
Amino Acid-Mediated Intracellular Ca2+ Rise Modulates mTORC1 by Regulating the TSC2-Rheb Axis through Ca2+/Calmodulin


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
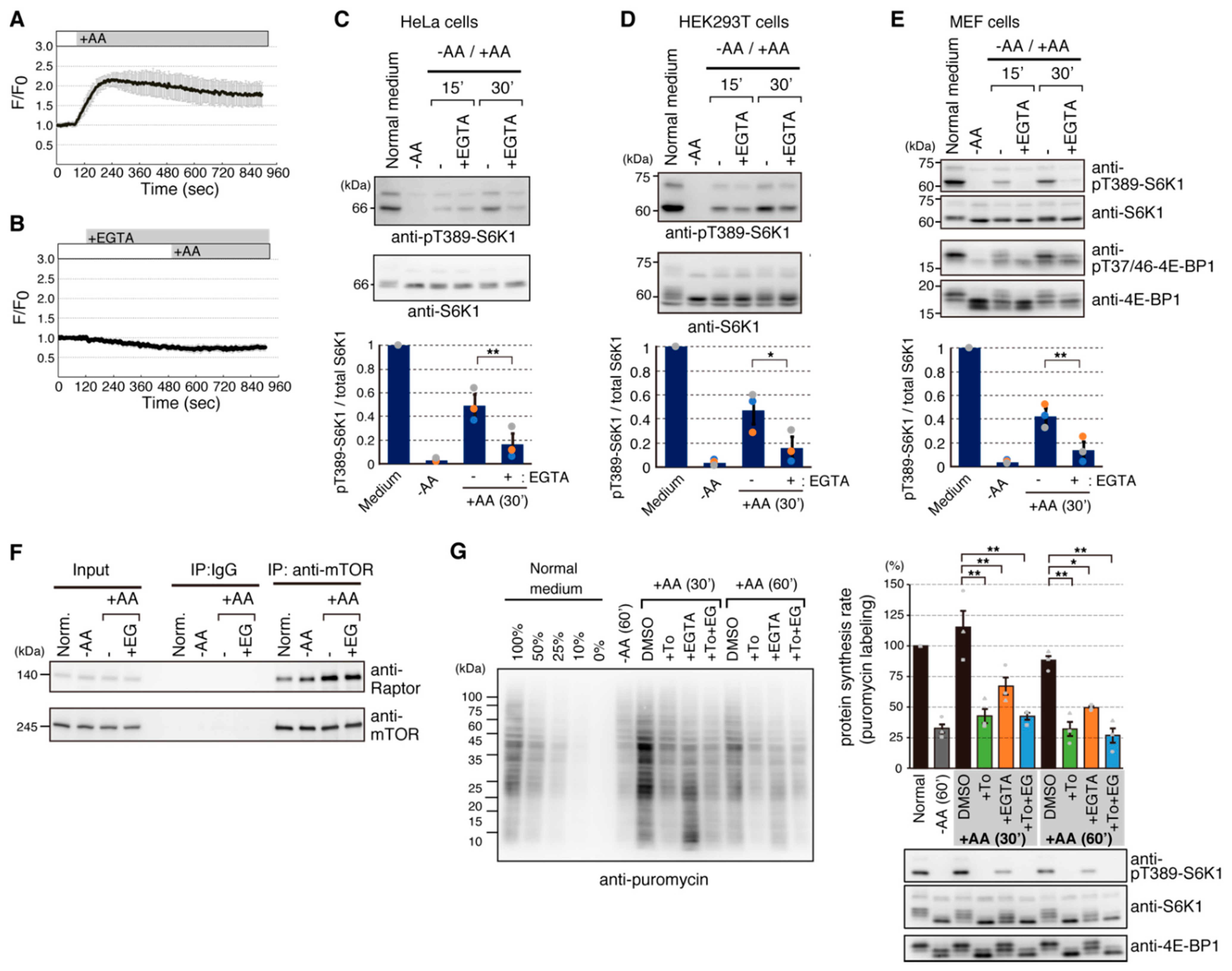
2.1. Addition of Amino Acids Induces Ca2+ Entry
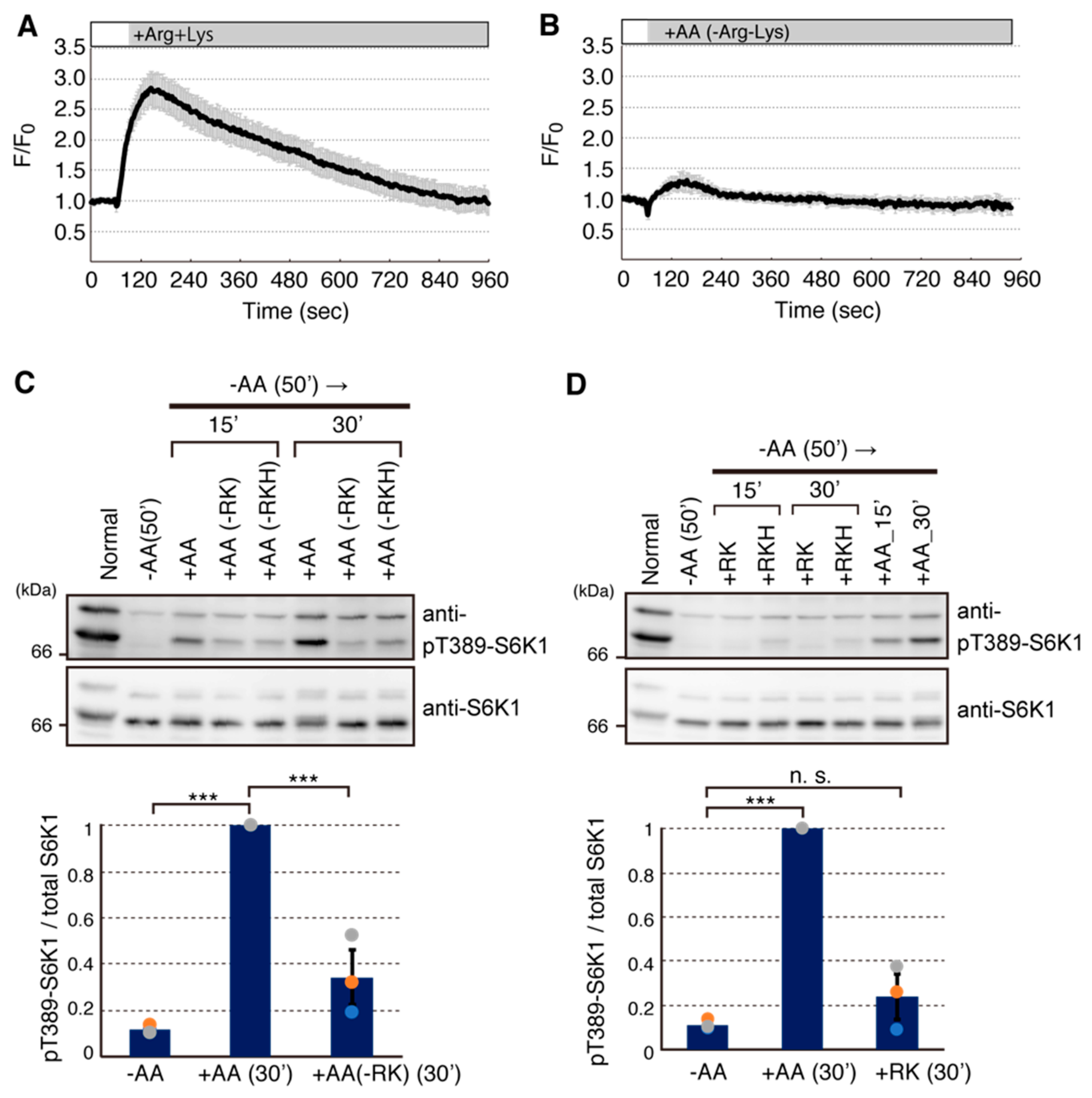
2.2. Arginine and Lysine Are Largely Responsible for Ca2+ Entry
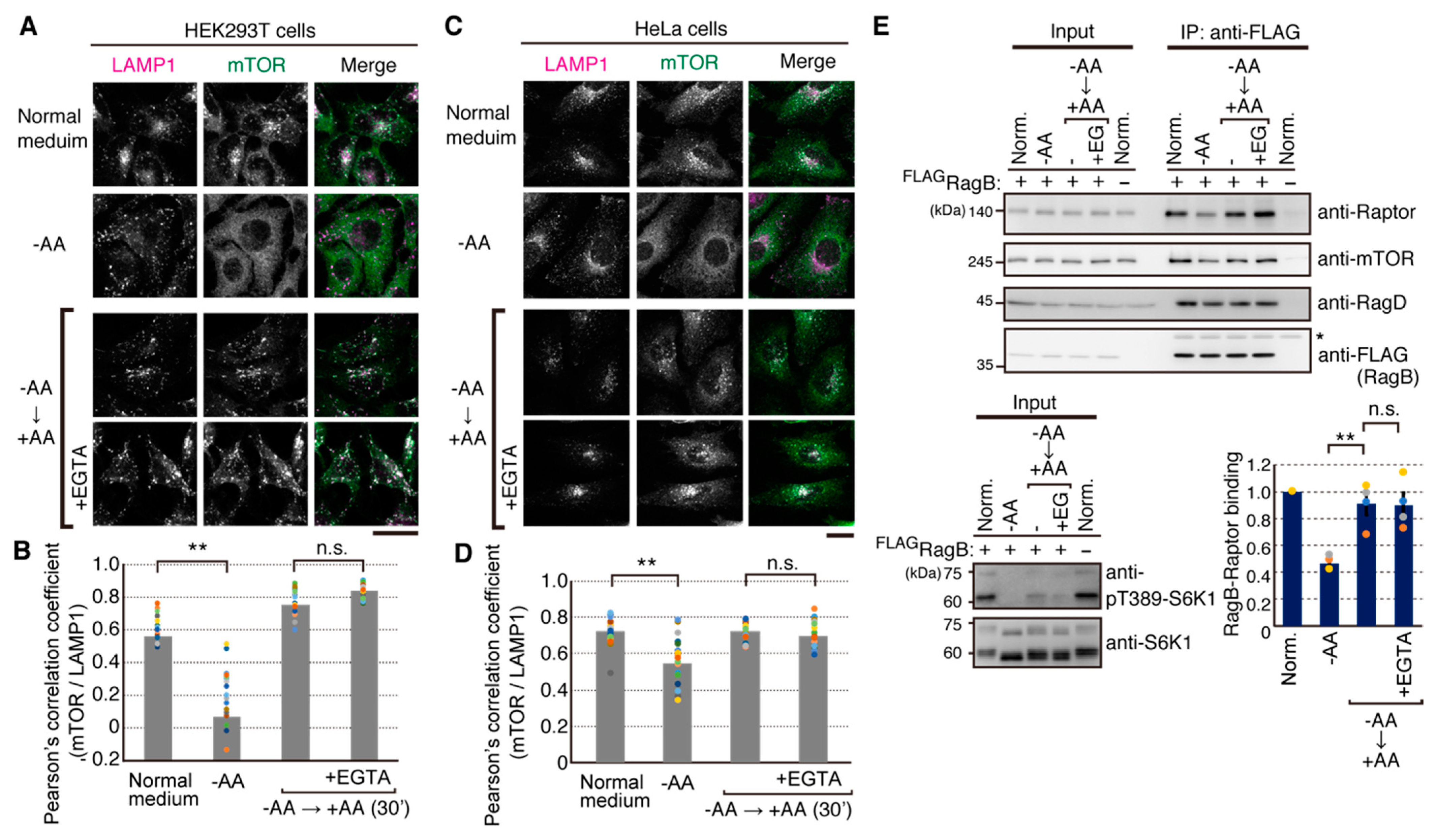
2.3. Amino Acid-Mediated Ca2+ Rise Does Not Affect Lysosomal Translocation of mTORC1
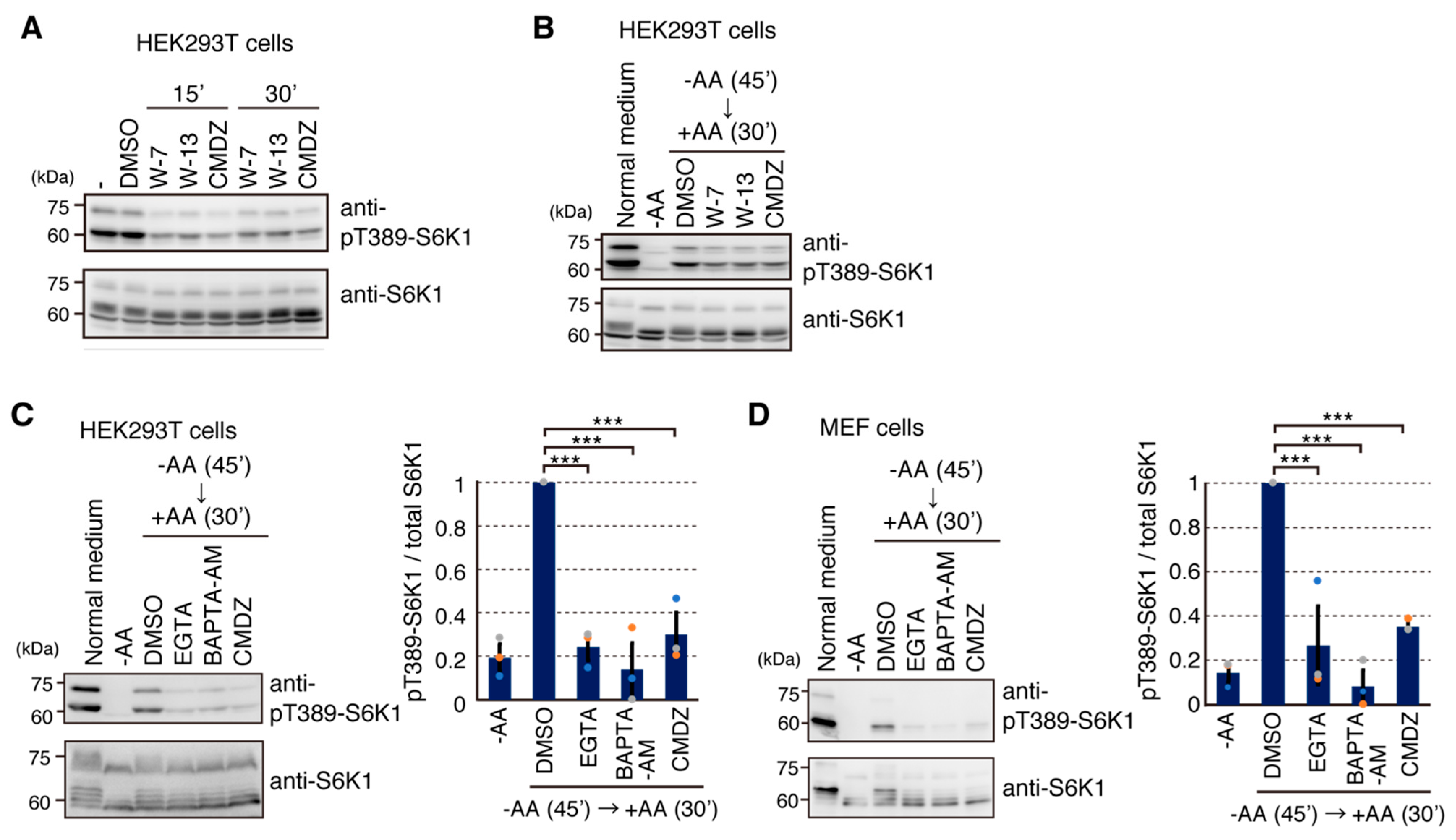
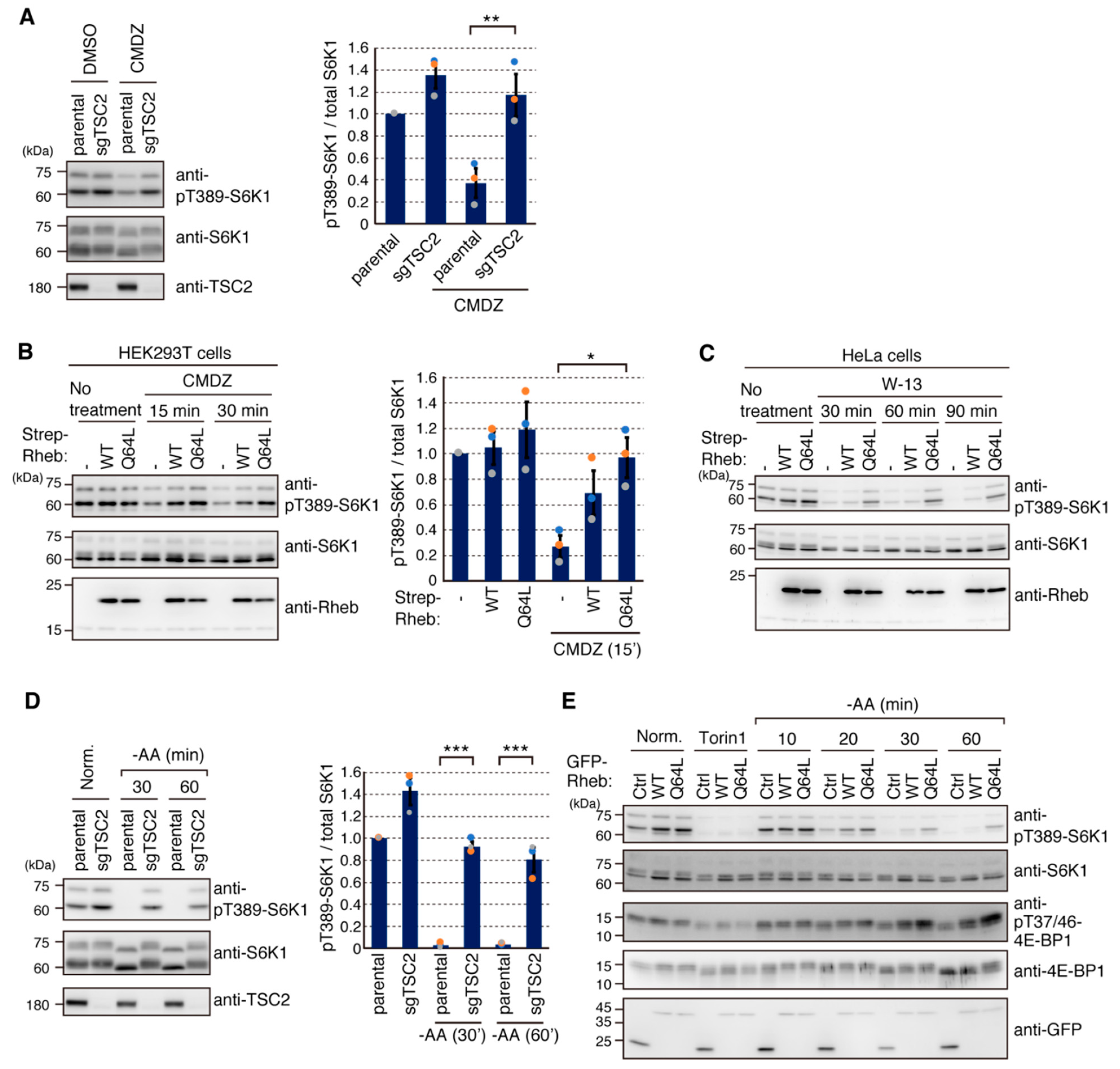
2.4. Calmodulin (CaM) Is Involved in mTORC1 Activation
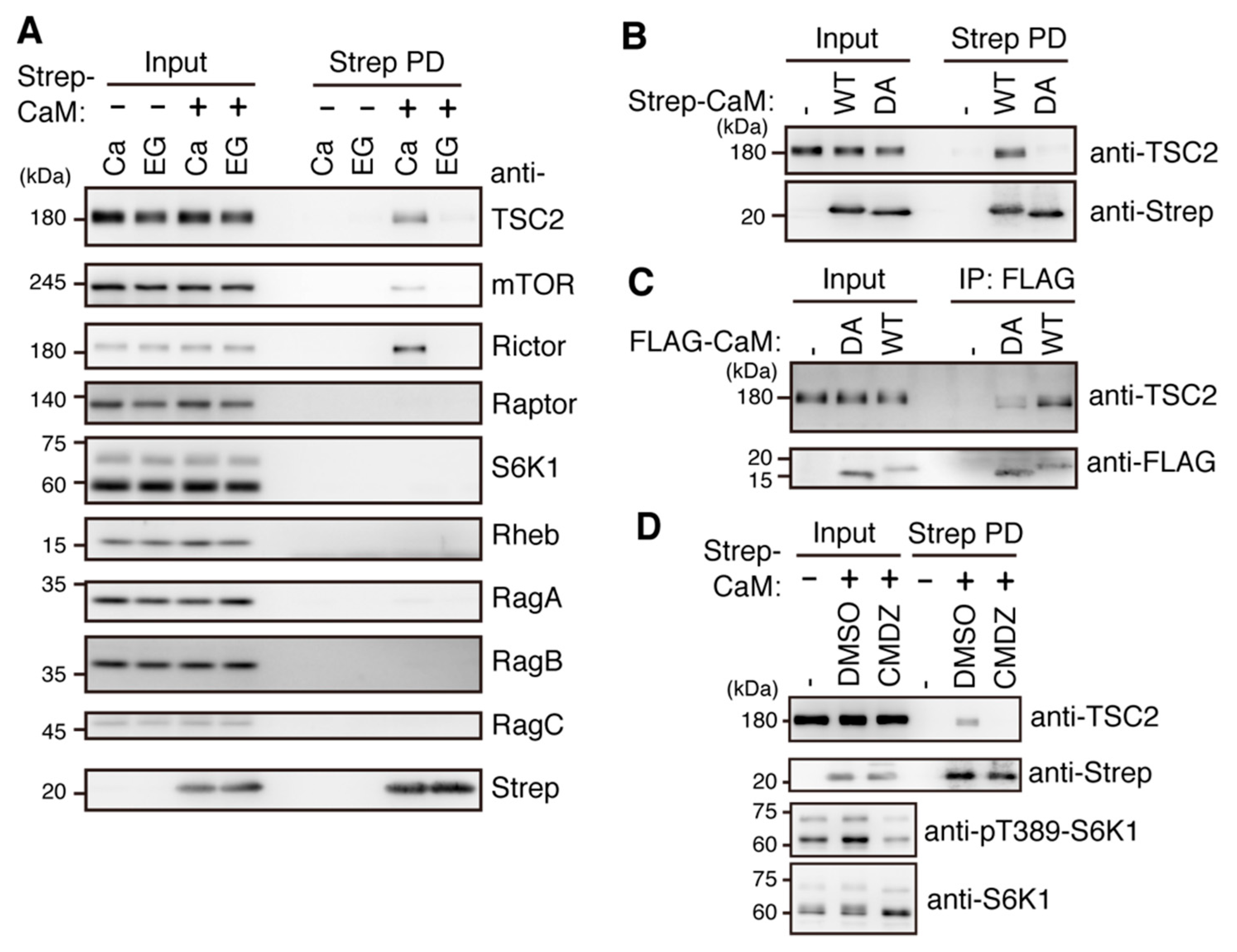
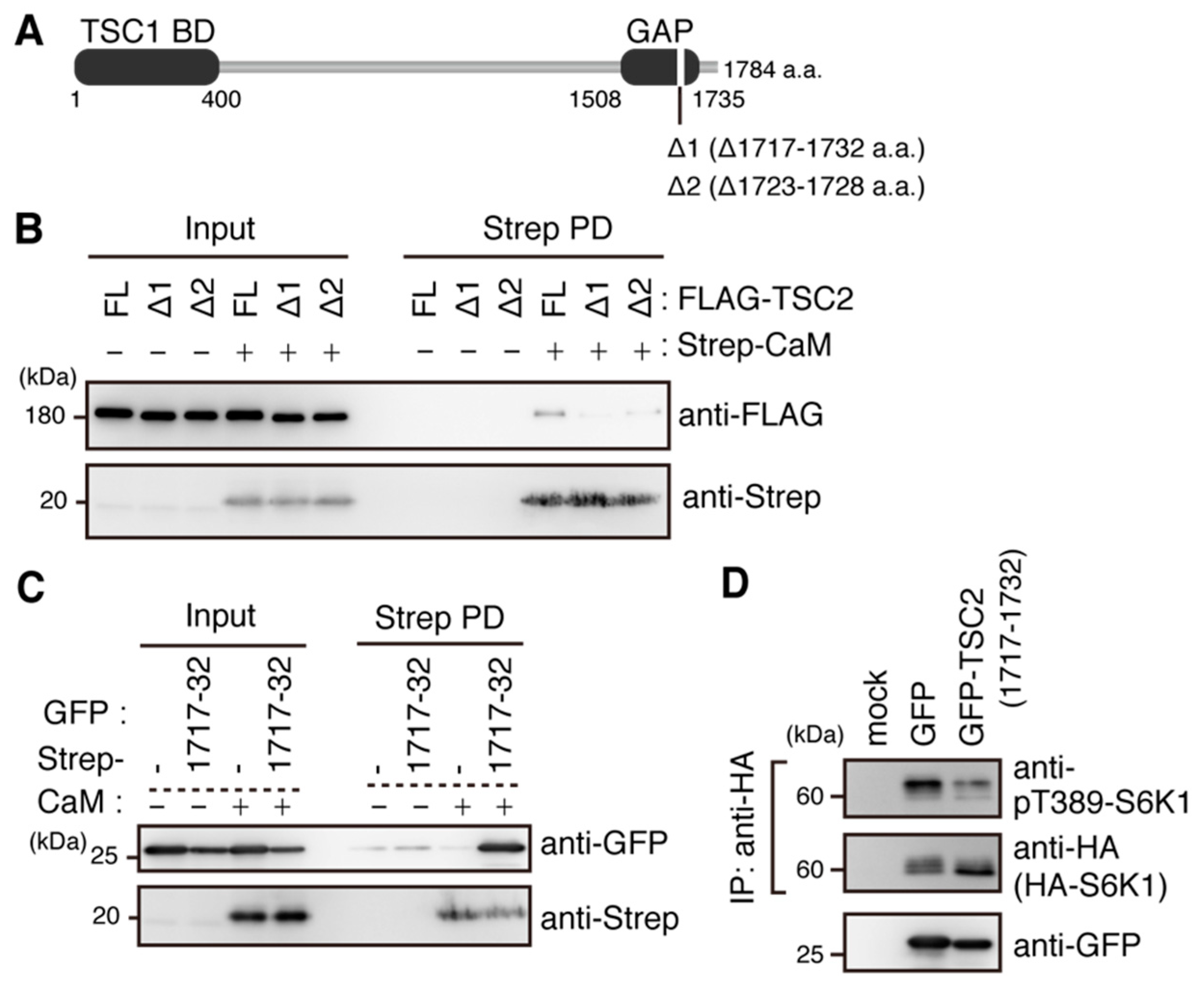
2.5. Ca2+/CaM Binds to TSC2
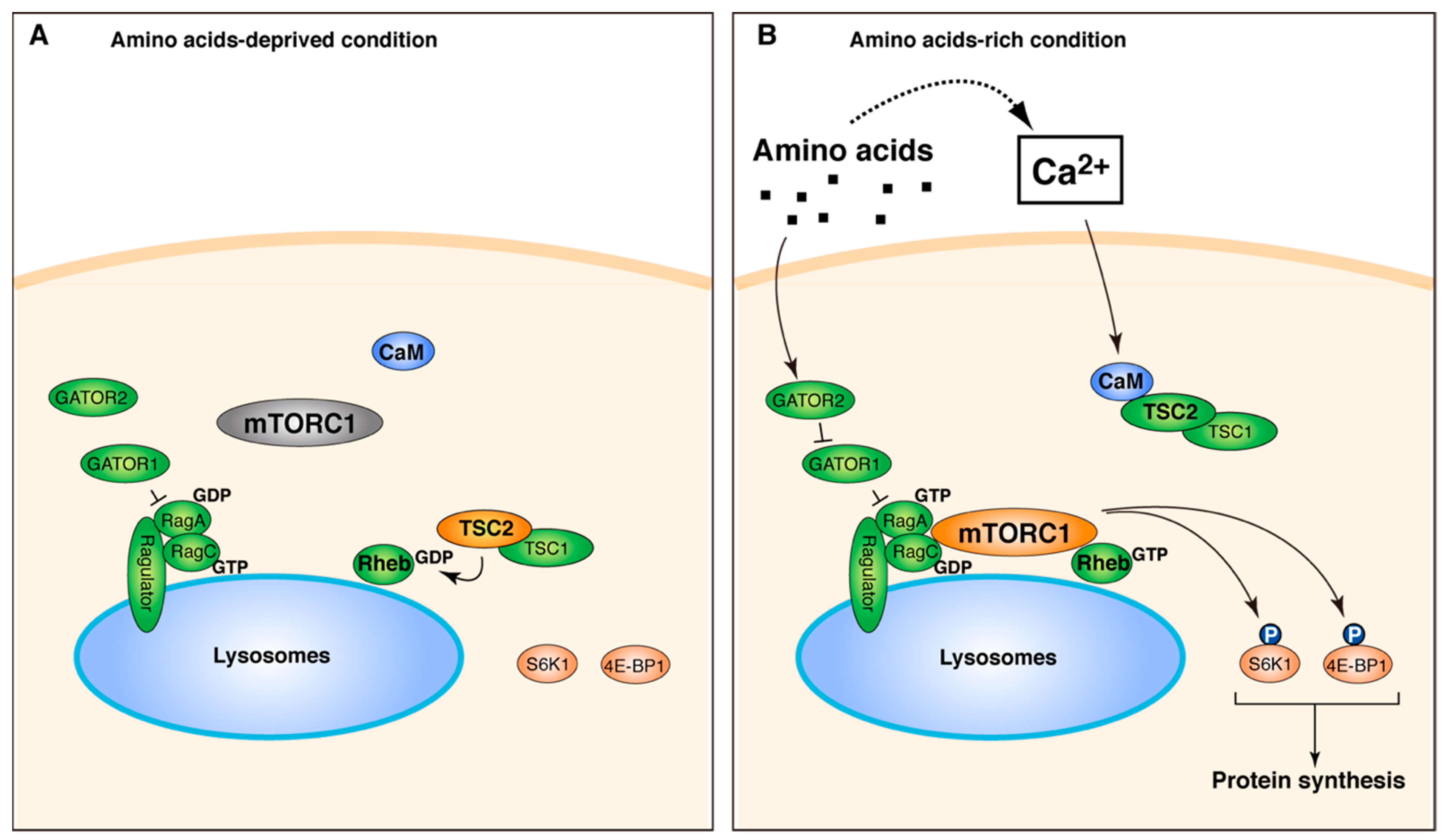
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Plasmids
4.3. Mammalian Cell Culture, Transfection and Retroviral Infection
4.4. Generation of TSC2 Knockout HEK293T Cells
4.5. Analysis of Protein Synthesis Rate
4.6. Live-Cell Imaging
4.7. Indirect Immunofluorescence
4.8. Colocalization Analysis
4.9. Cell Lysate Preparation, Pulldown and Western Blotting
4.10. In Vivo Crosslinking
4.11. Statistical Analysis
4.12. Research Ethics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CaM | calmodulin |
| CMDZ | calmidazolium |
| DMEM | Dulbecco’s modified Eagle’s medium |
| GAP | GTPase activating protein |
| HBSS | Hank’s balanced salt solution |
| HRP | horseradish peroxidase |
| mTORC1 | mechanistic target of rapamycin complex 1 |
| PI3P | phosphatidylinositol 3-phosphate |
| SOCE | store-operated Ca2+ entry |
