
Rutaecarpine Increases Nitric Oxide Synthesis via eNOS Phosphorylation by TRPV1-Dependent CaMKII and CaMKKβ/AMPK Signaling Pathway in Human Endothelial Cells

 ,
,  and
and 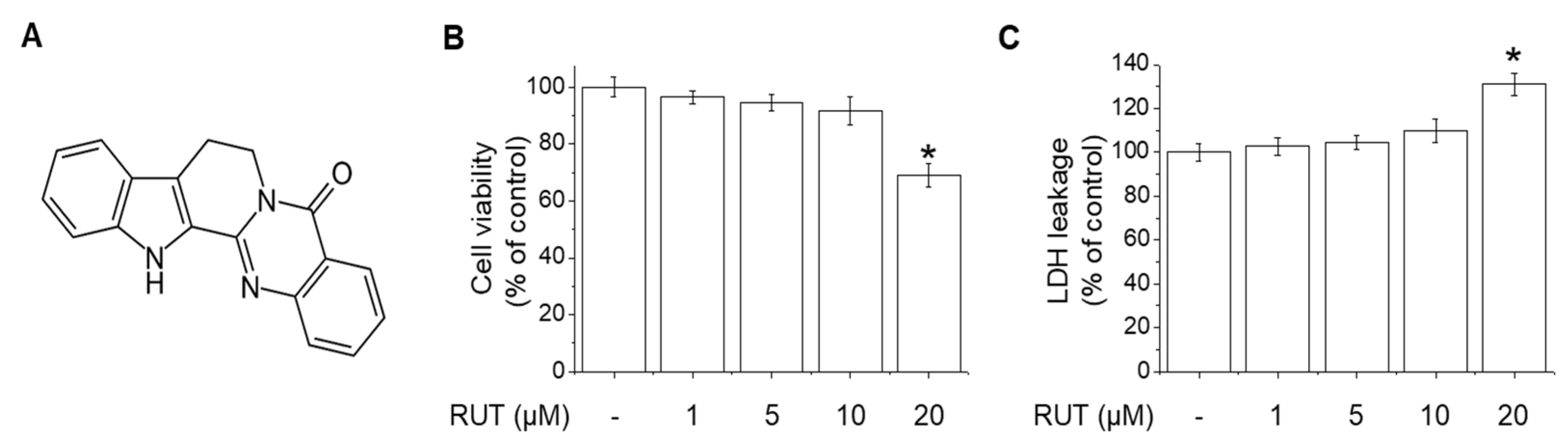{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Effects of RUT on the Cell Viability and Cytotoxicity in Endothelial Cells
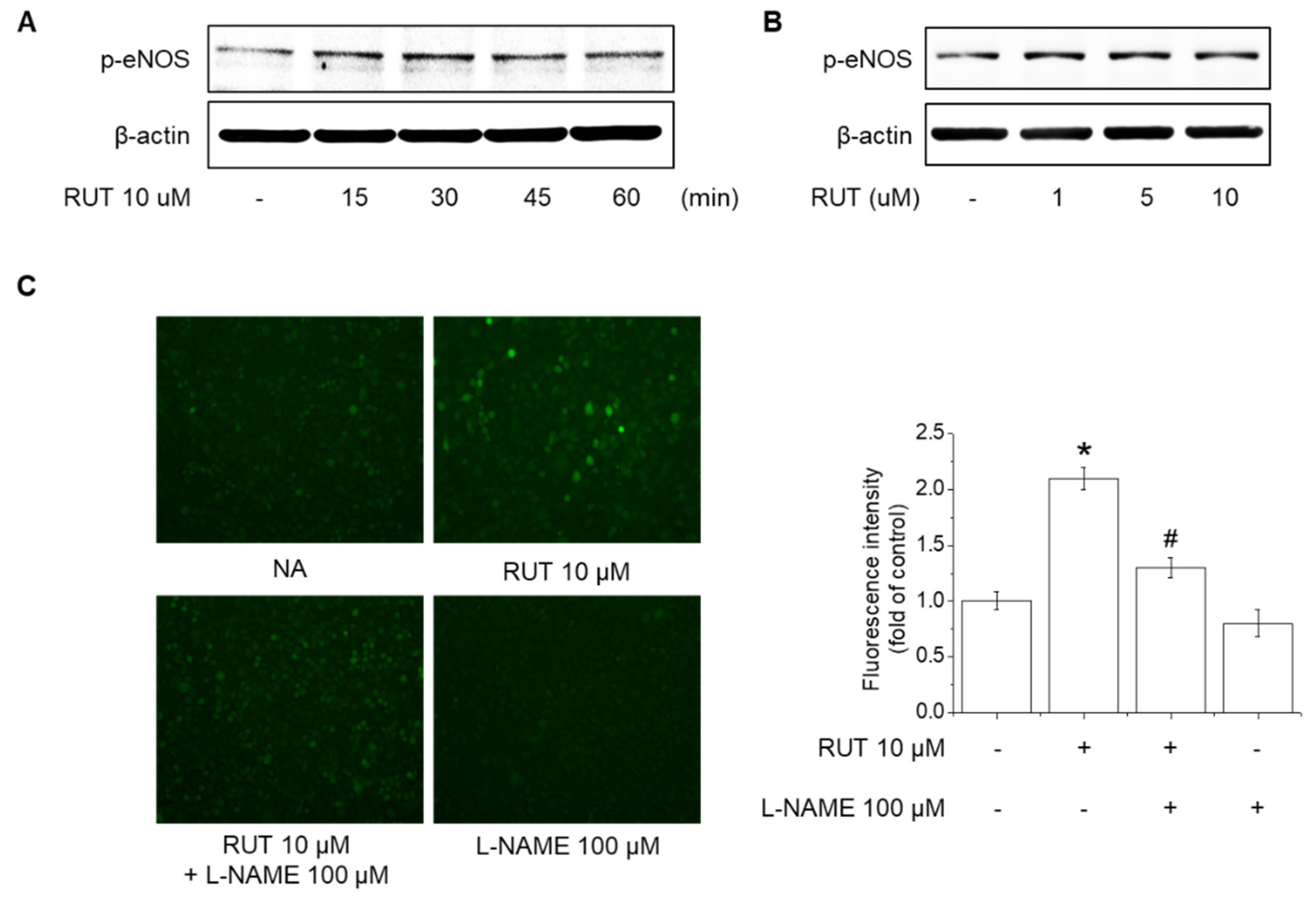
2.2. Effects of RUT on eNOS Phosphorylation and NO Generation
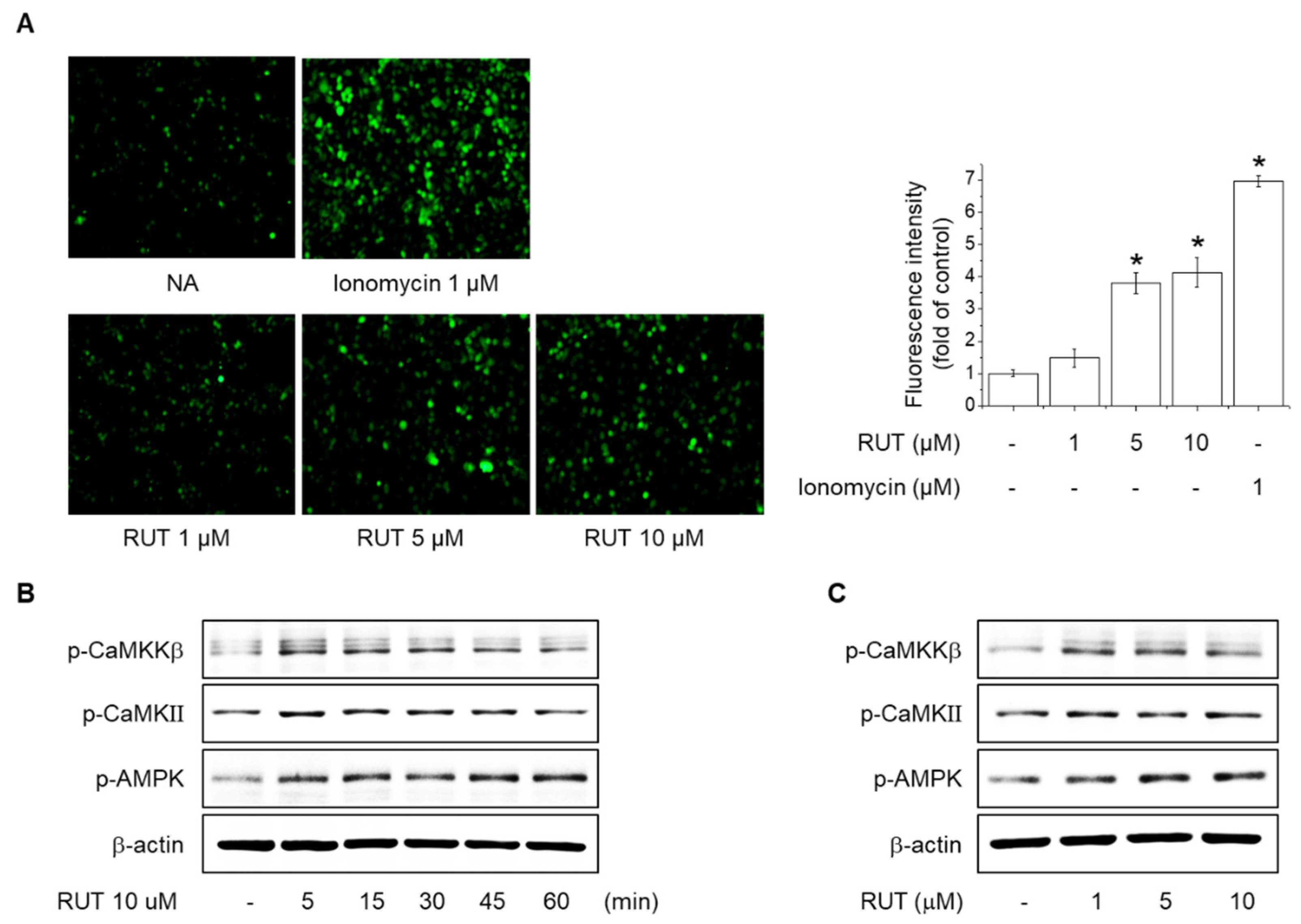
2.3. Effects of RUT on the Intracellular Ca2+ Levels and Phosphorylation of CaMKII, CaMKKβ and AMPK
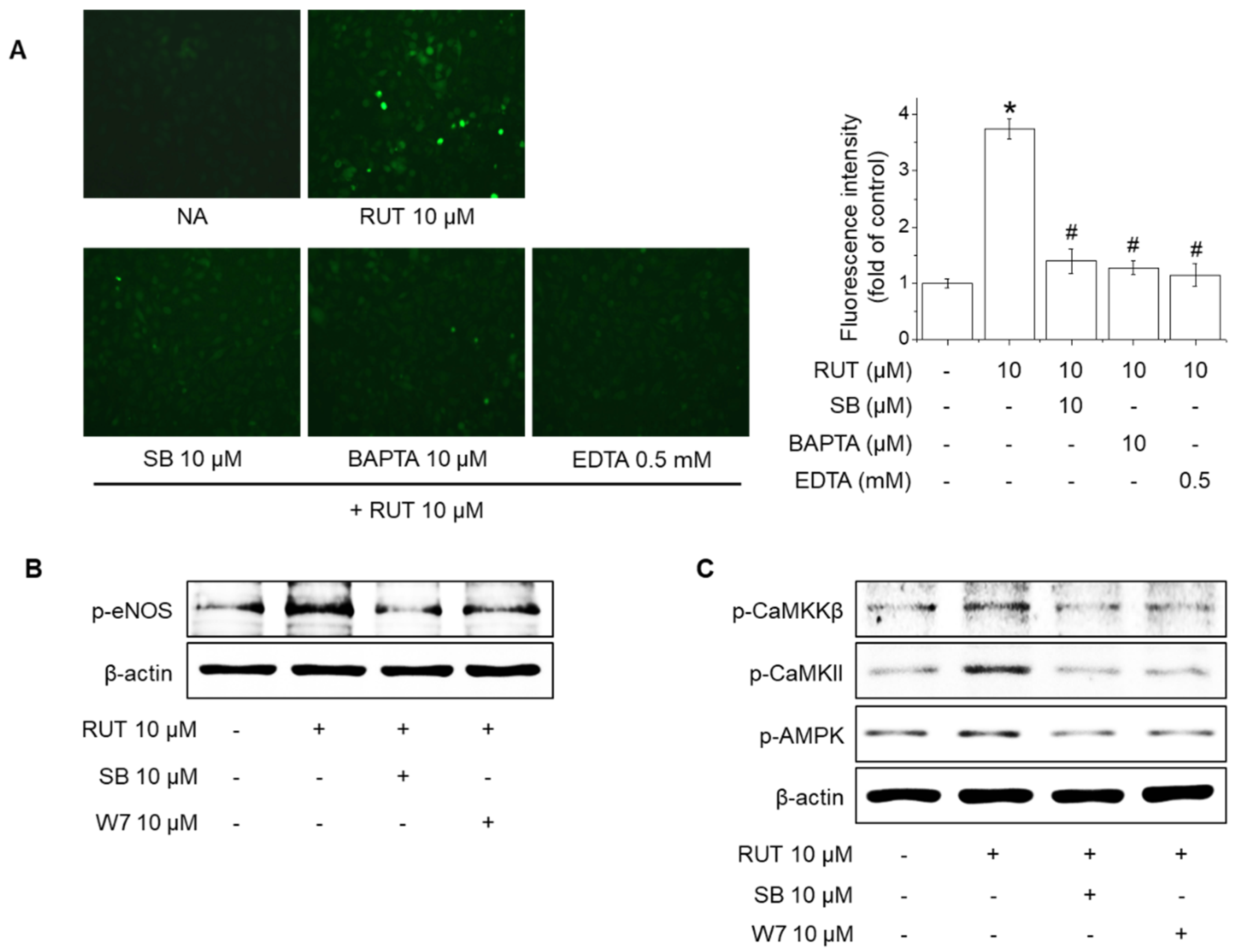
2.4. Effects of TRPV1 Blockade on Intracellular Ca2+ Levels and Phosphorylation of CaMKKβ, CaMKII, AMPK and eNOS in Response to RUT
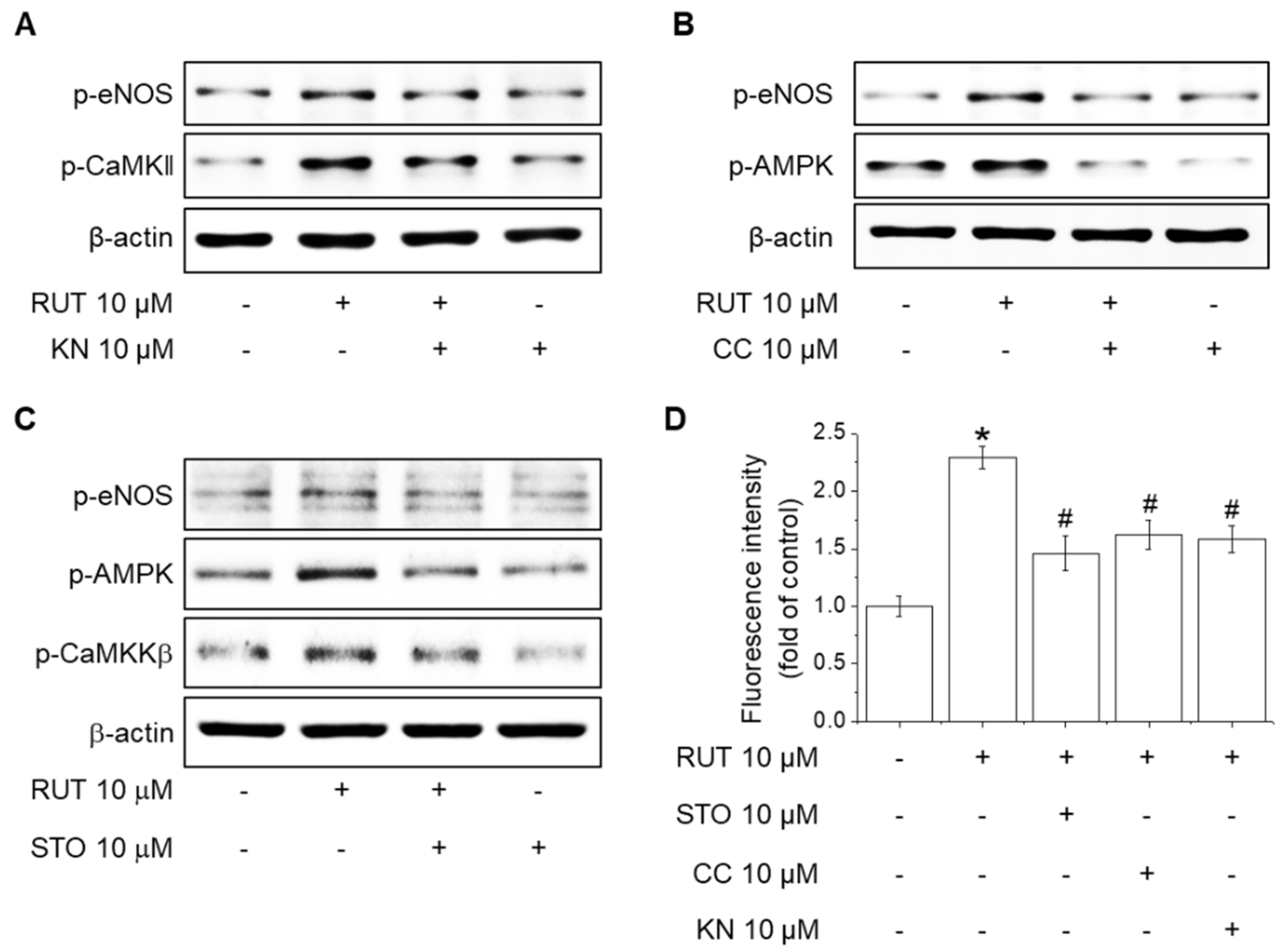
2.5. Roles of CaMKII and the CaMKKβ/AMPK Signaling Pathway in RUT-Induced eNOS Activation and NO Generation
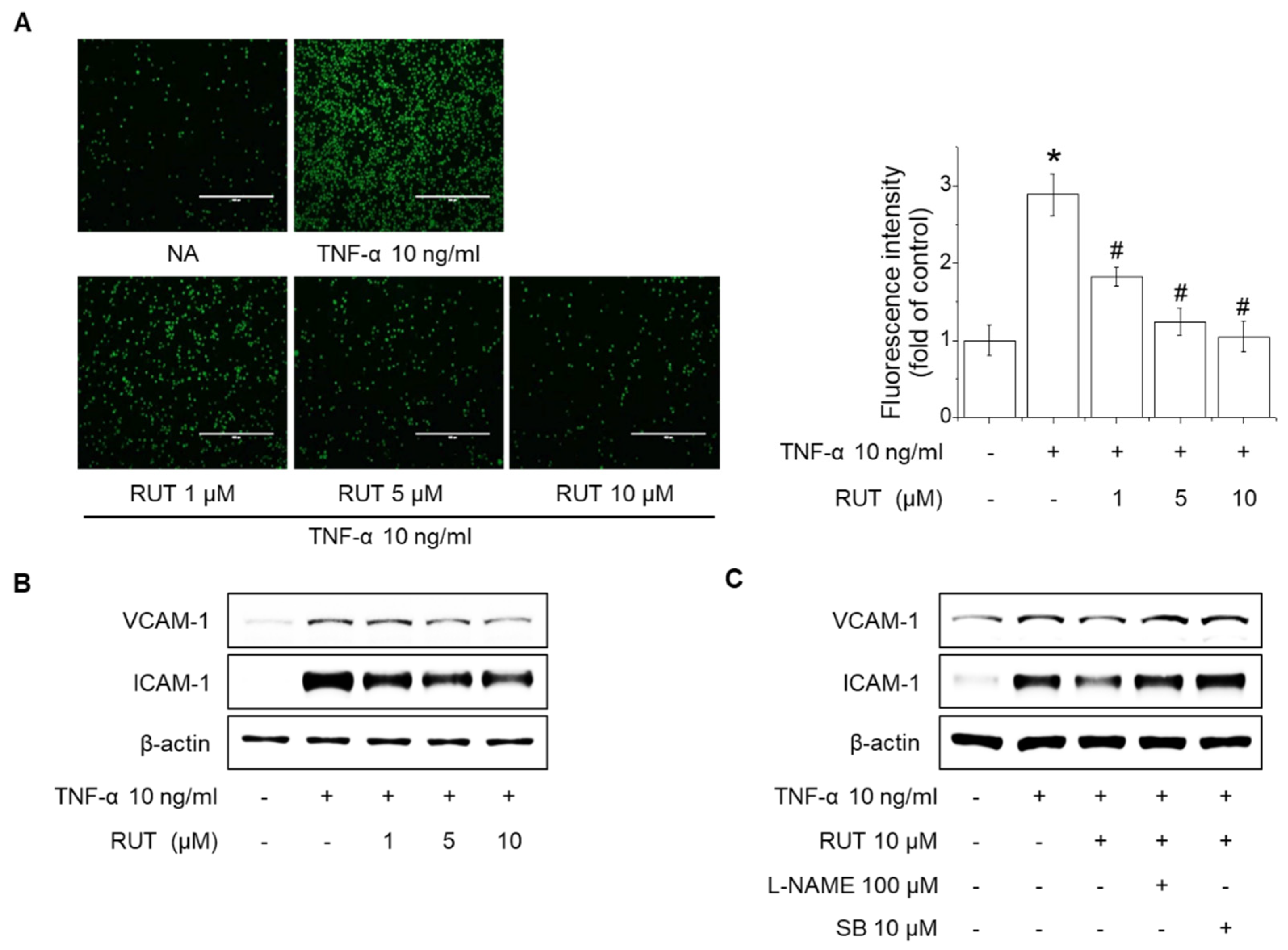
2.6. Effects of RUT on the Suppression of Monocyte Adhesion and Expression of Adhesion Molecules in Endothelial Cells
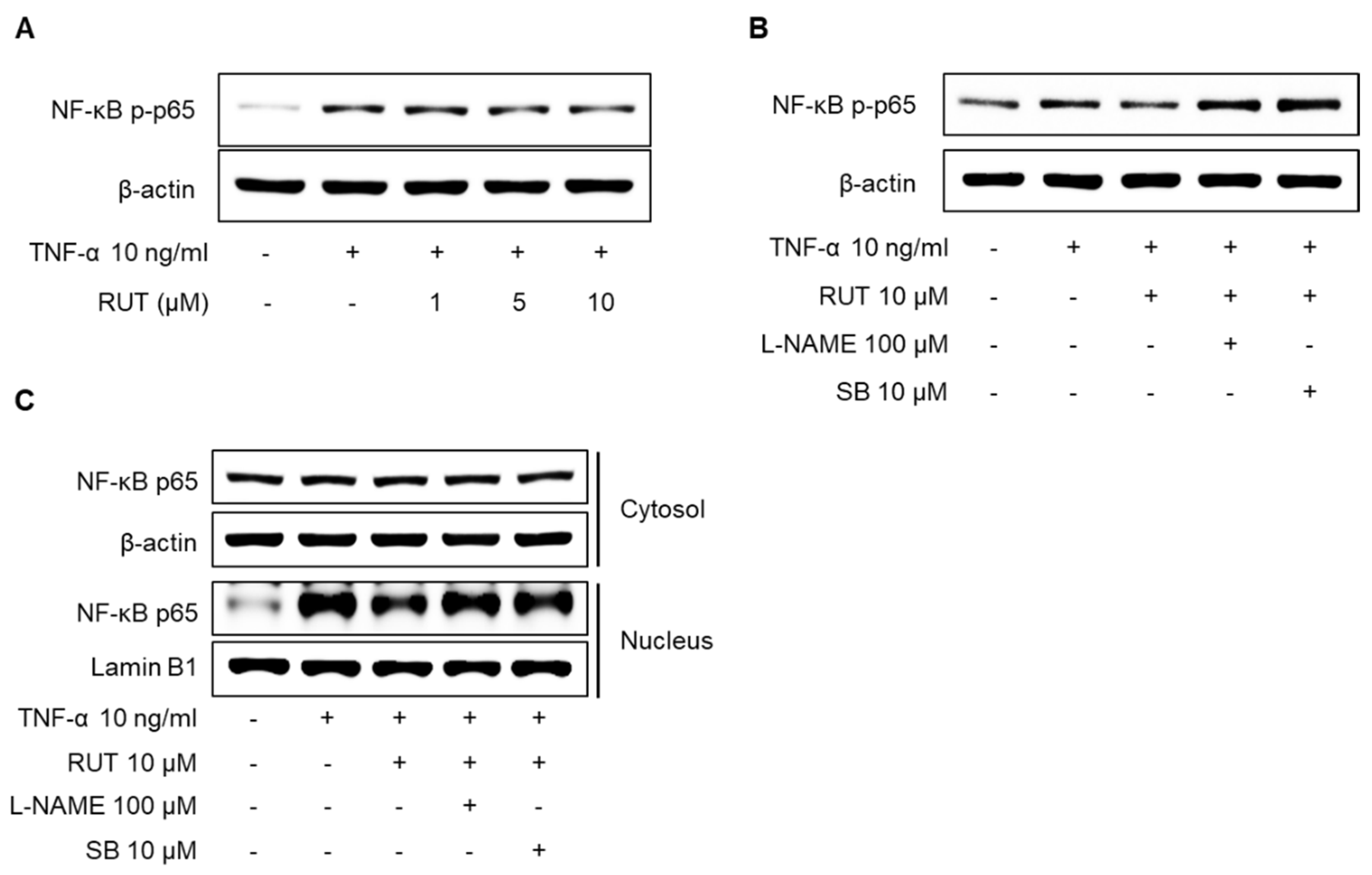
2.7. Effects of RUT on the Inhibition of NF-κB p65 Signaling Pathway Induced by TNF-α in Endothelial Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatment
4.2. MTT and LDH Assays
4.3. Western Blotting Analysis
4.4. Measurement of NO Production
4.5. Ca2+ Measurement
4.6. Monocyte Adhesion
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AMPK | AMP-activated protein kinase |
| BAPTA | BAPTA-AM |
| CaM | Calmodulin |
| CC | Compound C |
| CaMKII | Ca2+/calmodulin-dependent kinase II |
