
Pediatric Catecholaminergic Polymorphic Ventricular Tachycardia: A Translational Perspective for the Clinician-Scientist

 ,
,
Abstract
:1. Introduction
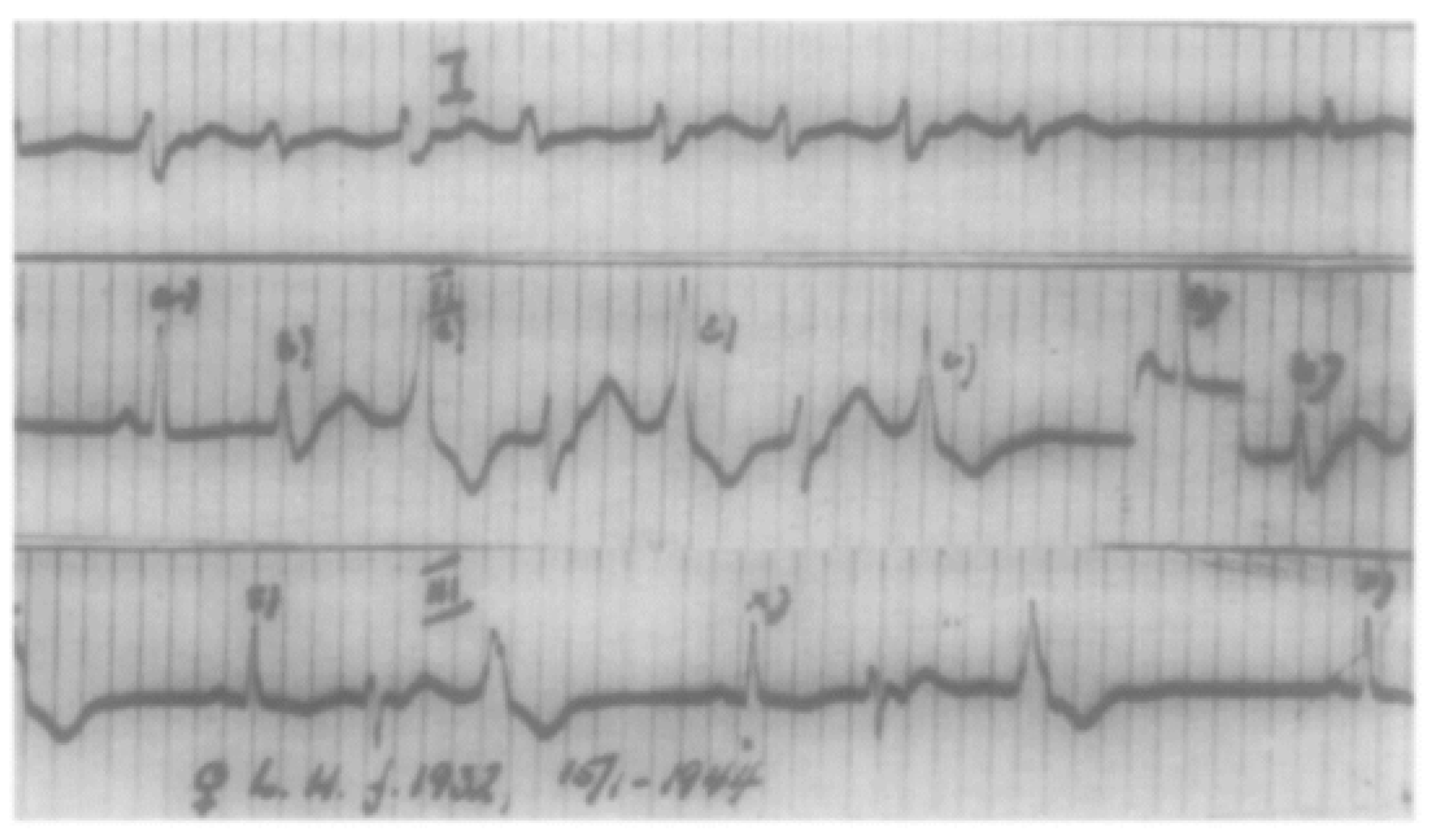
2. History of CPVT
3. Genetics
3.1. Ryanodine Receptor 2
3.2. Calsequestrin 2
3.3. Trans-2,3-enoyl-CoA Reductase-like
3.4. Calmodulin and Triadin
3.5. Expressivity and Penetrance
4. Pathophysiology
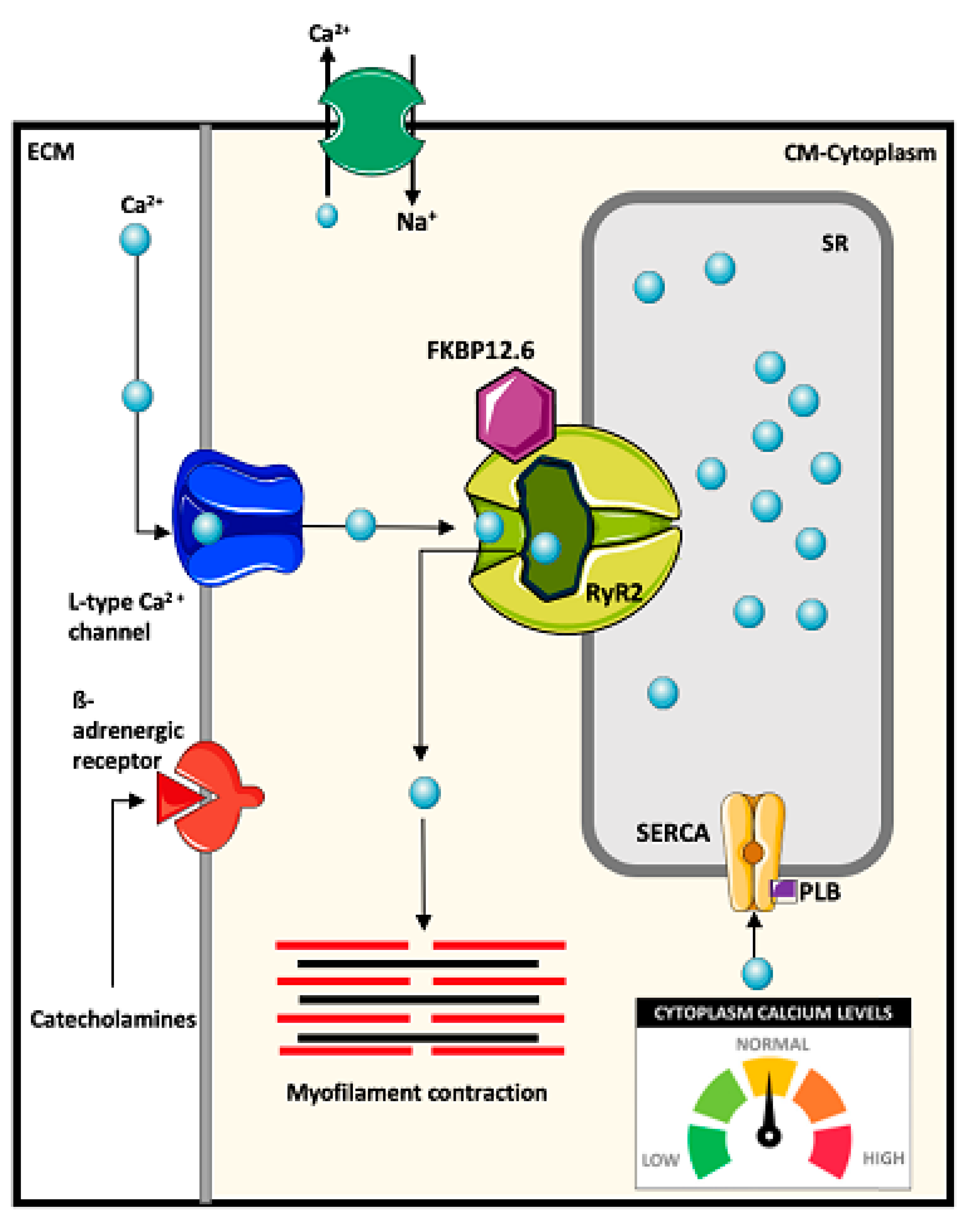
4.1. Mechanisms of Intracellular Ca2+ Regulation
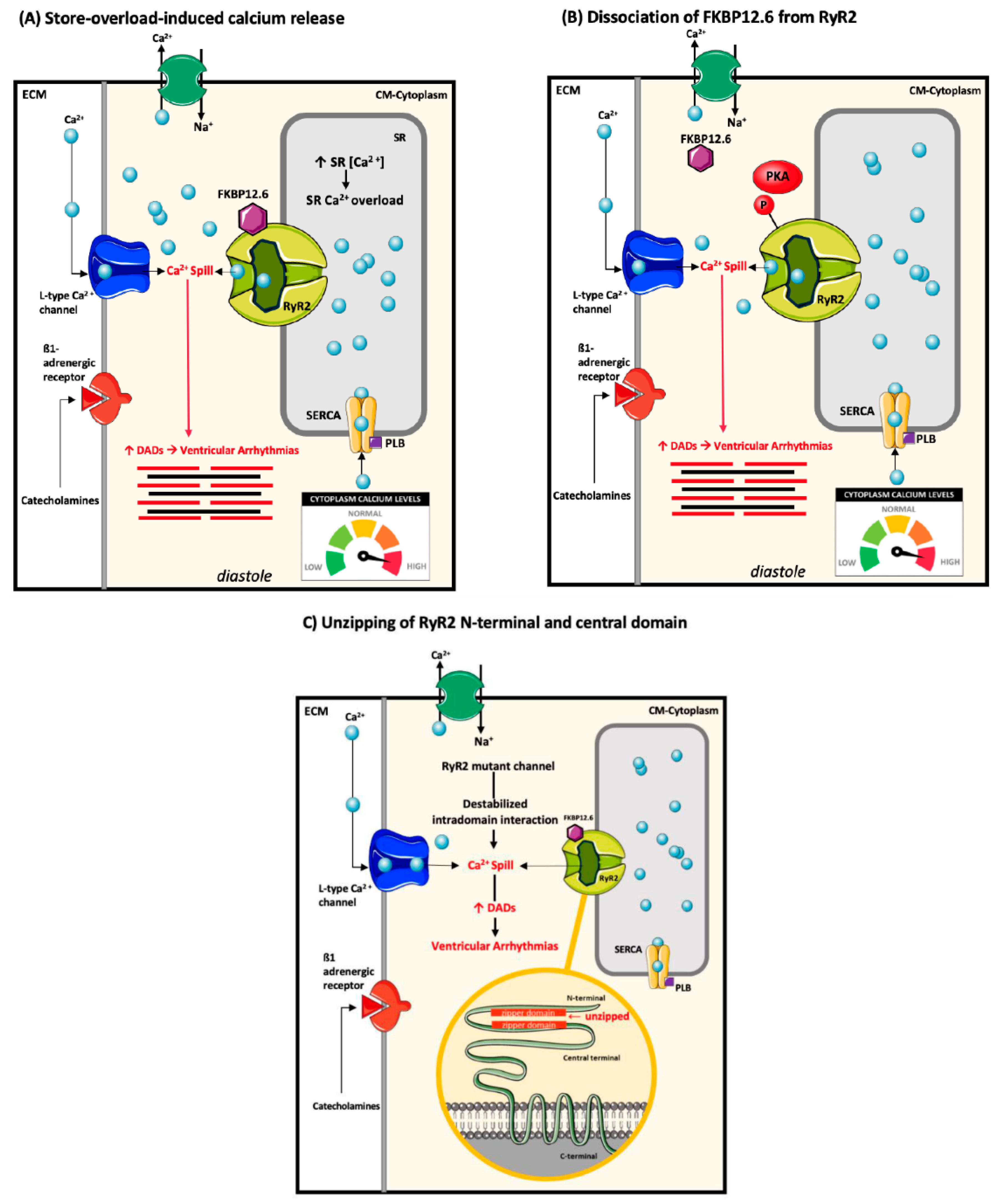
4.2. Ryanodine Receptor-2 Associated Arrhythmogenesis
4.3. Calsequestrin-2 Associated Arrhythmogenesis
4.4. TRDN, CALM1, and TECRL Associated Arrhythmogenesis
5. Clinical Presentation
6. Diagnosis
- -
- An unexplained catecholamine-induced bidirectional VT or polymorphic ventricular premature beats (VPBs), or VT in an individual younger than 40 years;
- -
- A patient (index case or family member) with a pathogenic variant in a CPVT-related gene (see Genetic Etiology for details); and
- -
- Family members of a CPVT index case with a normal heart who manifest exercise-induced PVCs or bidirectional or polymorphic VT.