| SUnSET | Surface Sensing of Translation |
References
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [Green Version]
- Bar-Peled, L.; Sabatini, D.M. Regulation of mTORC1 by amino acids. Trends Cell Biol. 2014, 24, 400–406. [Google Scholar] [CrossRef]
- Takahara, T.; Amemiya, Y.; Sugiyama, R.; Maki, M.; Shibata, H. Amino acid-dependent control of mTORC1 signaling: A variety of regulatory modes. J. Biomed. Sci. 2020, 27, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Guan, K.L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Carroll, B. Spatial regulation of mTORC1 signalling: Beyond the Rag GTPases. Semin. Cell Dev. Biol. 2020, 107, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial Control of the TSC Complex Integrates Insulin and Nutrient Regulation of mTORC1 at the Lysosome. Cell 2014, 156, 771–785. [Google Scholar] [CrossRef] [Green Version]
- Fawal, M.A.; Brandt, M.; Djouder, N. MCRS1 binds and couples rheb to amino acid-dependent mTORC1 activation. Dev. Cell 2015, 33, 67–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, F.; Kondo, K.; Itoh, T.; Ikari, S.; Nada, S.; Okada, M.; Noda, T. Rheb localized on the Golgi membrane activates lysosome-localized mTORC1 at the Golgi-lysosome contact site. J. Cell Sci. 2017, 131, jcs.208017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angarola, B.; Ferguson, S.M. Weak membrane interactions allow Rheb to activate mTORC1 signaling without major lysosome enrichment. Mol. Biol. Cell 2019, 30, 2750–2760. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.-L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Tee, A.R.; Logsdon, M.N.; Blenis, J.; Cantley, L.C. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol. Cell 2002, 10, 151–162. [Google Scholar] [CrossRef]