| CaMKKβ | Ca2+/calmodulin-dependent protein kinase kinase β |
| eNOS | Endothelial nitric oxide synthase |
| ICAM-1 | Intercellular adhesion molecule-1 |
| KN | KN-62 |
| L-NAME | L-NG-Nitro arginine methyl ester |
| NF-κB | Nuclear factor-κB |
| NO | Nitric oxide |
| RUT | Rutaecarpine |
| SB | SB366791 |
| TNF-α | Tumor necrosis factor-α |
| TRP | Transient receptor potential |
| TRPV1 | Transient receptor potential vanilloid type 1 |
| VCAM-1 | Vascular cell adhesion molecule-1 |
References
- Zeiher, A.M.; Drexler, H.; Wollschlager, H.; Just, H. Endothelial dysfunction of the coronary microvasculature is associated with coronary blood flow regulation in patients with early atherosclerosis. Circulation 1991, 84, 1984–1992. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Forstermann, U. Nitric oxide in the pathogenesis of vascular disease. J. Pathol. 2000, 190, 244–254. [Google Scholar] [CrossRef]
- Zhao, Y.; Vanhoutte, P.M.; Leung, S.W. Vascular nitric oxide: Beyond eNOS. J. Pharmacol. Sci. 2015, 129, 83–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.A.; Druhan, L.J.; Varadharaj, S.; Chen, Y.R.; Zweier, J.L. Phosphorylation of endothelial nitric-oxide synthase regulates superoxide generation from the enzyme. J. Biol. Chem. 2008, 283, 27038–27047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, J.C.; El Kebir, D.; Chereau, C.; Lanone, S.; Huang, X.L.; De Buys Roessingh, A.S.; Mercier, J.C.; Dall’Ava-Santucci, J.; Dinh-Xuan, A.T. Involvement of Ca2+/calmodulin-dependent protein kinase II in endothelial NO production and endothelium-dependent relaxation. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H2311–H2319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrow, V.A.; Foufelle, F.; Connell, J.M.; Petrie, J.R.; Gould, G.W.; Salt, I.P. Direct activation of AMP-activated protein kinase stimulates nitric-oxide synthesis in human aortic endothelial cells. J. Biol. Chem. 2003, 278, 31629–31639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, L.; Li, N.; He, Z.; Zeng, X.; Nan, Y.; Chen, J.; Miao, P.; Ying, Y.; Lin, W.; Zhao, X.; et al. Fibroblast growth factor 21 Ameliorates diabetes-induced endothelial dysfunction in mouse aorta via activation of the CaMKK2/AMPKalpha signaling pathway. Cell Death Dis. 2019, 10, 665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busse, R.; Mulsch, A. Calcium-dependent nitric oxide synthesis in endothelial cytosol is mediated by calmodulin. FEBS Lett. 1990, 265, 133–136. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.W.; Choi, C.Y.; Hwang, Y.P.; Kim, H.G.; Kim, S.J.; Chung, Y.C.; Lee, K.J.; Jeong, T.C.; Jeong, H.G. Betulinic Acid Increases eNOS Phosphorylation and NO Synthesis via the Calcium-Signaling Pathway. J. Agric. Food Chem. 2016, 64, 785–791. [Google Scholar] [CrossRef]
- Tominaga, M.; Tominaga, T. Structure and function of TRPV1. Pflügers Arch. 2005, 451, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cui, L.; Xu, H.; Liu, S.; Zhu, F.; Yan, F.; Shen, S.; Zhu, M. TRPV1 agonism inhibits endothelial cell inflammation via activation of eNOS/NO pathway. Atherosclerosis 2017, 260, 13–19. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Rosti, V.; Antognazza, M.R.; Lodola, F.; Moccia, F. Endothelial TRPV1 as an Emerging Molecular Target to Promote Therapeutic Angiogenesis. Cells 2020, 9, 1341. [Google Scholar] [CrossRef]