Genetic Testing Guidelines in CPVT
7. Therapy and Management
7.1. β-Blockers
7.2. Flecainide
7.3. Left Cardiac Sympathetic Denervation
7.4. Implantable Cardioverter Defibrillators
7.5. Lifestyle Management: Sport Participation
7.6. Other Therapeutic Interventions
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Leenhardt, A.; Lucet, V.; Denjoy, I.; Grau, F.; Ngoc, D.D.; Coumel, P. Catecholaminergic Polymorphic Ventricular Tachycardia in Children A 7-Year Follow-up of 21 Patients. Circulation 1995, 91, 1512–1519. [Google Scholar] [CrossRef]
- Swan, H.; Piippo, K.; Viitasalo, M.; Heikkilä, P.; Paavonen, T.; Kainulainen, K.; Kere, J.; Keto, P.; Kontula, K.; Toivonen, L. Arrhythmic disorder mapped to chromosome 1q42–q43 causes malignant polymorphic ventricular tachycardia in structurally normal hearts. J. Am. Coll. Cardiol. 1999, 34, 2035–2042. [Google Scholar] [CrossRef] [Green Version]
- Priori, S.G.; Napolitano, C.; Memmi, M.; Colombi, B.; Drago, F.; Gasparini, M.; DeSimone, L.; Coltorti, F.; Bloise, R.; Keegan, R.; et al. Clinical and Molecular Characterization of Patients With Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation 2002, 106, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauce, B.; Rampazzo, A.; Basso, C.; Bagattin, A.; Daliento, L.; Tiso, N.; Turrini, P.; Thiene, G.; Danieli, G.A.; Nava, A. Screening for ryanodine receptor type 2 mutations in families with effort-induced polymorphic ventricular arrhythmias and sudden death. J. Am. Coll. Cardiol. 2002, 40, 341–349. [Google Scholar] [CrossRef]
- Postma, A.V.; Denjoy, I.; Kamblock, J.; Alders, M.; Lupoglazoff, J.-M.; Vaksmann, G.; Dubosq-Bidot, L.; Sebillon, P.; Mannens, M.M.A.M.; Guicheney, P.; et al. Catecholaminergic polymorphic ventricular tachycardia: RYR2 mutations, bradycardia, and follow up of the patients. J. Med. Genet. 2005, 42, 863–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priori, S.G.; Wilde, A.A.; Horie, M.; Cho, Y.; Behr, E.; Berul, C.; Blom, N.; Brugada, J.; Chiang, C.-E.; Huikuri, H.; et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: Document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm 2013, 10, 1932–1963. [Google Scholar] [CrossRef]
- Priori, S.G.; Napolitano, C.; Tiso, N.; Memmi, M.; Vignati, G.; Bloise, R.; Sorrentino, V.; Danieli, G.A. Mutations in the Cardiac Ryanodine Receptor Gene (hRyR2) Underlie Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation 2001, 103, 196–200. [Google Scholar] [CrossRef] [Green Version]
- Lahat, H.; Pras, E.; Olender, T.; Avidan, N.; Ben-Asher, E.; Man, O.; Levy-Nissenbaum, E.; Khoury, A.; Lorber, A.; Goldman, B.; et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am. J. Hum. Genet. 2001, 69, 1378–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, M.; Denjoy, I.; Extramiana, F.; Maltret, A.; Buisson, N.R.; Lupoglazoff, J.M.; Klug, D.; Hayashi, M.; Takatsuki, S.; Villain, E.; et al. Incidence and Risk Factors of Arrhythmic Events in Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation 2009, 119, 2426–2434. [Google Scholar] [CrossRef] [Green Version]
- Sumitomo, N.; Harada, K.; Nagashima, M.; Yasuda, T.; Nakamura, Y.; Aragaki, Y.; Saito, A.; Kurosaki, K.; Jouo, K.; Koujiro, M.; et al. Catecholaminergic polymorphic ventricular tachycardia: Electrocardiographic characteristics and optimal therapeutic strategies to prevent sudden death. Heart 2003, 89, 66–70. [Google Scholar] [CrossRef] [Green Version]
- Sy, R.W.; Gollob, M.; Klein, G.J.; Yee, R.; Skanes, A.C.; Gula, L.J.; Leong-Sit, P.; Gow, R.M.; Green, M.S.; Birnie, D.H.; et al. Arrhythmia characterization and long-term outcomes in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2011, 8, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Roston, T.M.; Vinocur, J.M.; Maginot, K.R.; Mohammed, S.; Salerno, J.C.; Etheridge, S.P.; Cohen, M.; Hamilton, R.M.; Pflaumer, A.; Kanter, R.J.; et al. Catecholaminergic polymorphic ventricular tachycardia in children: Analysis of therapeutic strategies and outcomes from an international multicenter registry. Circ. Arrhythm. Electrophysiol. 2015, 8, 633–642. [Google Scholar] [CrossRef] [Green Version]
- van der Werf, C.; Kannankeril, P.J.; Sacher, F.; Krahn, A.D.; Viskin, S.; Leenhardt, A.; Shimizu, W.; Sumitomo, N.; Fish, F.A.; Bhuiyan, Z.A.; et al. Flecainide therapy reduces exercise-induced ventricular arrhythmias in patients with catecholaminergic polymorphic ventricular tachycardia. J. Am. Coll. Cardiol. 2011, 57, 2244–2254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannankeril, P.J.; Moore, J.P.; Cerrone, M.; Priori, S.G.; Kertesz, N.J.; Ro, P.S.; Batra, A.S.; Kaufman, E.S.; Fairbrother, D.L.; Saarel, E.V.; et al. Efficacy of Flecainide in the Treatment of Catecholaminergic Polymorphic Ventricular Tachycardia: A Randomized Clinical Trial. JAMA Cardiol. 2017, 2, 759–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, H.; Chopra, N.; Laver, D.; Hwang, H.S.; Davies, S.; E Roach, D.; Duff, H.; Roden, D.M.; Wilde, A.A.M.; Knollmann, B.C. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat. Med. 2009, 15, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Khoury, A.; Marai, I.; Suleiman, M.; Blich, M.; Lorber, A.; Gepstein, L.; Boulos, M. Flecainide therapy suppresses exercise-induced ventricular arrhythmias in patients with CASQ2-associated catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2013, 10, 1671–1675. [Google Scholar] [CrossRef]
- van der Werf, C.; Lieve, K.V.; Bos, J.M.; Lane, C.M.; Denjoy, I.; Roses-Noguer, F.; Aiba, T.; Wada, Y.; Ingles, J.; Leren, I.S.; et al. Implantable cardioverter-defibrillators in previously undiagnosed patients with catecholaminergic polymorphic ventricular tachycardia resuscitated from sudden cardiac arrest. Eur. Heart J. 2019, 40, 2953–2961. [Google Scholar] [CrossRef]
- De Ferrari, G.M.; Dusi, V.; Spazzolini, C.; Bos, J.M.; Abrams, D.J.; Berul, C.I.; Crotti, L.; Eldar, M.; Kharlap, M.; Khoury, A.; et al. Clinical Management of Catecholaminergic Polymorphic Ventricular Tachycardia: The Role of Left Cardiac Sympathetic Denervation. Circulation 2015, 131, 2185–2193. [Google Scholar] [CrossRef] [Green Version]
- van der Werf, C.; Nederend, I.; Hofman, N.; van Geloven, N.; Ebink, C.; Frohn-Mulder, I.M.; Alings, A.M.; Bosker, H.A.; Bracke, F.A.; van den Heuvel, F.; et al. Familial evaluation in catecholaminergic polymorphic ventricular tachycardia: Disease penetrance and expression in cardiac ryanodine receptor mutation-carrying relatives. Circ. Arrhythm. Electrophysiol. 2012, 5, 748–756. [Google Scholar] [CrossRef] [Green Version]