- Demetriades, C.; Plescher, M.; Teleman, A.A. Lysosomal recruitment of TSC2 is a universal response to cellular stress. Nat. Commun. 2016, 7, 10662. [Google Scholar] [CrossRef] [PubMed]
- Conus, N.M.; Hemmings, B.A.; Pearson, R.B. Differential regulation by calcium reveals distinct signaling requirements for the activation of Akt and p70S6k. J. Biol. Chem. 1998, 273, 4776–4782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graves, L.M.; He, Y.; Lambert, J.; Hunter, D.; Li, X.; Earp, H.S. An intracellular calcium signal activates p70 but not p90 ribosomal S6 kinase in liver epithelial cells. J. Biol. Chem. 1997, 272, 1920–1928. [Google Scholar] [CrossRef] [Green Version]
- Gulati, P.; Gaspers, L.D.; Dann, S.G.; Joaquin, M.; Nobukuni, T.; Natt, F.; Kozma, S.C.; Thomas, A.P.; Thomas, G. Amino acids activate mTOR complex 1 via Ca2+/CaM signaling to hVps34. Cell Metab. 2008, 7, 456–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byfield, M.P.; Murray, J.T.; Backer, J.M. hVps34 is a nutrient-regulated lipid kinase required for activation of p70 S6 kinase. J. Biol. Chem. 2005, 280, 33076–33082. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Flinn, R.J.; Wu, H.; Schnur, R.S.; Backer, J.M. hVps15, but not Ca2+/CaM, is required for the activity and regulation of hVps34 in mammalian cells. Biochem. J. 2009, 417, 747–755. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [Green Version]
