
Molecular Mechanisms and Animal Models of HBV-Related Hepatocellular Carcinoma: With Emphasis on Metastatic Tumor Antigen 1




Abstract
:1. Introduction
2. Oncogenic Mechanisms of HBV
2.1. HBV DNA Integration Promotes the Occurrence of HCC
2.2. HBx Is a Multifunctional Viral Protein with Versatile Oncogenic Activities
2.3. Roles of HBV Surface Protein in HCC
2.4. HBV-Induced Inflammation, Liver Injury, and Immunosuppressive Microenvironments Contribute to Hepatocarcinogenesis
3. Animal Models of HBV-HCC
3.1. Genetically Engineered Mouse Models
3.1.1. Conventional HBV Transgenic Mouse Model
3.1.2. Viral Vector-Mediated Transgenic Mouse Model
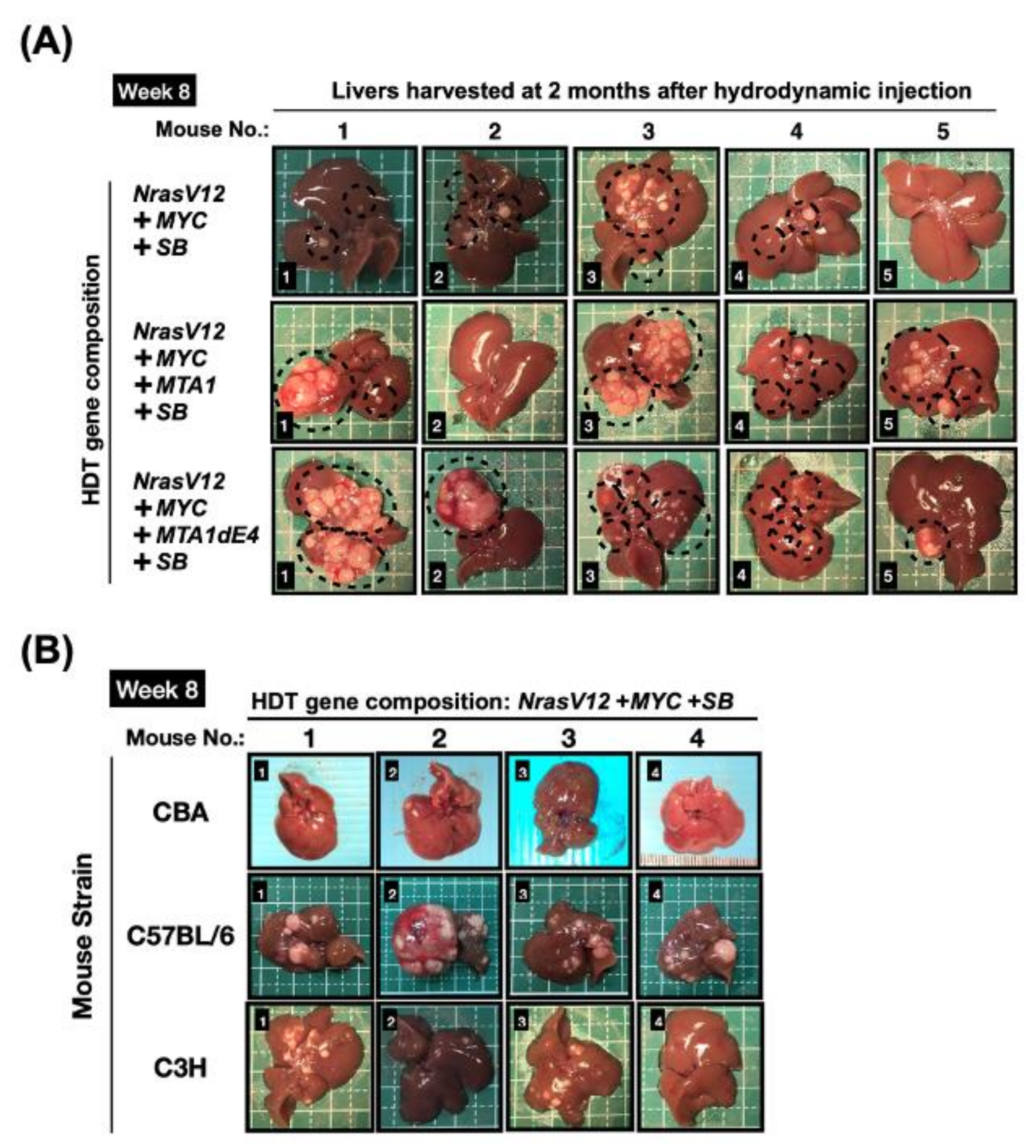
3.1.3. Hydrodynamics-Based Transfection (HDT) and Genetic Modification Systems
3.2. Hepadnavirus Natural Infection-Induced HCC Model
3.2.1. Tree Shrew Model of HBV-HCC
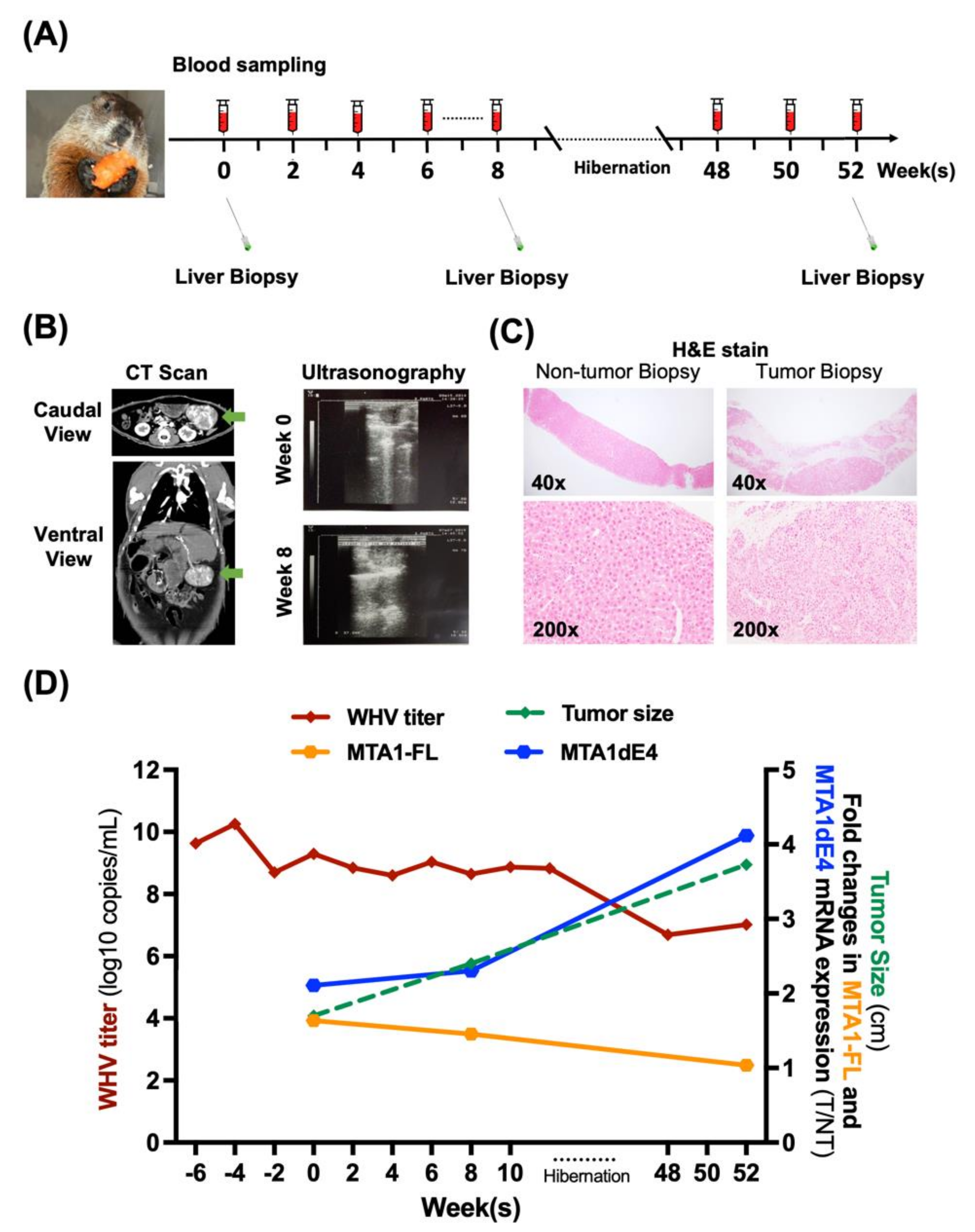
3.2.2. Woodchuck HCC Model: A Surrogate Model Based on HBV-Related Hepadnavirus
3.3. Human Liver-Chimeric Mice Model
4. Relationship between the MTA1 and HBV-HCC
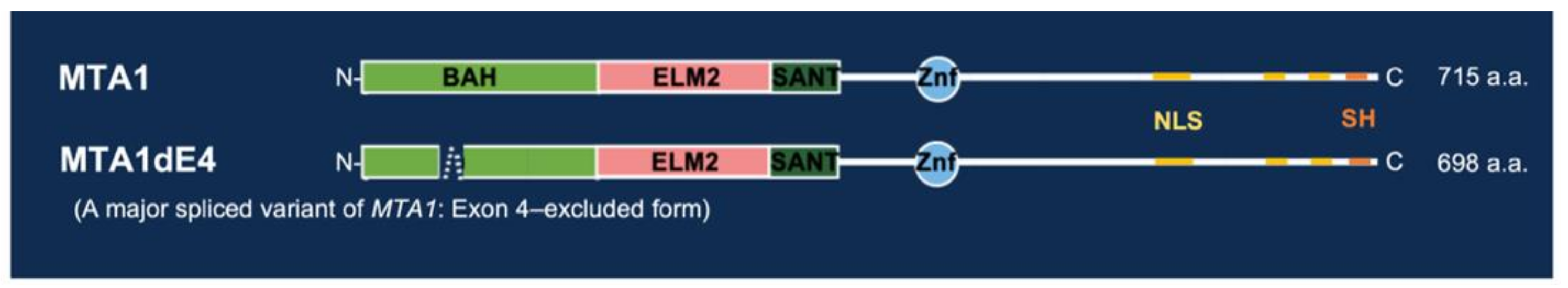
4.1. Structure and Molecular Function of MTA1: An Overview
4.2. Regulation of MTA1 Expression in HBV-HCC
4.3. MTA1 Is Overexpressed in HBV-HCC
4.4. Therapeutic Significance of MTA1
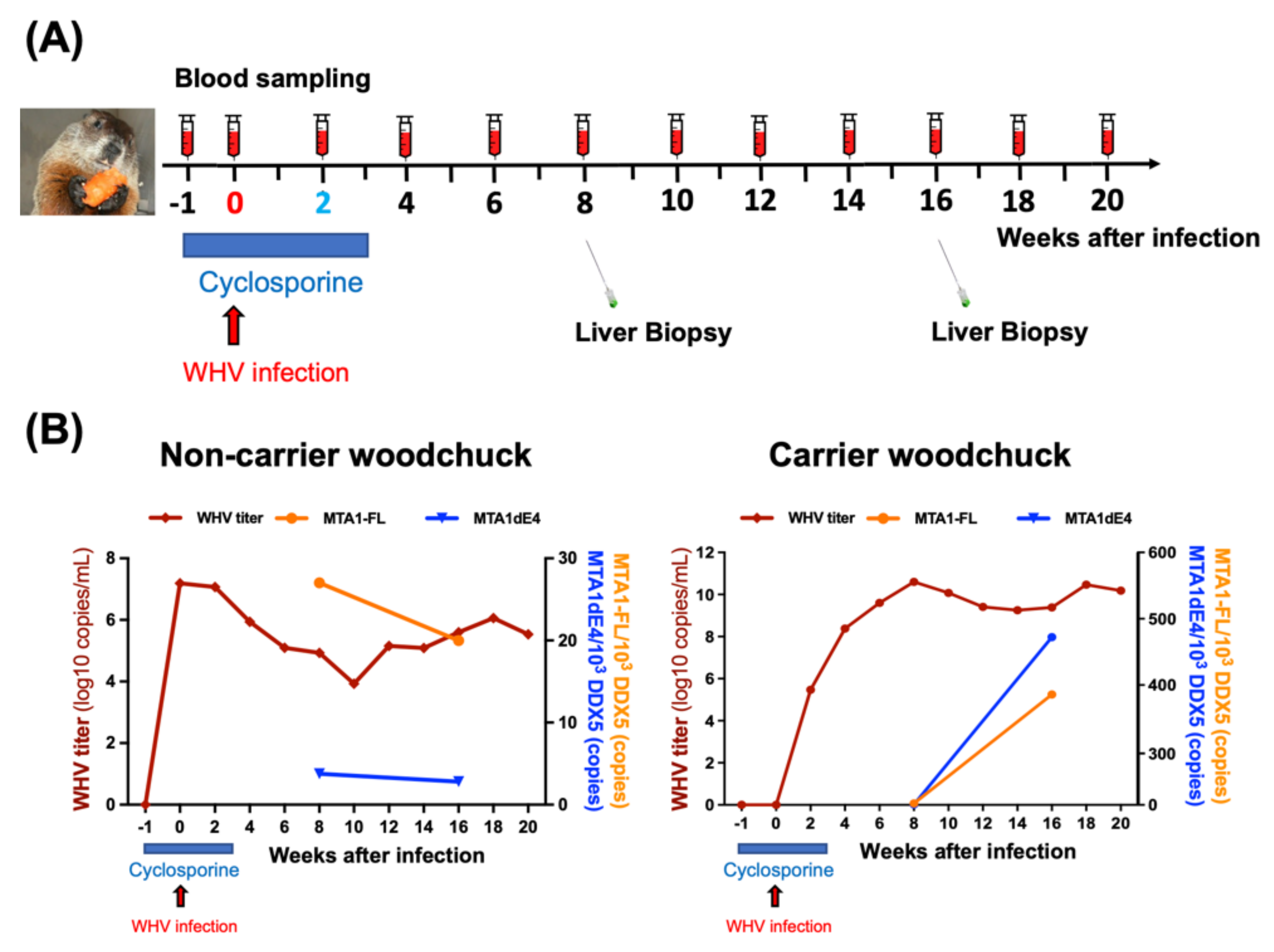
4.5. Application of Hepadnavirus-Induced Woodchuck HCC Model for Studying the Biological Functions and Clinical Significance of MTA1 in HBV-HCC
4.6. Translational Researches Based on the Woodchuck Model
4.7. Potential Issues on Application of HDT-Based Murine Models for Oncogenic Collaboration Studies in HBV-HCC
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.E.M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today; International Agency for Research on Cancer: Lyon, France, 2020. [Google Scholar]
- Wang, W.; Wei, C. Advances in the early diagnosis of hepatocellular carcinoma. Genes Dis. 2020, 7, 308–319. [Google Scholar] [CrossRef]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef]
- El-Serag, H.B. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012, 142, 1264–1273.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLachlan, J.H.; Locarnini, S.; Cowie, B.C. Estimating the global prevalence of hepatitis B. Lancet 2015, 386, 1515–1517. [Google Scholar] [CrossRef]
- Zhu, R.X.; Seto, W.K.; Lai, C.L.; Yuen, M.F. Epidemiology of Hepatocellular Carcinoma in the Asia-Pacific Region. Gut Liver 2016, 10, 332–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petruzziello, A. Epidemiology of Hepatitis B Virus (HBV) and Hepatitis C Virus (HCV) Related Hepatocellular Carcinoma. Open Virol. J. 2018, 12, 26–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, M. Risk of hepatocellular carcinoma in hepatitis B and prevention through treatment. Cleve Clin. J. Med. 2009, 76 (Suppl. 3), S6–S9. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.J.; Yang, H.I.; Su, J.; Jen, C.L.; You, S.L.; Lu, S.N.; Huang, G.T.; Iloeje, U.H.; Group, R.-H.S. Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA level. JAMA 2006, 295, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Kao, J.H. Role of viral factors in the natural course and therapy of chronic hepatitis B. Hepatol. Int. 2007, 1, 415–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.W.; Yeh, S.H.; Chen, P.J.; Liaw, Y.F.; Lin, C.L.; Liu, C.J.; Shih, W.L.; Kao, J.H.; Chen, D.S.; Chen, C.J. Hepatitis B virus genotype and DNA level and hepatocellular carcinoma: A prospective study in men. J. Natl. Cancer Inst. 2005, 97, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.J.; Kao, J.H. Hepatitis B virus-related hepatocellular carcinoma: Epidemiology and pathogenic role of viral factors. J. Chin. Med. Assoc. 2007, 70, 141–145. [Google Scholar] [CrossRef] [Green Version]
- Kao, J.H.; Chen, P.J.; Lai, M.Y.; Chen, D.S. Hepatitis B genotypes correlate with clinical outcomes in patients with chronic hepatitis B. Gastroenterology 2000, 118, 554–559. [Google Scholar] [CrossRef]
- Liu, C.J.; Chen, B.F.; Chen, P.J.; Lai, M.Y.; Huang, W.L.; Kao, J.H.; Chen, D.S. Role of hepatitis B viral load and basal core promoter mutation in hepatocellular carcinoma in hepatitis B carriers. J. Infect. Dis. 2006, 193, 1258–1265. [Google Scholar] [CrossRef] [PubMed]
- Pandyarajan, V.; Govalan, R.; Yang, J.D. Risk Factors and Biomarkers for Chronic Hepatitis B Associated Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 479. [Google Scholar] [CrossRef] [PubMed]
- Yano, Y.; Seo, Y.; Azuma, T.; Hayashi, Y. Hepatitis B virus and host factors. Hepatobiliary Surg. Nutr. 2013, 2, 121–123. [Google Scholar] [CrossRef]
- Liaw, Y.F. Natural history of chronic hepatitis B virus infection and long-term outcome under treatment. Liver Int. 2009, 29 (Suppl. 1), 100–107. [Google Scholar] [CrossRef]
- Singal, A.G.; El-Serag, H.B. Hepatocellular Carcinoma From Epidemiology to Prevention: Translating Knowledge into Practice. Clin. Gastroenterol. Hepatol. 2015, 13, 2140–2151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.J.; Kao, J.H. Global perspective on the natural history of chronic hepatitis B: Role of hepatitis B virus genotypes A to J. Semin. Liver Dis. 2013, 33, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.L.; Kao, J.H. Prevention of hepatitis B virus-related hepatocellular carcinoma. Hepatoma Res. 2021, 7, 9. [Google Scholar]
- Thiele, M.; Gluud, L.L.; Dahl, E.K.; Krag, A. Antiviral therapy for prevention of hepatocellular carcinoma and mortality in chronic hepatitis B: Systematic review and meta-analysis. BMJ Open 2013, 3, e003265. [Google Scholar] [CrossRef] [Green Version]
- Kao, J.H. Hepatitis B vaccination and prevention of hepatocellular carcinoma. Best Pract. Res. Clin. Gastroenterol. 2015, 29, 907–917. [Google Scholar] [CrossRef]
- El-Serag, H.B. Hepatocellular carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef]