- Kallas, D.; Roston, T.M.; Franciosi, S.; Brett, L.; Lieve, K.V.; Kwok, S.-Y.; Kannankeril, P.J.; Krahn, A.D.; LaPage, M.J.; Etheridge, S.; et al. An Evaluation of Age at Symptom-Onset, Proband Status and Sex as Predictors of Disease Severity in Pediatric Catecholaminergic Polymorphic Ventricular Tachycardia. Heart Rhythm 2021. [Google Scholar] [CrossRef]
- Berg, K.J. Multifocal ventricular extrasytoles with adams-stokes syndrome in siblings. Am. Heart J. 1960, 60, 965–970. [Google Scholar] [CrossRef]
- Reid, D.S.; Tynan, M.; Braidwood, L.; Fitzgerald, G.R. Bidirectional tachycardia in a child. A study using his bundle electrography. Heart 1975, 37, 339–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laitinen, P.J.; Brown, K.M.; Piippo, K.; Swan, H.; Devaney, J.M.; Brahmbhatt, B.; Donarum, E.A.; Marino, M.; Tiso, N.; Viitasalo, M.; et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation 2001, 103, 485–490. [Google Scholar] [CrossRef]
- Lanner, J.; Georgiou, D.K.; Joshi, A.D.; Hamilton, S.L. Ryanodine receptors: Structure, expression, molecular details, and function in calcium release. Cold Spring Harb Perspect. Biol. 2010, 2, a003996. [Google Scholar] [CrossRef] [Green Version]
- Ng, K.; Titus, E.W.; Lieve, K.V.; Roston, T.M.; Mazzanti, A.; Deiter, F.H.; Denjoy, I.; Ingles, J.; Till, J.; Robyns, T.; et al. An International Multicenter Evaluation of Inheritance Patterns, Arrhythmic Risks, and Underlying Mechanisms of CASQ2 -Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation 2020, 142, 932–947. [Google Scholar] [CrossRef]
- Garcia-Elias, A.; Benito, B. Ion Channel Disorders and Sudden Cardiac Death. Int. J. Mol. Sci. 2018, 19, 692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyegaard, M.; Overgaard, M.T.; Søndergaard, M.T.; Vranas, M.; Behr, E.; Hildebrandt, L.L.; Lund, J.; Hedley, P.; Camm, A.J.; Wettrell, G.; et al. Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am. J. Hum. Genet. 2012, 91, 703–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roux-Buisson, N.; Cacheux, M.; Fourest-Lieuvin, A.; Fauconnier, J.; Brocard, J.; Denjoy, I.; Durand, P.; Guicheney, P.; Kyndt, F.; Leenhardt, A.; et al. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum. Mol. Genet. 2012, 21, 2759–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirchhefer, U.; Wehrmeister, D.; Postma, A.; Pohlentz, G.; Mormann, M.; Kucerova, D.; Müller, F.U.; Schmitz, W.; Schulze-Bahr, E.; Wilde, A.A.; et al. The human CASQ2 mutation K206N is associated with hyperglycosylation and altered cellular calcium handling. J. Mol. Cell. Cardiol. 2010, 49, 95–105. [Google Scholar] [CrossRef]
- Gray, B.; Bagnall, R.D.; Lam, L.; Ingles, J.; Turner, C.; Haan, E.; Davis, A.; Yang, P.-C.; Clancy, C.E.; Sy, R.W.; et al. A novel heterozygous mutation in cardiac calsequestrin causes autosomal dominant catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2016, 13, 1652–1660. [Google Scholar] [CrossRef] [Green Version]
- Roston, T.M.; Yuchi, Z.; Kannankeril, P.J.; Hathaway, J.; Vinocur, J.M.; Etheridge, S.P.; E Potts, J.; Maginot, K.R.; Salerno, J.C.; Cohen, I.M.; et al. The clinical and genetic spectrum of catecholaminergic polymorphic ventricular tachycardia: Findings from an international multicentre registry. Europace 2018, 20, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Bhuiyan, Z.A.; Hamdan, M.A.; Shamsi, E.T.; Postma, A.V.; Mannens, M.M.; Wilde, A.A.M.; Al-Gazali, L. A novel early onset lethal form of catecholaminergic polymorphic ventricular tachycardia maps to chromosome 7p14-p22. J. Cardiovasc. Electrophysiol. 2007, 18, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Webster, G.; Aburawi, E.H.; A Chaix, M.; Chandler, S.; Foo, R.; Islam, A.K.M.M.; A E Kammeraad, J.; Rioux, J.D.; Al-Gazali, L.; Sayeed, Z.; et al. Life-threatening arrhythmias with autosomal recessive TECRL variants. Europace 2020, 23, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Kalscheur, M.M.; Vaidyanathan, R.; Orland, K.M.; Abozeid, S.; Fabry, N.; Maginot, K.R.; January, C.T.; Makielski, J.C.; Eckhardt, L.L. KCNJ2 mutation causes an adrenergic-dependent rectification abnormality with calcium sensitivity and ventricular arrhythmia. Heart Rhythm 2014, 11, 885–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumitomo, N. Current topics in catecholaminergic polymorphic ventricular tachycardia. J. Arrhythm. 2016, 32, 344–351. [Google Scholar] [CrossRef] [Green Version]
- Laurent, G.; Saal, S.; Amarouch, M.Y.; Béziau, D.M.; Marsman, R.F.; Faivre, L.; Barc, J.; Dina, C.; Bertaux, G.; Barthez, O.; et al. Multifocal ectopic Purkinje-related premature contractions: A new SCN5A-related cardiac channelopathy. J. Am. Coll. Cardiol. 2012, 60, 144–156. [Google Scholar] [CrossRef]
- Swan, H.; Amarouch, M.Y.; Leinonen, J.; Marjamaa, A.; Kucera, J.P.; Laitinen-Forsblom, P.J.; Lahtinen, A.M.; Palotie, A.; Kontula, K.; Toivonen, L.; et al. Gain-of-function mutation of the SCN5A gene causes exercise-induced polymorphic ventricular arrhythmias. Circ. Cardiovasc. Genet. 2014, 7, 771–781. [Google Scholar] [CrossRef] [Green Version]
- Imberti, J.F.; Underwood, K.; Mazzanti, A.; Priori, S.G. Clinical Challenges in Catecholaminergic Polymorphic Ventricular Tachycardia. Heart Lung. Circ. 2016, 25, 777–783. [Google Scholar] [CrossRef] [Green Version]
- Barajas-Martinez, H.; Hu, D.; Ontiveros, G.; Caceres, G.; Desai, M.; Burashnikov, E.; Scaglione, J.; Antzelevitch, C. Biophysical and molecular characterization of a novel de novo KCNJ2 mutation associated with Andersen-Tawil syndrome and catecholaminergic polymorphic ventricular tachycardia mimicry. Circ. Cardiovasc. Genet. 2011, 4, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Wangüemert, F.; Calero, C.B.; Pérez, C.; Campuzano, O.; Beltran-Alvarez, P.; Scornik, F.S.; Iglesias, A.; Berne, P.; Allegue, C.; Hernandez, P.M.R.; et al. Clinical and molecular characterization of a cardiac ryanodine receptor founder mutation causing catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2015, 12, 1636–1643. [Google Scholar] [CrossRef] [Green Version]