- Inoue, R.; Jensen, L.J.; Shi, J.; Morita, H.; Nishida, M.; Honda, A.; Ito, Y. Transient receptor potential channels in cardiovascular function and disease. Circ. Res. 2006, 99, 119–131. [Google Scholar] [CrossRef] [Green Version]
- Jia, S.; Hu, C. Pharmacological effects of rutaecarpine as a cardiovascular protective agent. Molecules 2010, 15, 1873–1881. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.H.; Jin, S.W.; Lee, G.H.; Han, E.H.; Hwang, Y.P.; Jeong, H.G. Rutaecarpine Protects against Acetaminophen-Induced Acute Liver Injury in Mice by Activating Antioxidant Enzymes. Antioxidants 2021, 10, 86. [Google Scholar] [CrossRef]
- Jin, S.W.; Hwang, Y.P.; Choi, C.Y.; Kim, H.G.; Kim, S.J.; Kim, Y.; Chung, Y.C.; Lee, K.J.; Jeong, T.C.; Jeong, H.G. Protective effect of rutaecarpine against t-BHP-induced hepatotoxicity by upregulating antioxidant enzymes via the CaMKII-Akt and Nrf2/ARE pathways. Food Chem. Toxicol. 2017, 100, 138–148. [Google Scholar] [CrossRef]
- Li, Y.J.; Zhang, F.; Gong, Q.H.; Wu, Q.; Yu, L.M.; Sun, A.S. Rutaecarpine inhibits angiotensin II-induced proliferation in rat vascular smooth muscle cells. Chin. J. Integr. Med. 2014, 20, 682–687. [Google Scholar] [CrossRef]
- Xu, Y.; Chen, X.P.; Zhang, F.; Hou, H.H.; Zhang, J.Y.; Lin, S.X.; Sun, A.S. Rutaecarpine Inhibits Intimal Hyperplasia in A Balloon-Injured Rat Artery Model. Chin. J. Integr. Med. 2018, 24, 429–435. [Google Scholar] [CrossRef]