- Demetriades, C.; Doumpas, N.; Teleman, A.A. Regulation of TORC1 in Response to Amino Acid Starvation via Lysosomal Recruitment of TSC2. Cell 2014, 156, 786–799. [Google Scholar] [CrossRef] [Green Version]
- Mercan, F.; Lee, H.; Kolli, S.; Bennett, A.M. Novel Role for SHP-2 in Nutrient-Responsive Control of S6 Kinase 1 Signaling. Mol. Cell. Biol. 2012, 33, 293–306. [Google Scholar] [CrossRef] [Green Version]
- Takahara, T.; Inoue, K.; Arai, Y.; Kuwata, K.; Shibata, H.; Maki, M. The calcium-binding protein ALG-2 regulates protein secretion and trafficking via interactions with MISSL and MAP1B proteins. J. Biol. Chem. 2017, 292, 17057–17072. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Araki, S.; Wu, J.; Teramoto, T.; Chang, Y.F.; Nakano, M.; Abdelfattah, A.S.; Fujiwara, M.; Ishihara, T.; Nagai, T.; et al. An expanded palette of genetically encoded Ca2+ indicators. Science 2011, 333, 1888–1891. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, E.K.; Clavarino, G.; Ceppi, M.; Pierre, P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 2009, 6, 275–277. [Google Scholar] [CrossRef]
- Goodman, C.A.; Hornberger, T.A. Measuring protein synthesis with SUnSET: A valid alternative to traditional techniques? Exerc. Sport Sci. Rev. 2013, 41, 107–115. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Muramatsu, A.; Matsuo, R.; Teranishi, N.; Kahara, Y.; Takahara, T.; Shibata, H.; Maki, M. The Penta-EF-Hand ALG-2 Protein Interacts with the Cytosolic Domain of the SOCE Regulator SARAF and Interferes with Ubiquitination. Int. J. Mol. Sci. 2020, 21, 6315. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Rydzewski, N.; Hider, A.; Zhang, X.; Yang, J.; Wang, W.; Gao, Q.; Cheng, X.; Xu, H. A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nat. Cell Biol. 2016, 18, 404–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.J.; Xu, J.; Fu, C.; Zhang, J.; Zheng, Y.G.; Jia, H.; Liu, J.O. Regulation of mTORC1 by lysosomal calcium and calmodulin. eLife 2016, 5. [Google Scholar] [CrossRef]
- Sun, X.; Yang, Y.; Zhong, X.Z.; Cao, Q.; Zhu, X.H.; Zhu, X.; Dong, X.P. A negative feedback regulation of MTORC1 activity by the lysosomal Ca2+ channel MCOLN1 (mucolipin 1) using a CALM (calmodulin)-dependent mechanism. Autophagy 2018, 14, 38–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noonan, D.J.; Lou, D.; Griffith, N.; Vanaman, T.C. A calmodulin binding site in the tuberous sclerosis 2 gene product is essential for regulation of transcription events and is altered by mutations linked to tuberous sclerosis and lymphangioleiomyomatosis. Arch. Biochem. Biophys. 2002, 398, 132–140. [Google Scholar] [CrossRef]
- Piazza, M.; Taiakina, V.; Dieckmann, T.; Guillemette, J.G. Structural Consequences of Calmodulin EF Hand Mutations. Biochemistry 2017, 56, 944–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rok, P.; Kasprzyk-Obara, J.; Domańska-Pakieła, D.; Jóźwiak, S. Clinical symptoms of tuberous sclerosis complex in patients with an identical TSC2 mutation. Med. Sci. Monit. 2005, 11, CR230–CR234. [Google Scholar] [PubMed]