- Llovet, J.M.; Fuster, J.; Bruix, J. Intention-to-treat analysis of surgical treatment for early hepatocellular carcinoma: Resection versus transplantation. Hepatology 1999, 30, 1434–1440. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Blanco, C.; Fondevila, F.; Garcia-Palomo, A.; Gonzalez-Gallego, J.; Mauriz, J.L. Sorafenib resistance in hepatocarcinoma: Role of hypoxia-inducible factors. Exp. Mol. Med. 2018, 50, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.J.; Zheng, B.; Wang, H.Y.; Chen, L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol. Sin. 2017, 38, 614–622. [Google Scholar] [CrossRef] [Green Version]
- Bouattour, M.; Mehta, N.; He, A.R.; Cohen, E.I.; Nault, J.C. Systemic Treatment for Advanced Hepatocellular Carcinoma. Liver Cancer 2019, 8, 341–358. [Google Scholar] [CrossRef] [PubMed]
- Jindal, A.; Thadi, A.; Shailubhai, K. Hepatocellular Carcinoma: Etiology and Current and Future Drugs. J. Clin. Exp. Hepatol. 2019, 9, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Chianese, A.; Santella, B.; Ambrosino, A.; Stelitano, D.; Rinaldi, L.; Galdiero, M.; Zannella, C.; Franci, G. Oncolytic Viruses in Combination Therapeutic Approaches with Epigenetic Modulators: Past, Present, and Future Perspectives. Cancers 2021, 13, 2761. [Google Scholar] [CrossRef]
- Pan, Y.; Chen, H.; Yu, J. Biomarkers in Hepatocellular Carcinoma: Current Status and Future Perspectives. Biomedicines 2020, 8, 576. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.T.; Wu, H.L.; Kao, J.H.; Cheng, H.R.; Ho, M.C.; Wang, C.C.; Chen, P.J.; Chen, D.S.; Liu, C.J. Expression of Metastatic Tumor Antigen 1 Splice Variant Correlates with Early Recurrence and Aggressive Features of Hepatitis B Virus-Associated Hepatocellular Carcinoma. Hepatology 2019, 70, 184–197. [Google Scholar] [CrossRef]
- Li, Y.T.; Liu, C.J.; Su, T.H.; Cheng, H.R.; Jeng, Y.M.; Lin, H.L.; Wang, C.C.; Kao, J.H.; Chen, P.J.; Chen, D.S.; et al. Characterization of metastatic tumor antigen 1 and its interaction with hepatitis B virus X protein in NF-kappaB signaling and tumor progression in a woodchuck hepatocellular carcinoma model. Oncotarget 2016, 7, 47173–47185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, T.; Budzinska, M.A.; Vondran, F.W.R.; Shackel, N.A.; Urban, S. Hepatitis B Virus DNA Integration Occurs Early in the Viral Life Cycle in an In Vitro Infection Model via Sodium Taurocholate Cotransporting Polypeptide-Dependent Uptake of Enveloped Virus Particles. J. Virol. 2018, 92, 6416–6421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimbi, G.C.; Kramvis, A.; Kew, M.C. Integration of hepatitis B virus DNA into chromosomal DNA during acute hepatitis B. World J. Gastroenterol. 2005, 11, 6416–6421. [Google Scholar] [CrossRef] [PubMed]
- Mason, W.S.; Gill, U.S.; Litwin, S.; Zhou, Y.; Peri, S.; Pop, O.; Hong, M.L.; Naik, S.; Quaglia, A.; Bertoletti, A.; et al. HBV DNA Integration and Clonal Hepatocyte Expansion in Chronic Hepatitis B Patients Considered Immune Tolerant. Gastroenterology 2016, 151, 986–998.e4. [Google Scholar] [CrossRef] [Green Version]
- Neuveut, C.; Wei, Y.; Buendia, M.A. Mechanisms of HBV-related hepatocarcinogenesis. J. Hepatol. 2010, 52, 594–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferber, M.J.; Yu, C.; Aderca, I.; McGee, A.; Thorland, E.C.; Nagorney, D.M.; Gostout, B.S.; Burgart, L.J.; Boix, L.; Bruix, J.; et al. Integrations of the hepatitis B virus (HBV) and human papillomavirus (HPV) into the human telomerase reverse transcriptase (hTERT) gene in liver and cervical cancers. Oncogene 2003, 22, 3813–3820. [Google Scholar] [CrossRef] [Green Version]
- Totoki, Y.; Tatsuno, K.; Yamamoto, S.; Arai, Y.; Hosoda, F.; Ishikawa, S.; Tsutsumi, S.; Sonoda, K.; Totsuka, H.; Shirakihara, T.; et al. High-resolution characterization of a hepatocellular carcinoma genome. Nat. Genet. 2011, 43, 464–469. [Google Scholar] [CrossRef]
- Li, M.; Zhao, H.; Zhang, X.; Wood, L.D.; Anders, R.A.; Choti, M.A.; Pawlik, T.M.; Daniel, H.D.; Kannangai, R.; Offerhaus, G.J.; et al. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat. Genet. 2011, 43, 828–829. [Google Scholar] [CrossRef] [Green Version]
- Furuta, T.; Kanematsu, T.; Matsumata, T.; Shirabe, K.; Yamagata, M.; Utsunomiya, T.; Sugimachi, K. Clinicopathologic features of hepatocellular carcinoma in young patients. Cancer 1990, 66, 2395–2398. [Google Scholar] [CrossRef]
- Su, J.J.; Li, Y.; Ban, K.C.; Qin, L.L.; Wang, H.Y.; Yang, C.; Ou, C.; Duan, X.X.; Lee Yl, Y.; Yan, R.Q. Alteration of the p53 gene during tree shrews’ hepatocarcinogenesis. Hepatobiliary Pancreat. Dis. Int. 2003, 2, 612–616. [Google Scholar]
- Sung, W.K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Meier, M.A.; Calabrese, D.; Suslov, A.; Terracciano, L.M.; Heim, M.H.; Wieland, S. Ubiquitous expression of HBsAg from integrated HBV DNA in patients with low viral load. J. Hepatol. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Bonilla Guerrero, R.; Roberts, L.R. The role of hepatitis B virus integrations in the pathogenesis of human hepatocellular carcinoma. J. Hepatol. 2005, 42, 760–777. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Liu, A.; Xia, Y. Insights into Hepatitis B Virus DNA Integration-55 Years after Virus Discovery. Innovation 2020, 1, 100034. [Google Scholar] [CrossRef]
- Sze, K.M.; Ho, D.W.; Chiu, Y.T.; Tsui, Y.M.; Chan, L.K.; Lee, J.M.; Chok, K.S.; Chan, A.C.; Tang, C.N.; Tang, V.W.; et al. Hepatitis B Virus-Telomerase Reverse Transcriptase Promoter Integration Harnesses Host ELF4, Resulting in Telomerase Reverse Transcriptase Gene Transcription in Hepatocellular Carcinoma. Hepatology 2021, 73, 23–40. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.-W.; Kim, H.-S.; Kim, J.-S.; Lee, S.-K.; Han, J.-W.; Sung, P.-S.; Bea, S.-H.; Choi, J.-Y.; Yoon, S.-K.; Han, D.-J.; et al. Distinct Patterns of HBV Integration and TERT Alterations between in Tumor and Non-Tumor Tissue in Patients with Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 7056. [Google Scholar] [PubMed]