- Kolder, I.; Tanck, M.; Postema, P.G.; Barc, J.; Sinner, M.F.; Zumhagen, S.; Husemann, A.; Stallmeyer, B.; Koopmann, T.T.; Hofman, N.; et al. Analysis for Genetic Modifiers of Disease Severity in Patients With Long-QT Syndrome Type 2. Circ. Cardiovasc. Genet. 2015, 8, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Crotti, L.; Lahtinen, A.M.; Spazzolini, C.; Mastantuono, E.; Monti, M.C.; Morassutto, C.; Parati, G.; Heradien, M.; Goosen, A.; Lichtner, P.; et al. Genetic Modifiers for the Long-QT Syndrome: How Important Is the Role of Variants in the 3’Untranslated Region of KCNQ1? Circ. Cardiovasc. Genet. 2016, 9, 330–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, S.; Wan, X.; Ramirez-Navarro, A.; Tesar, P.; Kaufman, E.S.; Ficker, E.; George, A.L.; Deschênes, I. Physiological genomics identifies genetic modifiers of long QT syndrome type 2 severity. J. Clin. Invest. 2018, 128, 1043–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marian, A.J. Modifier genes for hypertrophic cardiomyopathy. Curr. Opin. Cardiol. 2002, 17, 242–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roston, T.M.; Haji-Ghassemi, O.; LaPage, M.J.; Batra, A.S.; Bar-Cohen, Y.; Anderson, C.; Lau, Y.R.; Maginot, K.; Gebauer, R.A.; Etheridge, S.P.; et al. Catecholaminergic polymorphic ventricular tachycardia patients with multiple genetic variants in the PACES CPVT Registry. PLoS ONE 2018, 13, e0205925. [Google Scholar] [CrossRef] [PubMed]
- Lahrouchi, N.; Tadros, R.; Crotti, L.; Mizusawa, Y.; Postema, P.G.; Beekman, L.; Walsh, R.; Hasegawa, K.; Barc, J.; Ernsting, M.; et al. Transethnic Genome-Wide Association Study Provides Insights in the Genetic Architecture and Heritability of Long QT Syndrome. Circulation 2020, 142, 324–338. [Google Scholar] [CrossRef]
- Sutanto, H.; Lyon, A.; Lumens, J.; Schotten, U.; Dobrev, D.; Heijman, J. Cardiomyocyte calcium handling in health and disease: Insights from in vitro and in silico studies. Prog. Biophys. Mol. Biol. 2020, 157, 54–75. [Google Scholar] [CrossRef]
- Györke, I.; Hester, N.; Jones, L.R.; Györke, S. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys. J. 2004, 86, 2121–2128. [Google Scholar] [CrossRef] [Green Version]
- Priori, S.G.; Chen, S.R.W. Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ. Res. 2011, 108, 871–883. [Google Scholar] [CrossRef] [Green Version]
- Kass, R.S.; Tsien, R.W. Fluctuations in membrane current driven by intracellular calcium in cardiac Purkinje fibers. Biophys. J. 1982, 38, 259–269. [Google Scholar] [CrossRef] [Green Version]
- Orchard, C.H.; Eisner, D.A.; Allen, D.G. Oscillations of intracellular Ca2+ in mammalian cardiac muscle. Nature 1983, 304, 735–738. [Google Scholar] [CrossRef]
- Lohse, M.J.; Engelhardt, S.; Eschenhagen, T. What is the role of beta-adrenergic signaling in heart failure? Circ. Res. 2003, 93, 896–906. [Google Scholar] [CrossRef]
- Kamp, T.J.; Hell, J.W. Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ. Res. 2000, 87, 1095–1102. [Google Scholar] [CrossRef] [Green Version]
- Simmerman, H.K.; Jones, L.R. Phospholamban: Protein structure, mechanism of action, and role in cardiac function. Physiol. Rev. 1998, 78, 921–947. [Google Scholar] [CrossRef]
- Layland, J.; Solaro, R.J.; Shah, A.M. Regulation of cardiac contractile function by troponin I phosphorylation. Cardiovasc. Res. 2005, 66, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunst, G.; Kress, K.R.; Gruen, M.; Uttenweiler, D.; Gautel, M.; Fink, R.H.A. Myosin binding protein C, a phosphorylation-dependent force regulator in muscle that controls the attachment of myosin heads by its interaction with myosin S2. Circ. Res. 2000, 86, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Sampson, K.J.; Kass, R.S. Cardiac Delayed Rectifier Potassium Channels in Health and Disease. Card Electrophysiol. Clin. 2016, 8, 307–322. [Google Scholar] [CrossRef] [Green Version]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Kryshtal, D.O.; Blackwell, D.J.; Egly, C.L.; Smith, A.N.; Batiste, S.M.; Johnston, J.N.; Laver, D.R.; Knollmann, B.C. RYR2 Channel Inhibition Is the Principal Mechanism of Flecainide Action in CPVT. Circ. Res. 2021, 128, 321–331. [Google Scholar] [CrossRef]
- Wleklinski, M.J.; Kannankeril, P.J.; Knollmann, B.C. Molecular and tissue mechanisms of catecholaminergic polymorphic ventricular tachycardia. J. Physiol. 2020, 598, 2817–2834. [Google Scholar] [CrossRef] [PubMed]
- Willegems, K.; Efremov, R.G. Structural Details of the Ryanodine Receptor Calcium Release Channel and Its Gating Mechanism. Adv. Exp. Med. Biol. 2017, 981, 179–204. [Google Scholar]
- Jiang, D.; Xiao, B.; Yang, D.; Wang, R.; Choi, P.; Zhang, L.; Cheng, H.; Chen, S.R.W. RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR). Proc. Natl. Acad. Sci. USA 2004, 101, 13062–13067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Kimlicka, L.; Hiess, F.; Tian, X.; Wang, R.; Zhang, L.; Jones, P.P.; Van Petegem, F.; Chen, S.R.W. The CPVT-associated RyR2 mutation G230C enhances store overload-induced Ca2+ release and destabilizes the N-terminal domains. Biochem. J. 2013, 454, 123–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLennan, D.H.; Chen, S.R. Store overload-induced Ca2+ release as a triggering mechanism for CPVT and MH episodes caused by mutations in RYR and CASQ genes. J. Physiol. 2009, 587, 3113–3115. [Google Scholar] [CrossRef] [PubMed]
- Wehrens, X.H.; Lehnart, S.E.; Huang, F.; Vest, J.A.; Reiken, S.R.; Mohler, P.J.; Sun, J.; Guatimosim, S.; Song, L.-S.; Rosemblit, N.; et al. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell 2003, 113, 829–840. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Tian, X.; Jones, P.P.; Bolstad, J.; Kong, H.; Wang, R.; Zhang, L.; Duff, H.; Gillis, A.M.; Fleischer, S.; et al. Removal of FKBP12.6 does not alter the conductance and activation of the cardiac ryanodine receptor or the susceptibility to stress-induced ventricular arrhythmias. J. Biol. Chem. 2007, 282, 34828–34838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikemoto, N.; Yamamoto, T. Regulation of calcium release by interdomain interaction within ryanodine receptors. Front. Biosci. 2002, 7, d671–d683. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Velasco, M.; Gomez, A.M.; Richard, S. Unzipping RyR2 in adult cardiomyocytes: Getting closer to mechanisms of inherited ventricular arrhythmias? Cardiovasc. Res. 2006, 70, 407–409. [Google Scholar] [CrossRef]
- Liu, N.; Rizzi, N.; Boveri, L.; Priori, S.G. Ryanodine receptor and calsequestrin in arrhythmogenesis: What we have learnt from genetic diseases and transgenic mice. J. Mol. Cell. Cardiol. 2009, 46, 149–159. [Google Scholar] [CrossRef]