- Ito, N.; Takeda, S. Role of Ca2+ signaling in skeletal muscle hypertrophy and atrophy. J. Phys. Fit. Sport. Med. 2015, 4, 171–176. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Finch, E.A.; Graham, V.; Zhang, Z.-S.; Ding, J.-D.; Burch, J.; Oh-hora, M.; Rosenberg, P. STIM1- Ca2+ Signaling Is Required for the Hypertrophic Growth of Skeletal Muscle in Mice. Mol. Cell. Biol. 2012, 32, 3009–3017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, N.; Ruegg, U.T.; Takeda, S. ATP-Induced Increase in Intracellular Calcium Levels and Subsequent Activation of mTOR as Regulators of Skeletal Muscle Hypertrophy. Int. J. Mol. Sci. 2018, 19, 2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanlialp, A.; Schumacher, D.; Kiper, L.; Varma, E.; Riechert, E.; Ho, T.C.; Hofmann, C.; Kmietczyk, V.; Zimmermann, F.; Dlugosz, S.; et al. Saraf-dependent activation of mTORC1 regulates cardiac growth. J. Mol. Cell. Cardiol. 2020, 141, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hu, H.; Liu, W.; Yan, S.M.; Li, Y.; Tan, L.; Chen, Y.; Liu, J.; Peng, Z.; Yuan, Y.; et al. Amino acids and RagD potentiate mTORC1 activation in CD8+ T cells to confer antitumor immunity. J. Immunother. Cancer 2021, 9, 1–14. [Google Scholar] [CrossRef]
- Weidinger, C.; Shaw, P.J.; Feske, S. STIM1 and STIM2-mediated Ca2+ influx regulates antitumour immunity by CD8+ T cells. EMBO Mol. Med. 2013, 5, 1311–1321. [Google Scholar] [CrossRef]
- Weinhaus, A.J.; Poronnik, P.; Tuch, B.E.; Cook, D.I. Mechanisms of arginine-induced increase in cytosolic calcium concentration in the beta-cell line NIT-1. Diabetologia 1997, 40, 374–382. [Google Scholar] [CrossRef] [Green Version]