- Saigo, K.; Yoshida, K.; Ikeda, R.; Sakamoto, Y.; Murakami, Y.; Urashima, T.; Asano, T.; Kenmochi, T.; Inoue, I. Integration of hepatiti’s B virus DNA into the myeloid/lymphoid or mixed-lineage leukemia (MLL4) gene and rearrangements of MLL4 in human hepatocellular carcinoma. Hum. Mutat. 2008, 29, 703–708. [Google Scholar] [CrossRef]
- Mathkar, P.P.; Chen, X.; Sulovari, A.; Li, D. Characterization of Hepatitis B Virus Integrations Identified in Hepatocellular Carcinoma Genomes. Viruses 2021, 13, 245. [Google Scholar] [CrossRef]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol. 2016, 64, S84–S101. [Google Scholar] [CrossRef]
- Lee, W.Y.; Bachtiar, M.; Choo, C.C.; Lee, C.G. Comprehensive review of Hepatitis B Virus-associated hepatocellular carcinoma research through text mining and big data analytics. Biol. Rev. Camb. Philos. Soc. 2019, 94, 353–367. [Google Scholar] [CrossRef]
- Chen, X.; Kost, J.; Sulovari, A.; Wong, N.; Liang, W.S.; Cao, J.; Li, D. A virome-wide clonal integration analysis platform for discovering cancer viral etiology. Genome Res. 2019, 29, 819–830. [Google Scholar] [CrossRef] [Green Version]
- Brechot, C. Pathogenesis of hepatitis B virus-related hepatocellular carcinoma: Old and new paradigms. Gastroenterology 2004, 127, S56–S61. [Google Scholar] [CrossRef]
- Fako, V.; Wang, X.V. Molecular Carcinogenesis of HBV-Related HCC. In Hepatitis B Virus and Liver Disease; Kao, J.H., Chen, D.S., Eds.; Springer: Singapore, 2018. [Google Scholar]
- Zheng, B.Y.; Gao, W.Y.; Huang, X.Y.; Lin, L.Y.; Fang, X.F.; Chen, Z.X.; Wang, X.Z. HBx promotes the proliferative ability of HL7702 cells via the COX2/Wnt/betacatenin pathway. Mol. Med. Rep. 2018, 17, 8432–8438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, W.; Kim, M.; de la Monte, S.; Longato, L.; Carlson, R.; Slagle, B.L.; Dong, X.; Wands, J.R. Activation of signal transduction pathways during hepatic oncogenesis. Cancer Lett. 2016, 370, 1–9. [Google Scholar] [CrossRef]
- Arsura, M.; Cavin, L.G. Nuclear factor-kappaB and liver carcinogenesis. Cancer Lett. 2005, 229, 157–169. [Google Scholar] [CrossRef]
- Bui-Nguyen, T.M.; Pakala, S.B.; Sirigiri, R.D.; Xia, W.; Hung, M.C.; Sarin, S.K.; Kumar, V.; Slagle, B.L.; Kumar, R. NF-kappaB signaling mediates the induction of MTA1 by hepatitis B virus transactivator protein HBx. Oncogene 2010, 29, 1179–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.H.; Yun, Y. HBx protein of hepatitis B virus activates Jak1-STAT signaling. J. Biol. Chem. 1998, 273, 25510–25515. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, S.; Lau, K.C.; Coffin, C.S.; Patel, T.R. Molecular mechanisms of viral hepatitis induced hepatocellular carcinoma. World J. Gastroenterol. 2020, 26, 5759–5783. [Google Scholar] [CrossRef]
- Wang, S.H.; Yeh, S.H.; Chen, P.J. Unique Features of Hepatitis B Virus-Related Hepatocellular Carcinoma in Pathogenesis and Clinical Significance. Cancers 2021, 13, 2454. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wen, X.; Huang, C.; Wei, Y. Unraveling the complexity of hepatitis B virus: From molecular understanding to therapeutic strategy in 50 years. Int. J. Biochem. Cell Biol. 2013, 45, 1987–1996. [Google Scholar] [CrossRef]
- Xie, N.; Chen, X.; Zhang, T.; Liu, B.; Huang, C. Using proteomics to identify the HBx interactome in hepatitis B virus: How can this inform the clinic? Expert Rev. Proteom. 2014, 11, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, H.; Amisaki, M.; Takenaga, A.; Honjo, S.; Fujiwara, Y.; Shiota, G. HBx and c-MYC Cooperate to Induce URI1 Expression in HBV-Related Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 5714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cougot, D.; Wu, Y.; Cairo, S.; Caramel, J.; Renard, C.A.; Levy, L.; Buendia, M.A.; Neuveut, C. The hepatitis B virus X protein functionally interacts with CREB-binding protein/p300 in the regulation of CREB-mediated transcription. J. Biol. Chem. 2007, 282, 4277–4287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.K.; Yu, G.Z.; Li, X.D.; Ren, X.Q. Molecular mechanism of hepatitis B virus (HBV) on suppression of raf kinase inhibitor protein (RKIP) expression. Oncotarget 2017, 8, 1132–1140. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Nomura, T.; Cheong, J.H.; Dorjsuren, D.; Iida, K.; Murakami, S. Hepatitis B virus X protein is a transcriptional modulator that communicates with transcription factor IIB and the RNA polymerase II subunit 5. J. Biol. Chem. 1997, 272, 7132–7139. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Yang, W.; Song, J.; Wu, Y.; Ni, B. Hepatitis B virus X protein-induced aberrant epigenetic modifications contributing to human hepatocellular carcinoma pathogenesis. Mol. Cell Biol. 2013, 33, 2810–2816. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.W.; Jia, Z.C.; Tian, Y.; Yang, P.H.; Sun, H.; Wang, C.H.; Ding, Y.; Zhang, M.J.; Zhang, Y.; Yang, D.; et al. HBx Protein Contributes to Liver Carcinogenesis by H3K4me3 Modification Through Stabilizing WD Repeat Domain 5 Protein. Hepatology 2020, 71, 1678–1695. [Google Scholar] [CrossRef]
- Okamoto, Y.; Shinjo, K.; Shimizu, Y.; Sano, T.; Yamao, K.; Gao, W.; Fujii, M.; Osada, H.; Sekido, Y.; Murakami, S.; et al. Hepatitis virus infection affects DNA methylation in mice with humanized livers. Gastroenterology 2014, 146, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Minor, M.M.; Slagle, B.L. Hepatitis B Virus HBx Protein Interactions with the Ubiquitin Proteasome System. Viruses 2014, 6, 4683–4702. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.D.; Wang, X.B.; Ren, L.F.; Zeng, M.; Wang, S.; Weng, Y.D.; Tang, Z.Z.; Wang, X.; Tang, Y.X.; Hu, H.D.; et al. HBx affects CUL4-DDB1 function in both positive and negative manners. Biochem. Bioph. Res. Commun. 2014, 450, 1492–1497. [Google Scholar] [CrossRef] [PubMed]
- Lazar, C.; Uta, M.; Branza-Nichita, N. Modulation of the unfolded protein response by the human hepatitis B virus. Front. Microbiol. 2014, 5, 433. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.C.; Wu, H.C.; Chen, C.F.; Fausto, N.; Lei, H.Y.; Su, I.J. Different types of ground glass hepatocytes in chronic hepatitis B virus infection contain specific pre-S mutants that may induce endoplasmic reticulum stress. Am. J. Pathol. 2003, 163, 2441–2449. [Google Scholar] [CrossRef] [Green Version]