- Jin, S.W.; Pham, H.T.; Choi, J.H.; Lee, G.H.; Han, E.H.; Cho, Y.H.; Chung, Y.C.; Kim, Y.H.; Jeong, H.G. Impressic Acid, a Lupane-Type Triterpenoid from Acanthopanax koreanum, Attenuates TNF-alpha-Induced Endothelial Dysfunction via Activation of eNOS/NO Pathway. Int. J. Mol. Sci. 2019, 20, 5772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, K.H.; Lin, S.J.; Wei, J.; Lee, K.I.; Zhao, J.F.; Shyue, S.K.; Lee, T.S. The essential role of transient receptor potential vanilloid 1 in simvastatin-induced activation of endothelial nitric oxide synthase and angiogenesis. Acta Physiol. 2014, 212, 191–204. [Google Scholar] [CrossRef]
- Du, L.L.; Shen, Z.; Li, Z.; Ye, X.; Wu, M.; Hong, L.; Zhao, Y. TRPC1 Deficiency Impairs the Endothelial Progenitor Cell Function via Inhibition of Calmodulin/eNOS Pathway. J. Cardiovasc. Transl. Res. 2018, 11, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.P.; Kim, H.G.; Hien, T.T.; Jeong, M.H.; Jeong, T.C.; Jeong, H.G. Puerarin activates endothelial nitric oxide synthase through estrogen receptor-dependent PI3-kinase and calcium-dependent AMP-activated protein kinase. Toxicol. Appl. Pharmacol. 2011, 257, 48–58. [Google Scholar] [CrossRef]
- Wu, Y.W.; Chang, T.T.; Chang, C.C.; Chen, J.W. Fatty-Acid-Binding Protein 4 as a Novel Contributor to Mononuclear Cell Activation and Endothelial Cell Dysfunction in Atherosclerosis. Int. J. Mol. Sci. 2020, 21, 9245. [Google Scholar] [CrossRef]