- Uchinoumi, H.; Yano, M.; Suetomi, T.; Ono, M.; Xu, X.; Tateishi, H.; Oda, T.; Okuda, S.; Doi, M.; Kobayashi, S.; et al. Catecholaminergic polymorphic ventricular tachycardia is caused by mutation-linked defective conformational regulation of the ryanodine receptor. Circ. Res. 2010, 106, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Liu, X.; Gong, Y.; Zhang, P.; Qiang, S.; Zhao, Q.; Guo, R.; Qian, Y.; Wang, L.; Zhu, L.; et al. Pathogenic mechanism of a catecholaminergic polymorphic ventricular tachycardia causing-mutation in cardiac calcium release channel RyR2. J. Mol. Cell. Cardiol. 2018, 117, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Youn, B.; Kemper, L.; Campbell, C.; Milting, H.; Varsanyi, M.; Kang, C. Characterization of human cardiac calsequestrin and its deleterious mutants. J. Mol. Biol. 2007, 373, 1047–1057. [Google Scholar] [CrossRef]
- Valle, G.; Arad, M.; Volpe, P. Molecular adaptation to calsequestrin 2 (CASQ2) point mutations leading to catecholaminergic polymorphic ventricular tachycardia (CPVT): Comparative analysis of R33Q and D307H mutants. J. Muscle Res. Cell Motil. 2020, 41, 251–258. [Google Scholar] [CrossRef]
- Jalloul, Y.; Refaat, M.M. Novel variants in TECRL cause catecholaminergic polymorphic ventricular tachycardia. J. Cardiovasc. Electrophysiol. 2020, 31, 1536–1538. [Google Scholar] [CrossRef] [PubMed]
- Sumitomo, N.; Sakurada, H.; Taniguchi, K.; Matsumura, M.; Abe, O.; Miyashita, M.; Kanamaru, H.; Karasawa, K.; Ayusawa, M.; Fukamizu, S.; et al. Association of atrial arrhythmia and sinus node dysfunction in patients with catecholaminergic polymorphic ventricular tachycardia. Circ. J. 2007, 71, 1606–1609. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, M.; Ohno, S.; Naiki, N.; Nagaoka, I.; Dochi, K.; Wang, Q.; Hasegawa, K.; Kimura, H.; Miyamoto, A.; Mizusawa, Y.; et al. Genetic Background of Catecholaminergic Polymorphic Ventricular Tachycardia in Japan. Circ. J. 2020, 84, 2124–2126. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.; Hasegawa, K.; Horie, M. Gender Differences in the Inheritance Mode of RYR2 Mutations in Catecholaminergic Polymorphic Ventricular Tachycardia Patients. PLoS ONE 2015, 10, e0131517. [Google Scholar] [CrossRef]
- Jiang, H.; Li, X.-M.; Ge, H.-Y.; Zhang, Y.; Liu, H.-J.; Li, M.-T. Investigation of Catecholaminergic Polymorphic Ventricular Tachycardia Children in China: Clinical Characteristics, Delay to Diagnosis, and Misdiagnosis. Chin. Med. J. 2018, 131, 2864–2865. [Google Scholar] [CrossRef]
- Li, Q.; Guo, R.; Gao, L.; Cui, L.; Zhao, Z.; Yu, X.; Yuan, Y.; Xu, X. CASQ2 variants in Chinese children with catecholaminergic polymorphic ventricular tachycardia. Mol. Genet. Genomic. Med. 2019, 7, e949. [Google Scholar] [CrossRef] [Green Version]
- Tester, D.J.; Dura, M.; Carturan, E.; Reiken, S.; Wronska, A.; Marks, A.R.; Ackerman, M.J. A mechanism for sudden infant death syndrome (SIDS): Stress-induced leak via ryanodine receptors. Heart Rhythm 2007, 4, 733–739. [Google Scholar] [CrossRef] [Green Version]
- Rutter, N.; Southall, D.P. Cardiac arrhythmias misdiagnosed as epilepsy. Arch. Dis. Child. 1985, 60, 54–56. [Google Scholar] [CrossRef]
- Yap, S.M.; Smyth, S. Ryanodine receptor 2 (RYR2) mutation: A potentially novel neurocardiac calcium channelopathy manifesting as primary generalised epilepsy. Seizure 2019, 67, 11–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehnart, S.E.; Mongillo, M.; Bellinger, A.; Lindegger, N.; Chen, B.-X.; Hsueh, W.; Reiken, S.; Wronska, A.; Drew, L.J.; Ward, C.W.; et al. Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J. Clin. Invest. 2008, 118, 2230–2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozawa, J.; Ohno, S.; Fujii, Y.; Makiyama, T.; Suzuki, H.; Saitoh, A.; Horie, M. Differential Diagnosis Between Catecholaminergic Polymorphic Ventricular Tachycardia and Long QT Syndrome Type 1-Modified Schwartz Score. Circ. J. 2018, 82, 2269–2276. [Google Scholar] [CrossRef]
- Letsas, K.P.; Prappa, E.; Bazoukis, G.; Lioni, L.; Pantou, M.P.; Gourzi, P.; Degiannis, D.; Sideris, A. A novel variant of RyR2 gene in a family misdiagnosed as congenital long QT syndrome: The importance of genetic testing. J. Electrocardiol. 2020, 60, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Giudicessi, J.R.; Ackerman, M.J. Exercise testing oversights underlie missed and delayed diagnosis of catecholaminergic polymorphic ventricular tachycardia in young sudden cardiac arrest survivors. Heart Rhythm 2019, 16, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.A. Andersen-Tawil syndrome, scarier for the doctor than for the patient? Who, when, and how to treat. Europace 2013, 15, 1690–1692. [Google Scholar] [CrossRef] [Green Version]
- Pflaumer, A.; Davis, A. Guidelines for the diagnosis and management of Catecholaminergic Polymorphic Ventricular Tachycardia. Heart Lung. Circ. 2012, 21, 96–100. [Google Scholar] [CrossRef]
- Sy, R.W.; Krahn, A.D. Exercise testing: The catecholaminergic polymorphic ventricular tachycardia crystal ball? Europace 2012, 14, 1225–1227. [Google Scholar] [CrossRef]
- Roston, T.M.; Kallas, D.; Davies, B.; Franciosi, S.; De Souza, A.M.; Laksman, Z.W.; Sanatani, S.; Krahn, A.D. Burst Exercise Testing Can Unmask Arrhythmias in Patients With Incompletely Penetrant Catecholaminergic Polymorphic Ventricular Tachycardia. JACC Clin. Electrophysiol. 2021, 7, 437–441. [Google Scholar] [CrossRef]
- Van der Werf, C.; Wilde, A.A. Catecholaminergic polymorphic ventricular tachycardia: From bench to bedside. Heart 2013, 99, 497–504. [Google Scholar] [CrossRef]
- Marjamaa, A.; Hiippala, A.; Arrhenius, B.; Lahtinen, A.M.; Kontula, K.; Toivonen, L.; Happonen, J.; Swan, H.; Tuiskula, A. Intravenous epinephrine infusion test in diagnosis of catecholaminergic polymorphic ventricular tachycardia. J. Cardiovasc. Electrophysiol. 2012, 23, 194–199. [Google Scholar] [CrossRef]
- Watanabe, H.; Knollmann, B. Mechanism underlying catecholaminergic polymorphic ventricular tachycardia and approaches to therapy. J. Electrocardiol. 2011, 44, 650–655. [Google Scholar] [CrossRef]
- Jabbari, J.; Jabbari, R.; Nielsen, M.W.; Holst, A.G.; Nielsen, J.B.; Haunsø, S.; Tfelt-Hansen, J.; Svendsen, J.H.; Olesen, M.S. New exome data question the pathogenicity of genetic variants previously associated with catecholaminergic polymorphic ventricular tachycardia. Circ. Cardiovasc. Genet. 2013, 6, 481–489. [Google Scholar] [CrossRef] [Green Version]