- Su, I.J.; Wang, H.C.; Wu, H.C.; Huang, W.Y. Ground glass hepatocytes contain pre-S mutants and represent preneoplastic lesions in chronic hepatitis B virus infection. J. Gastroen. Hepatol. 2008, 23, 1169–1174. [Google Scholar] [CrossRef]
- Teng, C.F.; Wu, H.C.; Su, I.J.; Jeng, L.B. Hepatitis B Virus Pre-S Mutants as Biomarkers and Targets for the Development and Recurrence of Hepatocellular Carcinoma. Viruses 2020, 12, 945. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.L.; Hung, J.H.; Huang, W. Association of the Hepatitis B Virus Large Surface Protein with Viral Infectivity and Endoplasmic Reticulum Stress-mediated Liver Carcinogenesis. Cells 2020, 9, 2052. [Google Scholar] [CrossRef]
- Haynes, C.M.; Titus, E.A.; Cooper, A.A. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol. Cell 2004, 15, 767–776. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Corazzari, M.; Gagliardi, M.; Fimia, G.M.; Piacentini, M. Endoplasmic Reticulum Stress, Unfolded Protein Response, and Cancer Cell Fate. Front. Oncol. 2017, 7, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, Y.H.; Su, I.J.; Wang, H.C.; Chang, W.W.; Lei, H.Y.; Lai, M.D.; Chang, W.T.; Huang, W. Pre-S mutant surface antigens in chronic hepatitis B virus infection induce oxidative stress and DNA damage. Carcinogenesis 2004, 25, 2023–2032. [Google Scholar] [CrossRef] [Green Version]
- Hildt, E.; Munz, B.; Saher, G.; Reifenberg, K.; Hofschneider, P.H. The PreS2 activator MHBs(t) of hepatitis B virus activates c-raf-1/Erk2 signaling in transgenic mice. EMBO J. 2002, 21, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal Transduct. Target Ther. 2021, 6, 263. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, Y.; Guidotti, L.G.; Kuhlen, C.V.; Fowler, P.; Chisari, F.V. Immune pathogenesis of hepatocellular carcinoma. J. Exp. Med. 1998, 188, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.H.; Mei, M.H.; Fei, R.; Liu, F.; Wang, J.H.; Liao, W.J.; Qin, L.L.; Wei, L.; Chen, H.S. Regulatory T cells in chronic hepatitis B patients affect the immunopathogenesis of hepatocellular carcinoma by suppressing the anti-tumour immune responses. J. Viral. Hepat. 2010, 17 (Suppl. 1), 34–43. [Google Scholar] [CrossRef]
- Trehanpati, N.; Vyas, A.K. Immune Regulation by T Regulatory Cells in Hepatitis B Virus-Related Inflammation and Cancer. Scand. J. Immunol. 2017, 85, 175–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Han, Q.; Zhang, C.; Xiao, M.; Zhang, J. Hepatitis B virus antigens impair NK cell function. Int. Immunopharmacol. 2016, 38, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Stoop, J.N.; van der Molen, R.G.; Baan, C.C.; van der Laan, L.J.; Kuipers, E.J.; Kusters, J.G.; Janssen, H.L. Regulatory T cells contribute to the impaired immune response in patients with chronic hepatitis B virus infection. Hepatology 2005, 41, 771–778. [Google Scholar] [CrossRef]
- Bolukbas, C.; Bolukbas, F.F.; Horoz, M.; Aslan, M.; Celik, H.; Erel, O. Increased oxidative stress associated with the severity of the liver disease in various forms of hepatitis B virus infection. BMC Infect. Dis. 2005, 5, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duygu, F.; Karsen, H.; Aksoy, N.; Taskin, A. Relationship of Oxidative Stress in Hepatitis B Infection Activity with HBV DNA and Fibrosis. Ann. Lab. Med. 2012, 32, 113–118. [Google Scholar] [CrossRef]
- Macek Jilkova, Z.; Kurma, K.; Decaens, T. Animal Models of Hepatocellular Carcinoma: The Role of Immune System and Tumor Microenvironment. Cancers 2019, 11, 1487. [Google Scholar] [CrossRef] [Green Version]
- Santos, N.P.; Colaco, A.A.; Oliveira, P.A. Animal models as a tool in hepatocellular carcinoma research: A Review. Tumour Biol. 2017, 39, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Maya, S.; Ploss, A. Animal Models of Hepatitis B Virus Infection-Success, Challenges, and Future Directions. Viruses 2021, 13, 777. [Google Scholar] [CrossRef]
- Burk, R.D.; DeLoia, J.A.; elAwady, M.K.; Gearhart, J.D. Tissue preferential expression of the hepatitis B virus (HBV) surface antigen gene in two lines of HBV transgenic mice. J. Virol. 1988, 62, 649–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, K.; Miyazaki, J.; Hino, O.; Tomita, N.; Chisaka, O.; Matsubara, K.; Yamamura, K. Expression and replication of hepatitis B virus genome in transgenic mice. Proc. Natl. Acad. Sci. USA 1989, 86, 207–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega-Prieto, A.M.; Cherry, C.; Gunn, H.; Dorner, M. In Vivo Model Systems for Hepatitis B Virus Research. ACS Infect. Dis. 2019, 5, 688–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Tian, D.A.; Li, P.Y.; He, X.X. Mouse models of liver cancer: Progress and recommendations. Oncotarget 2015, 6, 23306–23322. [Google Scholar] [CrossRef]
- Lee, T.H.; Finegold, M.J.; Shen, R.F.; DeMayo, J.L.; Woo, S.; Butel, J.S. Hepatitis B virus transactivator X protein is not tumorigenic in transgenic mice. J. Virol. 1990, 64, 5939–5974. [Google Scholar] [CrossRef] [Green Version]
- Billet, O.; Grimber, G.; Levrero, M.; Seye, K.A.; Briand, P.; Joulin, V. In vivo activity of the hepatitis B virus core promoter: Tissue specificity and temporal regulation. J. Virol. 1995, 69, 5912–5916. [Google Scholar] [CrossRef] [Green Version]
- Dragani, T.A.; Manenti, G.; Farza, H.; Della Porta, G.; Tiollais, P.; Pourcel, C. Transgenic mice containing hepatitis B virus sequences are more susceptible to carcinogen-induced hepatocarcinogenesis. Carcinogenesis 1990, 11, 953–956. [Google Scholar] [CrossRef]
- Wang, C.; Yang, W.; Yan, H.X.; Luo, T.; Zhang, J.; Tang, L.; Wu, F.Q.; Zhang, H.L.; Yu, L.X.; Zheng, L.Y.; et al. Hepatitis B virus X (HBx) induces tumorigenicity of hepatic progenitor cells in 3,5-diethoxycarbonyl-1,4-dihydrocollidine-treated HBx transgenic mice. Hepatology 2012, 55, 108–120. [Google Scholar] [CrossRef]
