
Peptide Derivatives of the Zonulin Inhibitor Larazotide (AT1001) as Potential Anti SARS-CoV-2: Molecular Modelling, Synthesis and Bioactivity Evaluation

 ,
,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
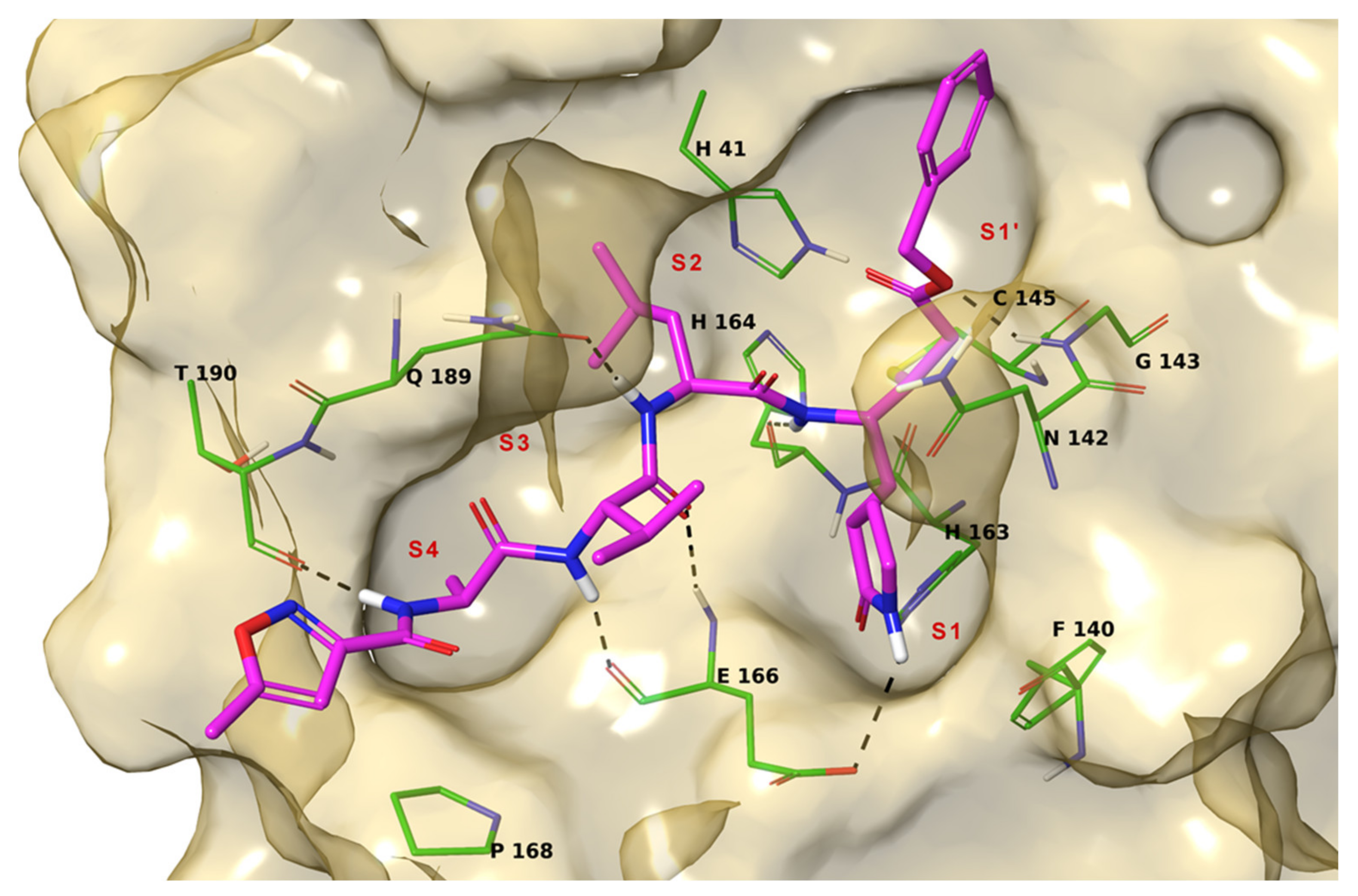
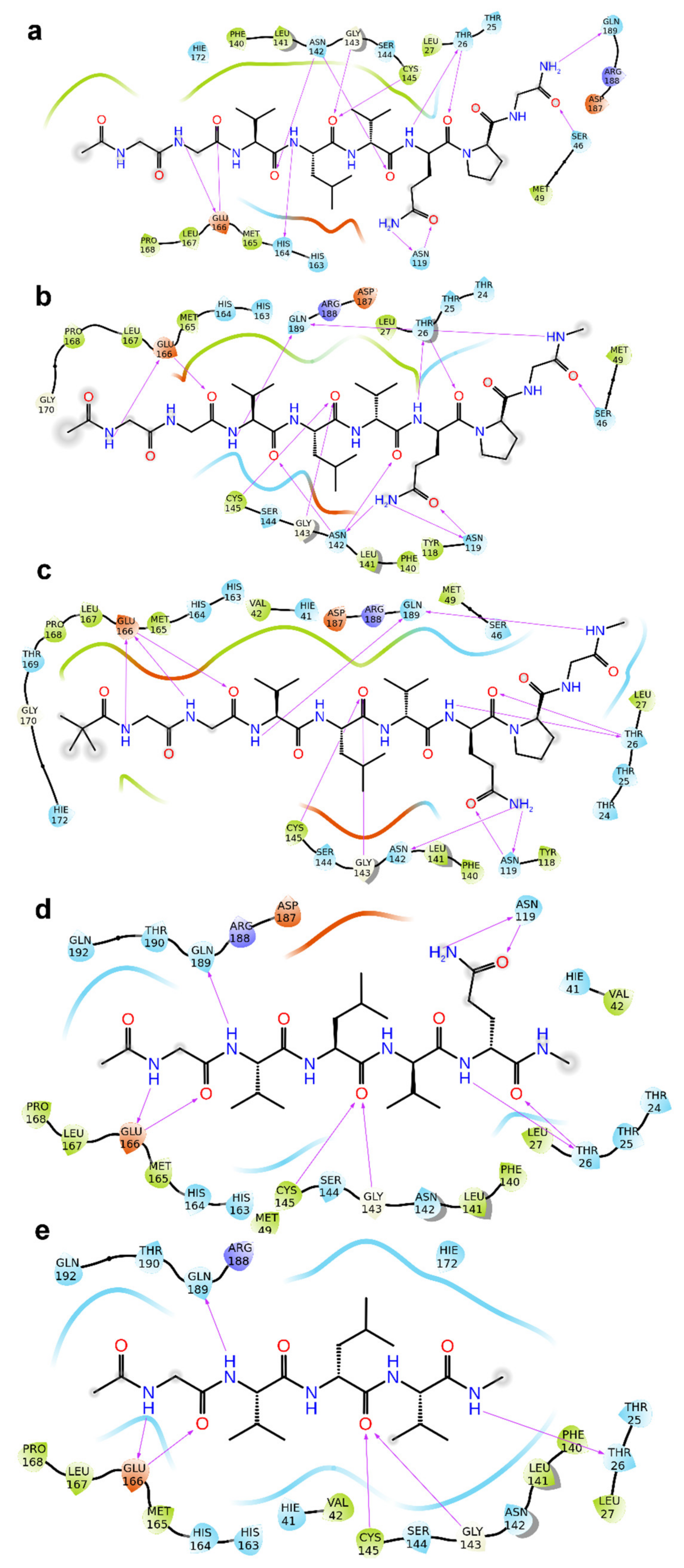
2. Results
3. Discussion
4. Materials and Methods
4.1. Molecular Docking
4.2. MM-GBSA
4.3. Molecular Dynamics
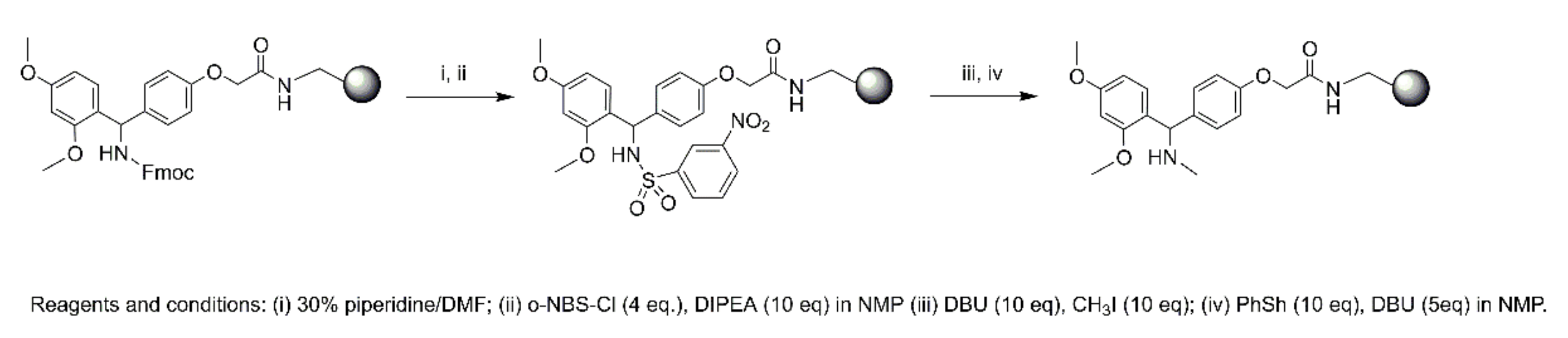
4.4. Synthesis
4.4.1. Material
4.4.2. Microwave Peptide Synthesis
Synthesis of 1
Synthesis of 2
Synthesis of 3–6
N-Terminal Capping
4.4.3. Purification and Characterization
4.5. Enzymatic Inhibition Assays
4.6. Biological Activity
4.6.1. SARS-CoV-2
4.6.2. Cytomegalovirus and Varicella-Zoster Virus
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Bontempia, E.; Vergalli, S.; Squazzoni, F. Understanding COVID-19 diffusion requires an interdisciplinary, multi-dimensional approach. Environ. Res. 2020, 188, 109814. [Google Scholar] [CrossRef]
- Mousavizadeh, L.; Ghasemi, S. Genotype and phenotype of COVID-19: Their roles in pathogenesis. J. Microbiol. Immunol. Infect. 2021, 54, 159–163. [Google Scholar] [CrossRef]
- Parlikar, A.; Kalia, K.; Sinha, S.; Patnaik, S.; Sharma, N.; Vemuri, S.G.; Sharma, G. Understanding genomic diversity, pan-genome, and evolution of SARS-CoV-2. PeerJ 2020, 8, e9576. [Google Scholar] [CrossRef]
- Kirtipal, N.; Bharadwaj, S.; Kang, S.G. From SARS to SARS-CoV-2, insights on structure, pathogenicity and immunity aspects of pandemic human coronaviruses. Infect. Genet. Evol. 2020, 85, 104502. [Google Scholar] [CrossRef]