- Sun, S.; Ji, Z.; Fu, J.; Wang, X.F.; Zhang, L.S. Endosulfan induces endothelial inflammation and dysfunction via IRE1alpha/NF-kappaB signaling pathway. Environ. Sci. Pollut. Res. Int. 2020, 27, 26163–26171. [Google Scholar] [CrossRef]
- Cheng, L.C.; Guo, B.C.; Chen, C.H.; Chang, C.J.; Yeh, T.S.; Lee, T.S. Endothelial Nitric Oxide Mediates the Anti-Atherosclerotic Action of Torenia concolor Lindley var. Formosama Yamazaki. Int. J. Mol. Sci. 2020, 21, 1532. [Google Scholar] [CrossRef] [Green Version]
- Scioli, M.G.; Storti, G.; D’Amico, F.; Rodriguez Guzman, R.; Centofanti, F.; Doldo, E.; Cespedes Miranda, E.M.; Orlandi, A. Oxidative Stress and New Pathogenetic Mechanisms in Endothelial Dysfunction: Potential Diagnostic Biomarkers and Therapeutic Targets. J. Clin. Med. 2020, 9, 1995. [Google Scholar] [CrossRef]
- Sandoo, A.; van Zanten, J.J.; Metsios, G.S.; Carroll, D.; Kitas, G.D. The endothelium and its role in regulating vascular tone. Open Cardiovasc. Med. J. 2010, 4, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.K. Regulation of endothelial nitric oxide synthase activity and gene expression. Ann. N. Y. Acad. Sci. 2002, 962, 122–130. [Google Scholar] [CrossRef]
- Koo, B.H.; Hwang, H.M.; Yi, B.G.; Lim, H.K.; Jeon, B.H.; Hoe, K.L.; Kwon, Y.G.; Won, M.H.; Kim, Y.M.; Berkowitz, D.E.; et al. Arginase II Contributes to the Ca(2+)/CaMKII/eNOS Axis by Regulating Ca(2+) Concentration Between the Cytosol and Mitochondria in a p32-Dependent Manner. J. Am. Heart Assoc. 2018, 7, e009579. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.P.; Mitchelhill, K.I.; Michell, B.J.; Stapleton, D.; Rodriguez-Crespo, I.; Witters, L.A.; Power, D.A.; Ortiz de Montellano, P.R.; Kemp, B.E. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. 1999, 443, 285–289. [Google Scholar] [CrossRef] [Green Version]