- Singh, M.; Kumar, V. Transgenic mouse models of hepatitis B virus-associated hepatocellular carcinoma. Rev. Med. Virol. 2003, 13, 243–253. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, F.; Lv, Y.; Li, C.; Xu, X.; Deng, C.; Wang, D.; Sun, Y.; Hu, G.; Lang, Z.; et al. HBsAg and HBx knocked into the p21 locus causes hepatocellular carcinoma in mice. Hepatology 2004, 39, 318–324. [Google Scholar]
- Dunsford, H.A.; Sell, S.; Chisari, F.V. Hepatocarcinogenesis Due to Chronic Liver-Cell Injury in Hepatitis-B Virus Transgenic Mice. Cancer Res. 1990, 50, 3400–3407. [Google Scholar] [PubMed]
- Chisari, F.V.; Klopchin, K.; Moriyama, T.; Pasquinelli, C.; Dunsford, H.A.; Sell, S.; Pinkert, C.A.; Brinster, R.L.; Palmiter, R.D. Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice. Cell 1989, 59, 1145–1156. [Google Scholar] [CrossRef]
- Flotte, T.R. Gene therapy progress and prospects: Recombinant adeno-associated virus (rAAV) vectors. Gene Ther. 2004, 11, 805–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Samulski, R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020, 21, 255–272. [Google Scholar] [CrossRef]
- Wang, D.; Tai, P.W.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Fang, C.C.; Tsuneyama, K.; Chou, H.Y.; Pan, W.Y.; Shih, Y.M.; Wu, P.Y.; Chen, Y.; Leung, P.S.; Gershwin, M.E.; et al. A murine model of hepatitis B-associated hepatocellular carcinoma generated by adeno-associated virus-mediated gene delivery. Int. J. Oncol. 2011, 39, 1511–1519. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Liu, L.; Zhu, D.; Peng, H.; Su, L.; Fu, Y.X.; Zhang, L. A mouse model for HBV immunotolerance and immunotherapy. Cell Mol. Immunol. 2014, 11, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Yu, H.; Li, C.; Hirsch, M.L.; Zhang, L.; Samulski, R.J.; Li, W.; Liu, Z. Adeno-Associated Virus Vector Mediated Delivery of the HBV Genome Induces Chronic Hepatitis B Virus Infection and Liver Fibrosis in Mice. PLoS ONE 2015, 10, e0130052. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Zhang, Y.; Duan, D.; Engelhardt, J.F. Trans-splicing vectors expand the utility of adeno-associated virus for gene therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 6716–6721. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Yue, Y.; Lai, Y.; Ye, C.; Qiu, J.; Pintel, D.J.; Duan, D. Trans-splicing adeno-associated viral vector-mediated gene therapy is limited by the accumulation of spliced mRNA but not by dual vector coinfection efficiency. Hum. Gene Ther. 2004, 15, 896–905. [Google Scholar] [CrossRef]
- Wu, L.L.; Wang, H.Y.; Chen, P.J. Hydrodynamic HBV Transfection Mouse Model. Methods Mol. Biol. 2017, 1540, 227–235. [Google Scholar] [CrossRef]
- Huang, L.R.; Wu, H.L.; Chen, P.J.; Chen, D.S. An immunocompetent mouse model for the tolerance of human chronic hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 2006, 103, 17862–17867. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.L.; Peng, W.H.; Wu, H.L.; Miaw, S.C.; Yeh, S.H.; Yang, H.C.; Liao, P.H.; Lin, J.S.; Chen, Y.R.; Hong, Y.T.; et al. Lymphocyte Antigen 6 Complex, Locus C(+) Monocytes and Kupffer Cells Orchestrate Liver Immune Responses Against Hepatitis B Virus in Mice. Hepatology 2019, 69, 2364–2380. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Wu, H.L.; Kao, J.H.; Hwang, L.H. Persistent hepatitis B viral replication in a FVB/N mouse model: Impact of host and viral factors. PLoS ONE 2012, 7, e36984. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.L.; Althage, A.; Chung, J.; Chisari, F.V. Hydrodynamic injection of viral DNA: A mouse model of acute hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 2002, 99, 13825–13830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Zhu, Y.; Shao, D.; Chang, H.; Zhang, X.; Zhou, D.; Gao, Y.; Lan, K.; Deng, Q. Recombinant covalently closed circular DNA of hepatitis B virus induces long-term viral persistence with chronic hepatitis in a mouse model. Hepatology 2018, 67, 56–70. [Google Scholar] [CrossRef]
- Ju, H.L.; Han, K.H.; Lee, J.D.; Ro, S.W. Transgenic mouse models generated by hydrodynamic transfection for genetic studies of liver cancer and preclinical testing of anti-cancer therapy. Int. J. Cancer 2016, 138, 1601–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keng, V.W.; Tschida, B.R.; Bell, J.B.; Largaespada, D.A. Modeling hepatitis B virus X-induced hepatocellular carcinoma in mice with the Sleeping Beauty transposon system. Hepatology 2011, 53, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239.e4. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.T.; Tseng, T.C.; Soong, R.S.; Peng, C.Y.; Cheng, Y.H.; Huang, S.F.; Chuang, T.H.; Kao, J.H.; Huang, L.R. A novel spontaneous hepatocellular carcinoma mouse model for studying T-cell exhaustion in the tumor microenvironment. J. Immunother Cancer 2018, 6, 144. [Google Scholar] [CrossRef]
- Chen, X.; Calvisi, D.F. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am. J. Pathol. 2014, 184, 912–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Qi, X.; Zeng, Z.; Wang, L.; Wang, J.; Zhang, T.; Xu, Q.; Shen, C.; Zhou, G.; Yang, S.; et al. CRISPR/Cas9-mediated p53 and Pten dual mutation accelerates hepatocarcinogenesis in adult hepatitis B virus transgenic mice. Sci. Rep. 2017, 7, 2796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, D.H.J.; Jeong, W.I.; Chung, J.Y.; An, M.Y.; Jung, C.Y.; Lee, G.J.; Kang, J.S.; Kang, B.C.; Jee, Y.H.; Williams, B.H.; et al. Hepatic cirrhosis occurring in a young woodchuck (Marmota monax) due to vertical transmission of woodchuck hepatitis virus (WHV). J. Vet. Sci. 2003, 4, 199–201. [Google Scholar] [CrossRef]
- Tennant, B.C.; Toshkov, I.A.; Peek, S.F.; Jacob, J.R.; Menne, S.; Hornbuckle, W.E.; Schinazi, R.D.; Korba, B.E.; Cote, P.J.; Gerin, J.L. Hepatocellular carcinoma in the woodchuck model of hepatitis B virus infection. Gastroenterology 2004, 127, S283–S293. [Google Scholar] [CrossRef] [Green Version]