- Giudicessi, J.R.; Lieve, K.V.; Rohatgi, R.K.; Koca, F.; Tester, D.J.; van der Werf, C.; Bos, J.M.; Wilde, A.A.; Ackerman, M.J. Assessment and Validation of a Phenotype-Enhanced Variant Classification Framework to Promote or Demote RYR2 Missense Variants of Uncertain Significance. Circ. Genom. Precis. Med. 2019, 12, e002510. [Google Scholar] [CrossRef] [Green Version]
- Ackerman, M.J.; Priori, S.G.; Willems, S.; Berul, C.; Brugada, R.; Calkins, H.; Camm, A.J.; Ellinor, P.; Gollob, M.; Hamilton, R.; et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm 2011, 8, 1308–1339. [Google Scholar] [CrossRef] [PubMed]
- Van der Werf, C.; Zwinderman, A.H.; Wilde, A.A. Therapeutic approach for patients with catecholaminergic polymorphic ventricular tachycardia: State of the art and future developments. Europace 2012, 14, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Leren, I.S.; Saberniak, J.; Majid, E.; Haland, T.F.; Edvardsen, T.; Haugaa, K.H. Nadolol decreases the incidence and severity of ventricular arrhythmias during exercise stress testing compared with β1-selective β-blockers in patients with catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2016, 13, 433–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padfield, G.J.; AlAhmari, L.; Lieve, K.V.; AlAhmari, T.; Roston, T.M.; Wilde, A.A.; Krahn, A.D.; Sanatani, S. Flecainide monotherapy is an option for selected patients with catecholaminergic polymorphic ventricular tachycardia intolerant of β-blockade. Heart Rhythm 2016, 13, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Bannister, M.L.; Thomas, N.L.; Sikkel, M.B.; Mukherjee, S.; Maxwell, C.; MacLeod, K.T.; George, C.H.; Williams, A.J. The mechanism of flecainide action in CPVT does not involve a direct effect on RyR2. Circ. Res. 2015, 116, 1324–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baltogiannis, G.G.; Lysitsas, D.N.; Di Giovanni, G.; Ciconte, G.; Sieira, J.; Conte, G.; Kolettis, T.M.; Chierchia, G.-B.; De Asmundis, C.; Brugada, P. CPVT: Arrhythmogenesis, Therapeutic Management, and Future Perspectives. A Brief Review of the Literature. Front. Cardiovasc. Med. 2019, 6, 92. [Google Scholar] [CrossRef]
- Schwartz, P.J. Cardiac sympathetic denervation to prevent life-threatening arrhythmias. Nat. Rev. Cardiol. 2014, 11, 346–353. [Google Scholar] [CrossRef]
- Priori, S.G.; Blomström-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 2015, 36, 2793–2867. [Google Scholar] [CrossRef] [Green Version]
- Al-Khatib, S.M.; Stevenson, W.G.; Ackerman, M.J.; Bryant, W.J.; Callans, D.J.; Curtis, A.B.; Deal, B.J.; Dickfeld, T.; Field, M.E.; Fonarow, G.C.; et al. 2017 AHA/ACC/HRS Guideline for Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. J. Am. Coll. Cardiol. 2018, 72, e91–e220. [Google Scholar] [CrossRef]
- Pizzale, S.; Gollob, M.; Gow, R.; Birnie, D.H. Sudden death in a young man with catecholaminergic polymorphic ventricular tachycardia and paroxysmal atrial fibrillation. J. Cardiovasc. Electrophysiol. 2008, 19, 1319–1321. [Google Scholar] [CrossRef]
- Mohamed, U.; Gollob, M.; Gow, R.M.; Krahn, A.D. Sudden cardiac death despite an implantable cardioverter-defibrillator in a young female with catecholaminergic ventricular tachycardia. Heart Rhythm 2006, 3, 1486–1489. [Google Scholar] [CrossRef]
- Marai, I.; Khoury, A.; Suleiman, M.; Gepstein, L.; Blich, M.; Lorber, A.; Boulos, M. Importance of ventricular tachycardia storms not terminated by implantable cardioverter defibrillators shocks in patients with CASQ2 associated catecholaminergic polymorphic ventricular tachycardia. Am. J. Cardiol. 2012, 110, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Roses-Noguer, F.; Jarman, J.W.; Clague, J.R.; Till, J. Outcomes of defibrillator therapy in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2014, 11, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Roston, T.M.; Jones, K.; Hawkins, N.M.; Bos, J.M.; Schwartz, P.J.; Perry, F.; Ackerman, M.J.; Laksman, Z.W.; Kaul, P.; Lieve, K.V.; et al. Implantable cardioverter-defibrillator use in catecholaminergic polymorphic ventricular tachycardia: A systematic review. Heart Rhythm 2018, 15, 1791–1799. [Google Scholar] [CrossRef] [PubMed]
- Miyake, C.Y.; Webster, G.; Czosek, R.J.; Kantoch, M.J.; Dubin, A.M.; Avasarala, K.; Atallah, J. Efficacy of implantable cardioverter defibrillators in young patients with catecholaminergic polymorphic ventricular tachycardia: Success depends on substrate. Circ. Arrhythm. Electrophysiol. 2013, 6, 579–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostby, S.A.; Bos, J.M.; Owen, H.J.; Wackel, P.L.; Cannon, B.C.; Ackerman, M.J. Competitive Sports Participation in Patients With Catecholaminergic Polymorphic Ventricular Tachycardia: A Single Center’s Early Experience. JACC Clin. Electrophysiol. 2016, 2, 253–262. [Google Scholar] [CrossRef]
- Kobayashi, S.; Yano, M.; Uchinoumi, H.; Suetomi, T.; Susa, T.; Ono, M.; Xu, X.; Tateishi, H.; Oda, T.; Okuda, S.; et al. Dantrolene, a therapeutic agent for malignant hyperthermia, inhibits catecholaminergic polymorphic ventricular tachycardia in a RyR2(R2474S/+) knock-in mouse model. Circ. J. 2010, 74, 2579–2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, S.; Bannister, M.; Gangopadhyay, J.P.; Hamada, T.; Parness, J.; Ikemoto, N. Dantrolene stabilizes domain interactions within the ryanodine receptor. J. Biol. Chem. 2005, 280, 6580–6587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suetomi, T.; Yano, M.; Uchinoumi, H.; Fukuda, M.; Hino, A.; Ono, M.; Xu, X.; Tateishi, H.; Okuda, S.; Doi, M.; et al. Mutation-linked defective interdomain interactions within ryanodine receptor cause aberrant Ca²⁺release leading to catecholaminergic polymorphic ventricular tachycardia. Circulation 2011, 124, 682–694. [Google Scholar] [CrossRef] [Green Version]
- Penttinen, K.; Swan, H.; Vanninen, S.; Paavola, J.; Lahtinen, A.M.; Kontula, K.; Aalto-Setälä, K. Antiarrhythmic Effects of Dantrolene in Patients with Catecholaminergic Polymorphic Ventricular Tachycardia and Replication of the Responses Using iPSC Models. PLoS ONE 2015, 10, e0125366. [Google Scholar]
- Laksman, Z.; Barichello, S.; Roston, T.M.; Deyell, M.W.; Krahn, A.D. Acute Management of Ventricular Arrhythmia in Patients With Suspected Inherited Heart Rhythm Disorders. JACC Clin. Electrophysiol. 2019, 5, 267–283. [Google Scholar] [CrossRef]
- De Rosa, G.; Delogu, A.B.; Piastra, M.; Chiaretti, A.; Bloise, R.; Priori, S.G. Catecholaminergic polymorphic ventricular tachycardia: Successful emergency treatment with intravenous propranolol. Pediatr. Emerg. Care. 2004, 20, 175–177. [Google Scholar] [CrossRef]