- Yoon, M.-S.; Du, G.; Backer, J.M.; Frohman, M.A.; Chen, J. Class III PI-3-kinase activates phospholipase D in an amino acid-sensing mTORC1 pathway. J. Cell Biol. 2011, 195, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Yu, Z.; Chen, X.; Li, J.; Li, N.; Cheng, J.; Gao, N.; Yuan, H.-X.; Ye, D.; Guan, K.-L.; et al. Structural insights into TSC complex assembly and GAP activity on Rheb. Nat. Commun. 2020, 12, 1–23. [Google Scholar] [CrossRef]
- Garami, A.; Zwartkruis, F.J.T.; Nobukuni, T.; Joaquin, M.; Roccio, M.; Stocker, H.; Kozma, S.C.; Hafen, E.; Bos, J.L.; Thomas, G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 2003, 11, 1457–1466. [Google Scholar] [CrossRef] [Green Version]
- Tee, A.R.; Fingar, D.C.; Manning, B.D.; Kwiatkowski, D.J.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 13571–13576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.M.; Finn, S.G.; Tee, A.R.; Browne, G.J.; Proud, C.G. The tuberous sclerosis protein TSC2 is not required for the regulation of the mammalian target of rapamycin by amino acids and certain cellular stresses. J. Biol. Chem. 2005, 280, 18717–18727. [Google Scholar] [CrossRef] [Green Version]
- Roccio, M.; Bos, J.L.; Zwartkruis, F.J.T. Regulation of the small GTPase Rheb by amino acids. Oncogene 2006, 25, 657–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohne, Y.; Takahara, T.; Hatakeyama, R.; Matsuzaki, T.; Noda, M.; Mizushima, N.; Maeda, T. Isolation of hyperactive mutants of mammalian target of rapamycin. J. Biol. Chem. 2008, 283, 31861–31870. [Google Scholar] [CrossRef] [Green Version]
- Kremers, G.-J.; Goedhart, J.; van den Heuvel, D.J.; Gerritsen, H.C.; Gadella, T.W.J. Improved green and blue fluorescent proteins for expression in bacteria and mammalian cells. Biochemistry 2007, 46, 3775–3783. [Google Scholar] [CrossRef]