- Gyorkey, F.; Melnick, J.L.; Mirkovic, R.; Cabral, G.A.; Gyorkey, P.; Hollinger, F.B. Experimental carcinoma of liver in macaque monkeys exposed to diethylnitrosamine and hepatitis B virus. J. Natl. Cancer Inst. 1977, 59, 1451–1467. [Google Scholar] [CrossRef]
- Marion, P.L.; Van Davelaar, M.J.; Knight, S.S.; Salazar, F.H.; Garcia, G.; Popper, H.; Robinson, W.S. Hepatocellular carcinoma in ground squirrels persistently infected with ground squirrel hepatitis virus. Proc. Natl. Acad. Sci. USA 1986, 83, 4543–4546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Ruan, P.; Ou, C.; Su, J.; Cao, J.; Luo, C.; Tang, Y.; Wang, Q.; Qin, H.; Sun, W.; et al. Chronic hepatitis B virus infection and occurrence of hepatocellular carcinoma in tree shrews (Tupaia belangeri chinensis). Virol. J. 2015, 12, 26. [Google Scholar] [CrossRef] [Green Version]
- Su, J.J.; Ban, K.C.; Li, Y.; Qin, L.L.; Wang, H.Y.; Yang, C.; Ou, C.; Duan, X.X.; Lee, Y.L.; Yang, R.Q. Alteration of p53 and p21 during hepatocarcinogenesis in tree shrews. World J. Gastroenterol. 2004, 10, 3559–3563. [Google Scholar] [CrossRef]
- Li, Y.; Wan, D.F.; Wei, W.; Su, J.J.; Cao, J.; Qiu, X.K.; Ou, C.; Ban, K.C.; Yang, C.; Yue, H.F. Candidate genes responsible for human hepatocellular carcinoma identified from differentially expressed genes in hepatocarcinogenesis of the tree shrew (Tupaia belangeri chinesis). Hepatol. Res. 2008, 38, 85–95. [Google Scholar] [CrossRef]
- Tennant, B.C. Animal models of hepadnavirus-associated hepatocellular carcinoma. Clin. Liver Dis. 2001, 5, 43–68. [Google Scholar] [CrossRef]
- Summers, J. Three recently described animal virus models for human hepatitis B virus. Hepatology 1981, 1, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Fourel, G.; Trepo, C.; Bougueleret, L.; Henglein, B.; Ponzetto, A.; Tiollais, P.; Buendia, M.A. Frequent activation of N-myc genes by hepadnavirus insertion in woodchuck liver tumours. Nature 1990, 347, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.; Moroy, T.; Etiemble, J.; Louise, A.; Trepo, C.; Tiollais, P.; Buendia, M.A. Activation of c-myc by woodchuck hepatitis virus insertion in hepatocellular carcinoma. Cell 1988, 55, 627–635. [Google Scholar] [CrossRef]
- Chen, H.S.; Kaneko, S.; Girones, R.; Anderson, R.W.; Hornbuckle, W.E.; Tennant, B.C.; Cote, P.J.; Gerin, J.L.; Purcell, R.H.; Miller, R.H. The Woodchuck Hepatitis Virus-X Gene Is Important for Establishment of Virus-Infection in Woodchucks. J. Virol. 1993, 67, 1218–1226. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.Y.; Yacoub, J.H.; Field, D.H.; Park, B.U.; Kallakury, B.; Korolowicz, K.E.; Menne, S. Suitability of the woodchuck HCC as a preclinical model for evaluation of intra-arterial therapies. Anim. Model Exp. Med. 2020, 3, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, L.R.; Stone, J.R.; Mata, J.; Hawrylack, A.; Kubicka, E.; Brautigan, D.L. The Use of the Woodchuck as an Animal Model for Evaluation of Transarterial Embolization. J. Vasc. Interv. Radiol. 2017, 28, 1467–1471. [Google Scholar] [CrossRef]
- Lai, F.; Wee, C.Y.Y.; Chen, Q.F. Establishment of Humanized Mice for the Study of HBV. Front. Immunol. 2021, 12, 638447. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Li, J. Humanized chimeric mouse models of hepatitis B virus infection. Int. J. Infect. Dis. 2017, 59, 131–136. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.Z.; Jiang, J.; Liu, X.; Zhang, Y.L.; Zhang, L.; Xin, J.J.; Wu, K.; Li, X.L.; Cao, J.L.; Guo, X.R.; et al. HBV infection-induced liver cirrhosis development in dual-humanised mice with human bone mesenchymal stem cell transplantation. Gut 2019, 68, 2044–2056. [Google Scholar] [CrossRef] [Green Version]
- Zacharakis, G.; Aleid, A.; Aldossari, K.K. New and old biomarkers of hepatocellular carcinoma. Hepatoma Res. 2018, 4, 65. [Google Scholar] [CrossRef] [Green Version]
- Ryu, S.H.; Chung, Y.H.; Lee, H.; Kim, J.A.; Shin, H.D.; Min, H.J.; Seo, D.D.; Jang, M.K.; Yu, E.; Kim, K.W. Metastatic tumor antigen 1 is closely associated with frequent postoperative recurrence and poor survival in patients with hepatocellular carcinoma. Hepatology 2008, 47, 929–936. [Google Scholar] [CrossRef]
- Toh, Y.; Nicolson, G.L. Properties and clinical relevance of MTA1 protein in human cancer. Cancer Metastasis. Rev. 2014, 33, 891–900. [Google Scholar] [CrossRef]
- Moon, W.S.; Chang, K.; Tarnawski, A.S. Overexpression of metastatic tumor antigen 1 in hepatocellular carcinoma: Relationship to vascular invasion and estrogen receptor-alpha. Hum. Pathol. 2004, 35, 424–429. [Google Scholar] [CrossRef]
- Li, D.Q.; Kumar, R. Unravelling the Complexity and Functions of MTA Coregulators in Human Cancer. Adv. Cancer Res. 2015, 127, 1–47. [Google Scholar] [CrossRef] [PubMed]
- Manavathi, B.; Kumar, R. Metastasis tumor antigens, an emerging family of multifaceted master coregulators. J. Biol. Chem. 2007, 282, 1529–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aasland, R.; Stewart, A.F.; Gibson, T. The SANT domain: A putative DNA-binding domain in the SWI-SNF and ADA complexes, the transcriptional co-repressor N-CoR and TFIIIB. Trends Biochem. Sci. 1996, 21, 87–88. [Google Scholar] [CrossRef]
- Sterner, D.E.; Wang, X.; Bloom, M.H.; Simon, G.M.; Berger, S.L. The SANT domain of Ada2 is required for normal acetylation of histones by the yeast SAGA complex. J. Biol. Chem. 2002, 277, 8178–8186. [Google Scholar] [CrossRef] [Green Version]
- Roche, A.E.; Bassett, B.J.; Samant, S.A.; Hong, W.; Blobel, G.A.; Svensson, E.C. The zinc finger and C-terminal domains of MTA proteins are required for FOG-2-mediated transcriptional repression via the NuRD complex. J. Mol. Cell Cardiol. 2008, 44, 352–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toh, Y.; Pencil, S.D.; Nicolson, G.L. A novel candidate metastasis-associated gene, mta1, differentially expressed in highly metastatic mammary adenocarcinoma cell lines. cDNA cloning, expression, and protein analyses. J. Biol. Chem. 1994, 269, 22958–22963. [Google Scholar] [CrossRef]