- Sirois, S.; Zhang, R.; Gao, W.; Gao, H.; Li, Y.; Zheng, H.; Wei, D.-Q. Discovery of Potent Anti-SARS-CoV Mpro Inhibitors. Curr. Comput. Aided Drug Des. 2007, 3, 191–200. [Google Scholar] [CrossRef]
- Bredenbeek, P.J.; Pachuk, C.J.; Noten, A.F.; Charité, J.; Luytjes, W.; Weiss, S.R.; Spaan, W.J. The primary structure and expression of the second open reading frame of the polymerase gene of the coronavirus MHV-A59; A highly conserved polymerase is expressed by an efficient ribosomal frameshifting mechanism. Nucleic Acids Res. 1990, 18, 1825–1832. [Google Scholar] [CrossRef] [Green Version]
- Anand, K.; Palm, G.J.; Mesters, J.R.; Siddell, S.G.; Ziebuhr, J.; Hilgenfeld, R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra a-helical domain. EMBO J. 2002, 21, 3213–3224. [Google Scholar] [CrossRef]
- Yang, H.; Yang, M.; Ding, Y.; Liu, Y.; Lou, Z.; Zhou, Z.; Sun, L.; Mo, L.; Ye, S.; Pang, H.; et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA 2003, 100, 13190–13195. [Google Scholar] [CrossRef] [Green Version]
- Báez-Santos, Y.M.; St John, S.E.; Mesecar, A.D. The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antivir. Res. 2015, 115, 21–38. [Google Scholar] [CrossRef]
- Ghahremanpour, M.M.; Tirado-Rives, J.; Deshmukh, M.; Ippolito, J.A.; Zhang, C.-H.; Cabeza de Vaca, I.; Liosi, M.-E.; Anderson, K.S.; Jorgensen, W.L. Identification of 14 Known Drugs as Inhibitors of the Main Protease of SARS-CoV-2. ACS Med. Chem. Lett. 2020, 11, 2526–2533. [Google Scholar] [CrossRef]
