Biol. Med. 2021, 135, 104568. [Google Scholar] [CrossRef] [PubMed]
- Uniyal, A.; Mahapatra, M.K.; Tiwari, V.; Sandhir, R.; Kumar, R. Targeting SARS-CoV-2 main protease: Structure based virtual screening, in silico ADMET studies and molecular dynamics simulation for identification of potential inhibitors. J. Biomol. Struct. Dyn. 2020, 4, 32. [Google Scholar] [CrossRef]
- Ram, T.S.; Munikumar, M.; Raju, V.N.; Devaraj, P.; Boiroju, N.K.; Hemalatha, R.; Prasad, P.V.V.; Gundeti, M.; Sisodia, B.S.; Pawar, S.; et al. In silico evaluation of the compounds of the ayurvedic drug, AYUSH-64, for the action against the SARS-CoV-2 main protease. J. Ayurveda Integr. Med. 2021. [Google Scholar] [CrossRef]
- Sabbah, D.A.; Hajjo, R.; Bardaweel, S.K.; Zhong, H.A. An Updated Review on SARS-CoV-2 Main Proteinase (M(Pro)): Protein Structure and Small-Molecule Inhibitors. Curr. Top. Med. Chem. 2021, 21, 442–460. [Google Scholar] [CrossRef] [PubMed]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Wong-Sam, A.; Wang, Y.F.; Zhang, Y.; Ghosh, A.K.; Harrison, R.W.; Weber, I.T. Drug Resistance Mutation L76V Alters Nonpolar Interactions at the Flap-Core Interface of HIV-1 Protease. ACS Omega 2018, 3, 12132–12140. [Google Scholar] [CrossRef]
- Olajuyigbe, F.; Demitri, N.; Geremia, S. Investigation of 2-Fold Disorder of Inhibitors and Relative Potency by Crystallizations of HIV-1 Protease in Ritonavir and Saquinavir Mixtures. Cryst. Growth Des. 2011, 11, 4378–4385. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molecular Operating Environment (MOE) 2018.0; Chemical Computing Group ULC: Montreal, QC, Canada, 2018.
- Yu, W.; He, X.; Vanommeslaeghe, K.; MacKerell, A.D., Jr. Extension of the CHARMM General Force Field to sulfonyl-containing compounds and its utility in biomolecular simulations. J. Comput. Chem. 2012, 33, 2451–2468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pronk, S.; Pall, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boonstra, S.; Onck, P.R.; Giessen, E. CHARMM TIP3P Water Model Suppresses Peptide Folding by Solvating the Unfolded State. J. Phys. Chem. B 2016, 120, 3692–3698. [Google Scholar] [CrossRef] [PubMed]
- Mooers, B.H.M. Shortcuts for faster image creation in PyMOL. Protein Sci. 2020, 29, 268–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The PyMOL Molecular Graphics System; Version 2.0; Schrödinger, LLC.: New York, NY, USA; DeLano Scientific: South San Francisco, CA, USA, 2020.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Residue | Ligand Atom | Bond Type | Energy |
|---|---|---|---|---|
| 3CLpro–PF | Cys145 | O2 | Hydrogen | −5.04 |
| Cys145 | H9 | Hydrogen | −5.04 | |
| Glu166 | O3 | Hydrogen | −4.41 | |
| Gln189 | H20 | Hydrogen | −3.01 | |
| 3CLpro–keto | Glu166 | O48 | Hydrogen | −0.70 |
| Cys145 | O41 | Hydrogen | −0.80 | |
| Met165 | O48 | Hydrogen | −0.80 | |
| His41 | H32 | Arene | −0.90 | |
| 3CLpro–lop | His41 | O38 | Hydrogen | −4.1 |
| Glu166 | O20 | Hydrogen | −1.6 | |
| 3CLpro–rit | His41 | H6 | Arene | −3.0 |
| Gly143 | O3 | Hydrogen | −2.2 | |
| Pro168 | Thiozyl group | Arene | −0.8 | |
| Thr190 | S2 | Hydrogen | −0.8 |
| Complex | VdW Energy | Electrostatic | Polar Solvation | SASA | Total Binding Energy (kJ/mol) |
|---|---|---|---|---|---|
| 3CLpro–PF | −135.294 +/− 16.258 | −39.293 +/− 25.780 | 96.026+/− 21.556 | −23.441 +/− 2.526 | −102.002 +/− 21.336 |
| 3CLpro–keto | −76.478+/− 25.052 | −40.348 +/− 30.471 | 50.648 +/− 49.365 | −7.887 +/− 3.036 | −74.065 +/− 22.579 |
| 3CLpro–lop | −110.529 +/− 28.28 | −65.740 +/−26.09 | 168.011 +/− 33.31 | −25.239 +/−2.40 | −33.497 +/− 26.52 |
| 3CLpro–rit | −75.994 +/− 60.285 | −55.115 +/− 23.09 | 91.803 +/− 18.798 | −4.219 +/− 8.798 | −43.525 +/− 29.171 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, B.; Batool, M.; Ain, Q.u.; Kim, M.S.; Choi, S. Exploring the Binding Mechanism of PF-07321332 SARS-CoV-2 Protease Inhibitor through Molecular Dynamics and Binding Free Energy Simulations. Int. J. Mol. Sci. 2021, 22, 9124. https://doi.org/10.3390/ijms22179124
Ahmad B, Batool M, Ain Qu, Kim MS, Choi S. Exploring the Binding Mechanism of PF-07321332 SARS-CoV-2 Protease Inhibitor through Molecular Dynamics and Binding Free Energy Simulations. International Journal of Molecular Sciences. 2021; 22(17):9124. https://doi.org/10.3390/ijms22179124
Chicago/Turabian StyleAhmad, Bilal, Maria Batool, Qurat ul Ain, Moon Suk Kim, and Sangdun Choi. 2021. "Exploring the Binding Mechanism of PF-07321332 SARS-CoV-2 Protease Inhibitor through Molecular Dynamics and Binding Free Energy Simulations" International Journal of Molecular Sciences 22, no. 17: 9124. https://doi.org/10.3390/ijms22179124
APA StyleAhmad, B., Batool, M., Ain, Q. u., Kim, M. S., & Choi, S. (2021). Exploring the Binding Mechanism of PF-07321332 SARS-CoV-2 Protease Inhibitor through Molecular Dynamics and Binding Free Energy Simulations. International Journal of Molecular Sciences, 22(17), 9124. https://doi.org/10.3390/ijms22179124