- Takahara, T.; Hara, K.; Yonezawa, K.; Sorimachi, H.; Maeda, T. Nutrient-dependent multimerization of the mammalian target of rapamycin through the N-terminal HEAT repeat region. J. Biol. Chem. 2006, 281, 28605–28614. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amemiya, Y.; Nakamura, N.; Ikeda, N.; Sugiyama, R.; Ishii, C.; Maki, M.; Shibata, H.; Takahara, T. Amino Acid-Mediated Intracellular Ca2+ Rise Modulates mTORC1 by Regulating the TSC2-Rheb Axis through Ca2+/Calmodulin. Int. J. Mol. Sci. 2021, 22, 6897. https://doi.org/10.3390/ijms22136897
Amemiya Y, Nakamura N, Ikeda N, Sugiyama R, Ishii C, Maki M, Shibata H, Takahara T. Amino Acid-Mediated Intracellular Ca2+ Rise Modulates mTORC1 by Regulating the TSC2-Rheb Axis through Ca2+/Calmodulin. International Journal of Molecular Sciences. 2021; 22(13):6897. https://doi.org/10.3390/ijms22136897
Chicago/Turabian StyleAmemiya, Yuna, Nao Nakamura, Nao Ikeda, Risa Sugiyama, Chiaki Ishii, Masatoshi Maki, Hideki Shibata, and Terunao Takahara. 2021. "Amino Acid-Mediated Intracellular Ca2+ Rise Modulates mTORC1 by Regulating the TSC2-Rheb Axis through Ca2+/Calmodulin" International Journal of Molecular Sciences 22, no. 13: 6897. https://doi.org/10.3390/ijms22136897
APA StyleAmemiya, Y., Nakamura, N., Ikeda, N., Sugiyama, R., Ishii, C., Maki, M., Shibata, H., & Takahara, T. (2021). Amino Acid-Mediated Intracellular Ca2+ Rise Modulates mTORC1 by Regulating the TSC2-Rheb Axis through Ca2+/Calmodulin. International Journal of Molecular Sciences, 22(13), 6897. https://doi.org/10.3390/ijms22136897