- Singh, R.R.; Kumar, R. MTA family of transcriptional metaregulators in mammary gland morphogenesis and breast cancer. J. Mammary Gland Biol. Neoplasia 2007, 12, 115–125. [Google Scholar] [CrossRef]
- Toh, Y.; Nicolson, G.L. Identification and characterization of metastasis-associated gene/protein 1 (MTA1). Cancer Metast. Rev. 2014, 33, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Suresh, M.; Menne, S. Application of the woodchuck animal model for the treatment of hepatitis B virus-induced liver cancer. World J. Gastrointest. Oncol. 2021, 13, 509–535. [Google Scholar] [CrossRef]
- Sen, N.; Gui, B.; Kumar, R. Role of MTA1 in cancer progression and metastasis. Cancer Metastasis Rev. 2014, 33, 879–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Han, Q.; Zhao, H.; Zhang, J. The Mechanisms of HBV-Induced Hepatocellular Carcinoma. J. Hepatocell. Carcinoma 2021, 8, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Hamatsu, T.; Rikimaru, T.; Yamashita, Y.; Aishima, S.; Tanaka, S.; Shirabe, K.; Shimada, M.; Toh, Y.; Sugimachi, K. The role of MTA1 gene expression in human hepatocellular carcinoma. Oncol. Rep. 2003, 10, 599–604. [Google Scholar]
- Jin, Y.J.; Chung, Y.H.; Kim, J.A.; Park, W.H.; Lee, D.; Seo, D.D.; Ryu, S.H.; Jang, M.K.; Yu, E.; Lee, Y.J. Factors predisposing metastatic tumor antigen 1 overexpression in hepatitis B virus associated hepatocellular carcinoma. Dig. Dis. Sci. 2012, 57, 2917–2923. [Google Scholar] [CrossRef]
- Zhang, X.Y.; DeSalle, L.M.; Patel, J.H.; Capobianco, A.J.; Yu, D.N.; Thomas-Tikhonenko, A.; McMahon, S.B. Metastasis-associated protein 1 (MTA1) is an essential downstream effector of the c-MYC oncoprotein. Proc. Natl. Acad. Sci. USA 2005, 102, 13968–13973. [Google Scholar] [CrossRef] [Green Version]
- Pakala, S.B.; Bui-Nguyen, T.M.; Reddy, S.D.; Li, D.Q.; Peng, S.; Rayala, S.K.; Behringer, R.R.; Kumar, R. Regulation of NF-kappaB circuitry by a component of the nucleosome remodeling and deacetylase complex controls inflammatory response homeostasis. J. Biol. Chem. 2010, 285, 23590–23597. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Liu, J.; Xue, H.; Li, C.; Liu, Q.; Zhou, Y.; Wang, T.; Wang, H.; Qian, H.; Wen, T. A TGF-beta-MTA1-SOX4-EZH2 signaling axis drives epithelial-mesenchymal transition in tumor metastasis. Oncogene 2020, 39, 2125–2139. [Google Scholar] [CrossRef]
- Pakala, S.B.; Singh, K.; Reddy, S.D.; Ohshiro, K.; Li, D.Q.; Mishra, L.; Kumar, R. TGF-beta1 signaling targets metastasis-associated protein 1, a new effector in epithelial cells. Oncogene 2011, 30, 2230–2241. [Google Scholar] [CrossRef] [Green Version]
- Yoo, Y.G.; Na, T.Y.; Seo, H.W.; Seong, J.K.; Park, C.K.; Shin, Y.K.; Lee, M.O. Hepatitis B virus X protein induces the expression of MTA1 and HDAC1, which enhances hypoxia signaling in hepatocellular carcinoma cells. Oncogene 2008, 27, 3405–3413. [Google Scholar] [CrossRef] [Green Version]
- Yoo, Y.G.; Kong, G.; Lee, M.O. Metastasis-associated protein 1 enhances stability of hypoxia-inducible factor-1alpha protein by recruiting histone deacetylase 1. EMBO J. 2006, 25, 1231–1241. [Google Scholar] [CrossRef] [Green Version]
- Friedmann Angeli, J.P.; Krysko, D.V.; Conrad, M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Cancer 2019, 19, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Konieczkowski, D.J.; Johannessen, C.M.; Garraway, L.A. A Convergence-Based Framework for Cancer Drug Resistance. Cancer Cell 2018, 33, 801–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.H.; Koh, D.; Na, H.; Ka, N.L.; Kim, S.; Kim, H.J.; Hong, S.; Shin, Y.K.; Seong, J.K.; Lee, M.O. MTA1 is a novel regulator of autophagy that induces tamoxifen resistance in breast cancer cells. Autophagy 2018, 14, 812–824. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Lee, M.H.; Kang, H.L.; Kim, S.H.; Ahn, J.R.; Na, H.; Na, T.Y.; Kim, Y.N.; Seong, J.K.; Lee, M.O. Differential regulation of estrogen receptor alpha expression in breast cancer cells by metastasis-associated protein 1. Cancer Res. 2014, 74, 1484–1494. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Hu, Y.; Chen, B.; Li, D.; Liang, R.; Shen, M.; Wu, M.; Tao, M. Metastasis-associated gene 1 (MTA1) enhances cisplatin resistance of malignant pleural mesothelioma by ATR-Chk1-mediated DNA repair. Ann. Transl. Med. 2021, 9, 670. [Google Scholar] [CrossRef]
- Li, P.; Cao, G.; Huang, Y.; Wu, W.; Chen, B.; Wang, Z.; Xiong, M. siMTA1-Loaded Exosomes Enhanced Chemotherapeutic Effect of Gemcitabine in Luminal-b Type Breast Cancer by Inhibition of EMT/HIF-alpha and Autophagy Pathways. Front. Oncol. 2020, 10, 541262. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Wu, Q.; Zhang, K.; Liu, Y.; Zhang, N.; Chen, Q.; Wang, L.; Li, W.; Zhang, J.; Liu, Y. O-GlcNAc modification regulates MTA1 transcriptional activity during breast cancer cell genotoxic adaptation. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129930. [Google Scholar] [CrossRef] [PubMed]
- Goody, D.; Gupta, S.K.; Engelmann, D.; Spitschak, A.; Marquardt, S.; Mikkat, S.; Meier, C.; Hauser, C.; Gundlach, J.P.; Egberts, J.H.; et al. Drug Repositioning Inferred from E2F1-Coregulator Interactions Studies for the Prevention and Treatment of Metastatic Cancers. Theranostics 2019, 9, 1490–1509. [Google Scholar] [CrossRef]
- Gadkari, K.; Kolhatkar, U.; Hemani, R.; Campanelli, G.; Cai, Q.; Kumar, A.; Levenson, A.S. Therapeutic Potential of Gnetin C in Prostate Cancer: A Pre-Clinical Study. Nutrients 2020, 12, 3631. [Google Scholar] [CrossRef]
- Banik, A.; Ahmed, S.R.; Sajib, E.H.; Deb, A.; Sinha, S.; Azim, K. Identification of potential inhibitory analogs of metastasis tumor antigens (MTAs) using bioactive compounds: Revealing therapeutic option to prevent malignancy. bioRxiv 2020. [Google Scholar] [CrossRef]