- Bezzerides, V.J.; Caballero, A.; Wang, S.; Ai, Y.; Hylind, R.J.; Lu, F.; Heims-Waldron, D.A.; Chambers, K.D.; Zhang, D.; Abrams, D.; et al. Gene Therapy for Catecholaminergic Polymorphic Ventricular Tachycardia by Inhibition of Ca. Circulation 2019, 140, 405–419. [Google Scholar] [CrossRef]
- Zhang, X.-H.; Wei, H.; Xia, Y.; Morad, M. Calcium signaling consequences of RyR2 mutations associated with CPVT1 introduced via CRISPR/Cas9 gene editing in human-induced pluripotent stem cell-derived cardiomyocytes: Comparison of RyR2-R420Q, F2483I, and Q4201R. Heart Rhythm 2021, 18, 250–260. [Google Scholar] [CrossRef]
- Meraviglia, V.; Arendzen, C.H.; Freund, C.; Atsma, D.E.; Mummery, C.L.; Bellin, M. Generation of two human induced pluripotent stem cell lines, LUMCi020-A and LUMCi021-A, from two patients with Catecholaminergic Polymorphic Ventricular Tachycardia carrying heterozygous mutations in the RYR2 gene. Stem. Cell Res. 2020, 45, 101764. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| CPVT Subtype | Gene | Protein Name | Chromosome Locus | Inheritance Pattern | Proportion of Cases Associated with This Gene | Year Discovered |
|---|---|---|---|---|---|---|
| CPVT1 | RYR2 | Ryanodine Receptor 2 | 1q43 | Autosomal Dominant | 55–60% [7] | 2001 |
| CPVT2 | CASQ2 | Calsequestrin 2 | 1p13.1 | Autosomal Recessive/ Autosomal Dominant [25] | 2–5% [8] | 2001 |
| CPVT3 * | TECRL | Trans-2,3-enoyl-CoA reductase-like | 7p22–p14 | Autosomal Recessive | <1% [26] | 2016 |
| CPVT4 * | CALM1 | Calmodulin | 14q32.11 | Autosomal Dominant | <1% [27] | 2012 |
| CPVT5 * | TRDN | Triadin | 6q22.31 | Autosomal Recessive | 1–2% [28] | 2012 |
| Author | Year Published | CPVT Cases (n) | Mean/Median Age of Symptom-Onset (Years) | Cardiac Events at Follow-Up † | Conclusions/Significance | |
|---|---|---|---|---|---|---|
| Proband | Family Member | |||||
| Leenhardt et al. [1] | 1995 | 20 | 1 | 7.8 ± 4.0 | 2 SCDs over a mean of 7 years | First large description of CPVT as adrenergic-induced lethal tachyarrhythmias in the absence of structural heart disease |
| Swan et al. [2] | 1999 | 14 (in 2 families) | 21 ± 10 | 1 patient with syncope and cardiac arrest over 8 ± 6 years | Arrhythmia disease mapped to chromosome 1q42-q43. | |
| Lahat et al. [8] | 2001 | 13 (in 7 families) | 6 ± 3 | 1 SCD over 40 months | First CASQ2-associated autosomal recessive CPVT case reported. | |
| Bauce et al. [4] | 2002 | 43 (in 8 families) | Not reported | No events during follow-up | Genetic screening is important for early diagnosis of asymptomatic carriers. | |
| Priori et al. [3] | 2002 | 30 | 9 | 8 ± 2, RYR2-associated cases; 20 ± 12, Non-genotyped cases | Not reported | Non-genotyped CPVT cases are often women with late-onset of symptoms. RYR2-associated CPVT often have an early-onset of symptoms. Men are at higher risk of cardiac events. |
| Sumitomo et al. [10] | 2003 | 25 | 4 | 10.3 ± 6.1 | 7 (25%) SCD over mean 6.8 ± 4.9 years | Prognosis is poor. |
| Postma et al. [5] | 2005 | 12 | 42 | 12, RYR2-associated cases | 1 (8%) SCD over median of 6 years | Patients with RYR2 variants have a significant resting sinus bradycardia. |
| Hayashi et al. [9] | 2009 | 50 | 51 | 12 ± 8 | 27 patients over 7.9 ± 4.9 years | Risk factors for arrhythmic events include younger age at diagnosis and absence of β-blocker therapy |
| van der Werf et al. [19] | 2012 | 24 | 116 (61 from 1 family) | Not reported | 4 (22%) probands over a median of 7.8 years; 2 relatives over a period of 6.7 years | Relatives have marked phenotypic diversity and less severe phenotypes compared with probands. |
| Kawamura et al. [76] | 2013 | 50 | 0 | 10.2 ± 7.3 | 5 (19%) experienced syncope over mean follow-up of 48 months | Penetrance of CPVT phenotype was significantly higher in patients with RYR2 variants among Japanese CPVT patients. |
| Ohno et al. [77] | 2015 | 36 | 0 | 9.7 ± 4.6 * | Not reported | Almost half of RYR2 variants are de novo, and others are more often inherited from mothers than fathers. |
| Roston et al. [12] | 2015 | 170 | 56 | 10.8 (6.8–13.2) | Not reported | CPVT can have a malignant phenotype and lengthy delay to diagnosis, probands are typically severely affected. |
| Jiang et al. [78] | 2018 | 12 | 8.4 ± 3.2 | 1 SCD over 0.92 ± 0.8 years | Severe delay to diagnosis and misdiagnosis of CPVT are not uncommon in China. | |
| Li et al. [79] | 2019 | 5 | 2 | 6.2 ± 1.3 | No events over mean follow-up of 16.5 months * | First systematic study to examine CASQ2-CPVT in Chinese children identifying three novel variants. |
| Ng et al. [25] | 2020 | 36 | 76 | 7.9±3.3, probands only | Not reported | Patients with pathogenic CASQ2 heterozygous variants may manifest CPVT phenotype. |
| Kallas et al. [20] | 2021 | 106 | 27 | 11 (7–13.5) | 44 (33%) inclusive of a 3% mortality rate over 6-years (3–11) after time of symptom-onset | Proband status, but not age of symptom-onset or male sex, independently predicted an earlier-onset of cardiac events. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kallas, D.; Lamba, A.; Roston, T.M.; Arslanova, A.; Franciosi, S.; Tibbits, G.F.; Sanatani, S. Pediatric Catecholaminergic Polymorphic Ventricular Tachycardia: A Translational Perspective for the Clinician-Scientist. Int. J. Mol. Sci. 2021, 22, 9293. https://doi.org/10.3390/ijms22179293
Kallas D, Lamba A, Roston TM, Arslanova A, Franciosi S, Tibbits GF, Sanatani S. Pediatric Catecholaminergic Polymorphic Ventricular Tachycardia: A Translational Perspective for the Clinician-Scientist. International Journal of Molecular Sciences. 2021; 22(17):9293. https://doi.org/10.3390/ijms22179293
Chicago/Turabian StyleKallas, Dania, Avani Lamba, Thomas M. Roston, Alia Arslanova, Sonia Franciosi, Glen F. Tibbits, and Shubhayan Sanatani. 2021. "Pediatric Catecholaminergic Polymorphic Ventricular Tachycardia: A Translational Perspective for the Clinician-Scientist" International Journal of Molecular Sciences 22, no. 17: 9293. https://doi.org/10.3390/ijms22179293
APA StyleKallas, D., Lamba, A., Roston, T. M., Arslanova, A., Franciosi, S., Tibbits, G. F., & Sanatani, S. (2021). Pediatric Catecholaminergic Polymorphic Ventricular Tachycardia: A Translational Perspective for the Clinician-Scientist. International Journal of Molecular Sciences, 22(17), 9293. https://doi.org/10.3390/ijms22179293