- Pham, T.H.; Jin, S.W.; Lee, G.H.; Park, J.S.; Kim, J.Y.; Thai, T.N.; Han, E.H.; Jeong, H.G. Sesamin Induces Endothelial Nitric Oxide Synthase Activation via Transient Receptor Potential Vanilloid Type 1. J. Agric. Food Chem. 2020, 68, 3474–3484. [Google Scholar] [CrossRef]
- Kwan, H.Y.; Huang, Y.; Yao, X. TRP channels in endothelial function and dysfunction. Biochim. Biophys. Acta 2007, 1772, 907–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.M.; Gu, J.A.; Rau, T.G.; Wang, C.; Yen, C.H.; Huang, S.H.; Lin, F.Y.; Lin, C.M.; Huang, S.T. Synthetic Fluororutaecarpine Inhibits Inflammatory Stimuli and Activates Endothelial Transient Receptor Potential Vanilloid-Type 1. Molecules 2017, 22, 656. [Google Scholar] [CrossRef] [Green Version]
- Cook-Mills, J.M.; Deem, T.L. Active participation of endothelial cells in inflammation. J. Leukoc. Biol. 2005, 77, 487–495. [Google Scholar] [CrossRef]
- Dorland, Y.L.; Huveneers, S. Cell-cell junctional mechanotransduction in endothelial remodeling. Cell Mol. Life Sci. 2017, 74, 279–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprague, A.H.; Khalil, R.A. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem. Pharmacol. 2009, 78, 539–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bath, P.M.; Hassall, D.G.; Gladwin, A.M.; Palmer, R.M.; Martin, J.F. Nitric oxide and prostacyclin. Divergence of inhibitory effects on monocyte chemotaxis and adhesion to endothelium in vitro. Arterioscler. Thromb. 