- Kneller, D.W.; Galanie, S.; Phillips, G.; O’Neill, H.M.; Coates, L.; Kovalevsky, A. Malleability of the SARS-CoV-2 3CL Mpro active-site cavity facilitates binding of clinical antivirals. Structure 2020, 28, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zha, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from 1 COVID-19 virus and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Xie, W.; Xue, X.; Yang, K.; Ma, J.; Liang, W.; Zhao, Q.; Zhou, Z.; Pei, D.; Ziebuhr, J.; et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005, 3, e324. [Google Scholar]
- Giordano, A.; Forte, G.; Massimo, L.; Riccio, R.; Bifulco, G.; Di Micco, S. Discovery of new erbB4 inhibitors: Repositioning an orphan chemical library by inverse virtual screening. Eur. J. Med. Chem. 2018, 152, 253–263. [Google Scholar] [CrossRef]
- Troisi, J.; Venutolo, G.; Terracciano, C.; Delli Carri, M.; Di Micco, S.; Landolfi, A.; Fasano, A. The therapeutic use of the zonulin inhibitor AT-1001 (Larazotide) for a variety of acute and chronic inflammatory diseases. Curr. Med. Chem. 2021, 28. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, S.; Musella, S.; Scala, M.C.; Sala, M.; Campiglia, P.; Bifulco, G.; Fasano, A. In silico Analysis Revealed Potential Anti-SARS-CoV-2 Main Protease Activity by the Zonulin Inhibitor Larazotide Acetate. Front. Chem. 2021, 8, 628609. [Google Scholar] [CrossRef]
- Schrödinger Release 2017-1, QikProp; Schrödinger, LLC.: New York, NY, USA, 2017.
- Günther, S.; Reinke, P.Y.A.; Fernández-García, Y.; Lieske, J.; Lane, T.J.; Ginn, H.M.; Koua, F.H.M.; Ehrt, C.; Ewert, W.; Oberthuer, D.; et al. X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease. Science 2021, 372, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Biron, E.; Kessler, H. Convenient synthesis of N-methylamino acids compatible with Fmoc solid-phase peptide synthesis. J. Org. Chem. 2005, 70, 5183–5189. [Google Scholar] [CrossRef] [PubMed]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.J.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Still, W.C.; Tempczyk, A.; Hawley, R.C.; Hendrickson, T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 1990, 112, 6127–6129. [Google Scholar] [CrossRef]
- Schrödinger Release 2017-1, LigPrep; Schrödinger, LLC.: New York, NY, USA, 2017.
- Protein Preparation Wizard Schrödinger LLC; Schrödinger LLC.: New York, NY, USA, 2017.
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Tubert-Brohman, I.; Sherman, W.; Repasky, M.; Beuming, T. Improved docking of polypeptides with glide. J. Chem. Inf. Model. 2013, 53, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Prime, Schrödinger, LLC; Prime, Version 3.1; Schrödinger, LLC.: New York, NY, USA, 2012.
- Knight, J.L.; Krilov, G.; Borrelli, K.W.; Williams, J.; Gunn, J.R.; Clowes, A.; Cheng, L.; Friesner, R.A.; Abel, R. Leveraging data fusion strategies in multireceptor lead optimization MM/GBSA end-point methods. J. Chem. Theory Comput. 2014, 10, 3207–3220. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, E.G.; Thornton, J.M. PROMOTIF-a program to identify and analyze structural motifs in proteins. Protein Sci. 1996, 5, 212–220. [Google Scholar] [CrossRef] [Green Version]
- System Builder, Schrödinger LLC; Schrödinger LLC.: New York, NY, USA, 2015.
- Desmond; DE Shaw Research: New York, NY, USA, 2017.