- Qian, Y.Y.; Liu, Z.S.; Yan, H.J.; Yuan, Y.F.; Levenson, A.S.; Li, K. Pterostilbene inhibits MTA1/HDAC1 complex leading to PTEN acetylation in hepatocellular carcinoma. Biomed. Pharmacother. 2018, 101, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.Y.; Liu, Z.S.; Pan, D.Y.; Li, K. Tumoricidal activities of pterostilbene depend upon destabilizing the MTA1-NuRD complex and enhancing P53 acetylation in hepatocellular carcinoma. Exp. Ther. Med. 2017, 14, 3098–3104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhar, S.; Kumar, A.; Zhang, L.; Rimando, A.M.; Lage, J.M.; Lewin, J.R.; Atfi, A.; Zhang, X.; Levenson, A.S. Dietary pterostilbene is a novel MTA1-targeted chemopreventive and therapeutic agent in prostate cancer. Oncotarget 2016, 7, 18469–18484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, H.; Sotiropoulos, G.C.; Brokalaki, E.I.; Schmitz, K.J.; Bertona, C.; Meyer, G.; Frilling, A.; Paul, A.; Malago, M.; Broelsch, C.E. Survival and recurrence rates after resection for hepatocellular carcinoma in noncirrhotic livers. J. Am. Coll. Surg. 2007, 205, 27–36. [Google Scholar] [CrossRef]
- Sempokuya, T.; Wong, L.L. Ten-year survival and recurrence of hepatocellular cancer. Hepatoma Res. 2019, 5, 38. [Google Scholar] [CrossRef]
- Moon, H.; Ju, H.L.; Chung, S.I.; Cho, K.J.; Eun, J.W.; Nam, S.W.; Han, K.H.; Calvisi, D.F.; Ro, S.W. Transforming Growth Factor-beta Promotes Liver Tumorigenesis in Mice via Up-regulation of Snail. Gastroenterology 2017, 153, 1378–1391.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | HCC Development | Advantages | Disadvantages | Features |
|---|---|---|---|---|
| Conventional HBV transgenic mice | HBV Tg: not observed | No genetic variation in established mouse line | Costly, laborious, and time-consuming | Inbred |
| HBx Tg: within 18 mo., but not every line developed HCC | Technically challenging | HBx Tg: no inflammation | ||
| LHBs/pre-S mut. Tg: within 18 mo. | Resource-demanding | LHBs/pre-S mut. Tg: inflammation | ||
| Viral vector-mediated transgenic mice | Varied (depending on the properties of the transgene), e.g., rAAV/HBV Tg: 12–16 mo. | Easy and simple procedure | Potential biohazards | Inbred rAAV/HBV Tg: occasionally displayed mild to moderate liver fibrosis |
| Hydrodynamics-based transfection (HDT) | Sleeping Beauty: varied (depending on the properties of the transgenes) | Easy and simple procedure Flexibility in gene delivery Combination of various genetic modifications possible | Random integration of transgene Genetic variation within a model | Inbred Different HCC latency periods were observed among distinct mouse strains |
| CRISPR/Cas9: varied | Easy and simple procedure Combination of various genetic modifications possible | Possible genetic variation (e.g., off-target effect) | Inbred Modification of endogenous genes (e.g., tumor suppressor) | |
| Tree shrew HCC model | Within 5–6 years after an HBV infection in the background of chronic viral infection | Only non-primates susceptible to HBV A greater propensity of progressing to chronic hepatitis when infected as neonates | Overall low viral titer and persistent rate Long period of time to develop the tumors Resource-demanding Lack of many research tools and reagents | Outbred Similar pathological changes in the liver as those in humans |
| Woodchuck HCC Model | Within 1–4 years after a WHV infection in the background of chronic viral infection | Naturally or experimentally infected by WHV Longitudinal monitoring possible (e.g., CT, MRI, US, etc.) Serial sample collection available (e.g., serum, liver biopsy, etc.) | Costly Research tools and reagents are limited | Outbred Comparable to human HBV-HCCs in tumor size, morphology, pathological changes, and molecular gene signature |
| Humanized mice (human hepatocytes and/or immune cells engraftment) | HCC not found HBV-associated liver pathology was observed (e.g., chronic hepatitis, fibrosis, and cirrhosis) | Supporting HBV infection and replication Capable of engrafting clinical specimens and genetically modified liver cells Functional human immune system | High costs Technically challenging Resource-demanding | Inbred Similar to CHB patients in respect of high viremia, inflammatory and immune responses |
| Properties | MTA1 | MTA1dE4 | ||
|---|---|---|---|---|
| Structural insight | Protein size (amino acids) | 715 | 698 | |
| Structure of BAH domain | Long disordered region within the BAH domain (Without regular secondary structure) | Deletion at the disordered region may cause conformational changes of BAH domain of MTA1dE4 * | ||
| Molecular insight | Protein-Protein interaction | With common and unique interacting partners to that of MTA1dE4 * | Interacting affinity and partners may alter due to Conformational changes * | |
| Effects on the aggressiveness of tumor cells | Migration | Promotion | Promotion (Higher activity) | |
| Invasion | Promotion | Promotion (Higher activity) | ||
| Clinical insight | Correlation with aggressive tumor characteristics (e.g., large tumor size and large number of tumors, etc.) | Yes [32,145] | Yes (Stronger) [32] | |
| Correlation with early HCC recurrence | Yes [32] (Studied at mRNA level) | Yes [32] (Studied at mRNA level) (A more sensitive marker) | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.-T.; Wu, H.-L.; Liu, C.-J. Molecular Mechanisms and Animal Models of HBV-Related Hepatocellular Carcinoma: With Emphasis on Metastatic Tumor Antigen 1. Int. J. Mol. Sci. 2021, 22, 9380. https://doi.org/10.3390/ijms22179380
Li Y-T, Wu H-L, Liu C-J. Molecular Mechanisms and Animal Models of HBV-Related Hepatocellular Carcinoma: With Emphasis on Metastatic Tumor Antigen 1. International Journal of Molecular Sciences. 2021; 22(17):9380. https://doi.org/10.3390/ijms22179380
Chicago/Turabian StyleLi, Yung-Tsung, Hui-Lin Wu, and Chun-Jen Liu. 2021. "Molecular Mechanisms and Animal Models of HBV-Related Hepatocellular Carcinoma: With Emphasis on Metastatic Tumor Antigen 1" International Journal of Molecular Sciences 22, no. 17: 9380. https://doi.org/10.3390/ijms22179380
APA StyleLi, Y. -T., Wu, H. -L., & Liu, C. -J. (2021). Molecular Mechanisms and Animal Models of HBV-Related Hepatocellular Carcinoma: With Emphasis on Metastatic Tumor Antigen 1. International Journal of Molecular Sciences, 22(17), 9380. https://doi.org/10.3390/ijms22179380