1991, 11, 254–260. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, G.H.; Kim, C.Y.; Zheng, C.; Jin, S.W.; Kim, J.Y.; Lee, S.Y.; Kim, M.Y.; Han, E.H.; Hwang, Y.P.; Jeong, H.G. Rutaecarpine Increases Nitric Oxide Synthesis via eNOS Phosphorylation by TRPV1-Dependent CaMKII and CaMKKβ/AMPK Signaling Pathway in Human Endothelial Cells. Int. J. Mol. Sci. 2021, 22, 9407. https://doi.org/10.3390/ijms22179407
Lee GH, Kim CY, Zheng C, Jin SW, Kim JY, Lee SY, Kim MY, Han EH, Hwang YP, Jeong HG. Rutaecarpine Increases Nitric Oxide Synthesis via eNOS Phosphorylation by TRPV1-Dependent CaMKII and CaMKKβ/AMPK Signaling Pathway in Human Endothelial Cells. International Journal of Molecular Sciences. 2021; 22(17):9407. https://doi.org/10.3390/ijms22179407
Chicago/Turabian StyleLee, Gi Ho, Chae Yeon Kim, Chuanfeng Zheng, Sun Woo Jin, Ji Yeon Kim, Seung Yeon Lee, Mi Yeon Kim, Eun Hee Han, Yong Pil Hwang, and Hye Gwang Jeong. 2021. "Rutaecarpine Increases Nitric Oxide Synthesis via eNOS Phosphorylation by TRPV1-Dependent CaMKII and CaMKKβ/AMPK Signaling Pathway in Human Endothelial Cells" International Journal of Molecular Sciences 22, no. 17: 9407. https://doi.org/10.3390/ijms22179407
APA StyleLee, G. H., Kim, C. Y., Zheng, C., Jin, S. W., Kim, J. Y., Lee, S. Y., Kim, M. Y., Han, E. H., Hwang, Y. P., & Jeong, H. G. (2021). Rutaecarpine Increases Nitric Oxide Synthesis via eNOS Phosphorylation by TRPV1-Dependent CaMKII and CaMKKβ/AMPK Signaling Pathway in Human Endothelial Cells. International Journal of Molecular Sciences, 22(17), 9407. https://doi.org/10.3390/ijms22179407