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the ACM/IEEE Conference on Supercomputing (SC06), Tampa, FL, USA, 11–17 November 2006. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Peptide | RMSD (Å) | ΔGbind (kcal/mol) | Inhibition a (%) | Antiviral Activity (EC50, µM) b | Cytotoxicity (µM) | ||
|---|---|---|---|---|---|---|---|
| UC-1074 | UC-1075 | Cell Morphology (MCC) c | Cell Growth (CC50) d | ||||
| AT1001 (1) | 0.000 | −106.26 | 27.7 ± 0.2 | >20 | >20 | 100 | 82.5 ± 6.2 |
| 2 | 0.864 | −112.34 | 27.1 ± 0.5 | ≥17.6 ± 2.4 | >20 | 100 | 83.0 ± 17.0 |
| 3 | 0.664 | −109.41 | 27.4 ± 1.1 | >20 | >20 | 100 | 81.8 ± 2.1 |
| 4 | 0.696 | −105.79 | 26.2 ± 0.3 | ≥20 | >20 | 100 | 74.9 ± 4.9 |
| 5 | 0.614 | −95.86 | 27.0 ± 1.1 | >20 | >20 | 100 | 75.0 ± 2.2 |
| 6 | 1.081 | −80.38 | 25.0 ± 0.5 | ≥20 | >20 | 100 | 77.4 ± 0.7 |
| Remdesivir | - | - | - | 0.89 ± 0.44 | 1.06 ± 0.44 | 100 | 70.6 ± 4.9 |
| Peptide | Antiviral Activity (EC50, µM) a | Cytotoxicity (µM) | ||
|---|---|---|---|---|
| AD-169 Strain | Davis Strain | Cell Morphology (MCC) b | Cell Growth (CC50) c | |
| AT1001 (1) | >20 | >20 | ≥100 | ND d |
| 2 | >20 | >20 | ≥20 | ND d |
| 3 | >20 | >100 | ≥20 | ND d |
| 4 | >20 | 100 | 100 | ND d |
| 5 | >20 | >100 | ≥100 | ND d |
| 6 | >4 | >20 | ≥20 | ND d |
| Ganciclovir | 2.56 ± 0.23 | 1.18 | ≥394 | 350.23 |
| Cidofovir | 1.27 ± 0.18 | 0.67 ± 0.49 | ≥317 | 159.22 ± 76.5 |
| Peptide | Antiviral Activity (EC50, µM) a | Cytotoxicity (µM) | ||
|---|---|---|---|---|
| TK+ VZV Strain OKA | TK-VZV Strain 07–1 | Cell Morphology (MCC) b | Cell Growth (CC50) c | |
| AT1001 (1) | 44.14 | 59.06 | ≥100 | ND d |
| 2 | 50.05 | 48.42 | >100 | ND d |
| 3 | 58.09 | 81.09 | >100 | ND d |
| 4 | 78.20 | >100 | ≥100 | ND d |
| 5 | 59.80 | >100 | >100 | ND d |
| 6 | >20 | >20 | 100 | ND d |
| Aciclovir | 8.39 | 116 ± 93 | >444 | >444 |
| Birivudine | 0.23 ± 0.04 | 2.13 | >300.3 | >300.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Micco, S.; Musella, S.; Sala, M.; Scala, M.C.; Andrei, G.; Snoeck, R.; Bifulco, G.; Campiglia, P.; Fasano, A. Peptide Derivatives of the Zonulin Inhibitor Larazotide (AT1001) as Potential Anti SARS-CoV-2: Molecular Modelling, Synthesis and Bioactivity Evaluation. Int. J. Mol. Sci. 2021, 22, 9427. https://doi.org/10.3390/ijms22179427
Di Micco S, Musella S, Sala M, Scala MC, Andrei G, Snoeck R, Bifulco G, Campiglia P, Fasano A. Peptide Derivatives of the Zonulin Inhibitor Larazotide (AT1001) as Potential Anti SARS-CoV-2: Molecular Modelling, Synthesis and Bioactivity Evaluation. International Journal of Molecular Sciences. 2021; 22(17):9427. https://doi.org/10.3390/ijms22179427
Chicago/Turabian StyleDi Micco, Simone, Simona Musella, Marina Sala, Maria C. Scala, Graciela Andrei, Robert Snoeck, Giuseppe Bifulco, Pietro Campiglia, and Alessio Fasano. 2021. "Peptide Derivatives of the Zonulin Inhibitor Larazotide (AT1001) as Potential Anti SARS-CoV-2: Molecular Modelling, Synthesis and Bioactivity Evaluation" International Journal of Molecular Sciences 22, no. 17: 9427. https://doi.org/10.3390/ijms22179427
APA StyleDi Micco, S., Musella, S., Sala, M., Scala, M. C., Andrei, G., Snoeck, R., Bifulco, G., Campiglia, P., & Fasano, A. (2021). Peptide Derivatives of the Zonulin Inhibitor Larazotide (AT1001) as Potential Anti SARS-CoV-2: Molecular Modelling, Synthesis and Bioactivity Evaluation. International Journal of Molecular Sciences, 22(17), 9427. https://doi.org/10.3390/ijms22179427