
Copper, Iron, Selenium and Lipo-Glycemic Dysmetabolism in Alzheimer’s Disease

 ,
,  ,
,  and
and 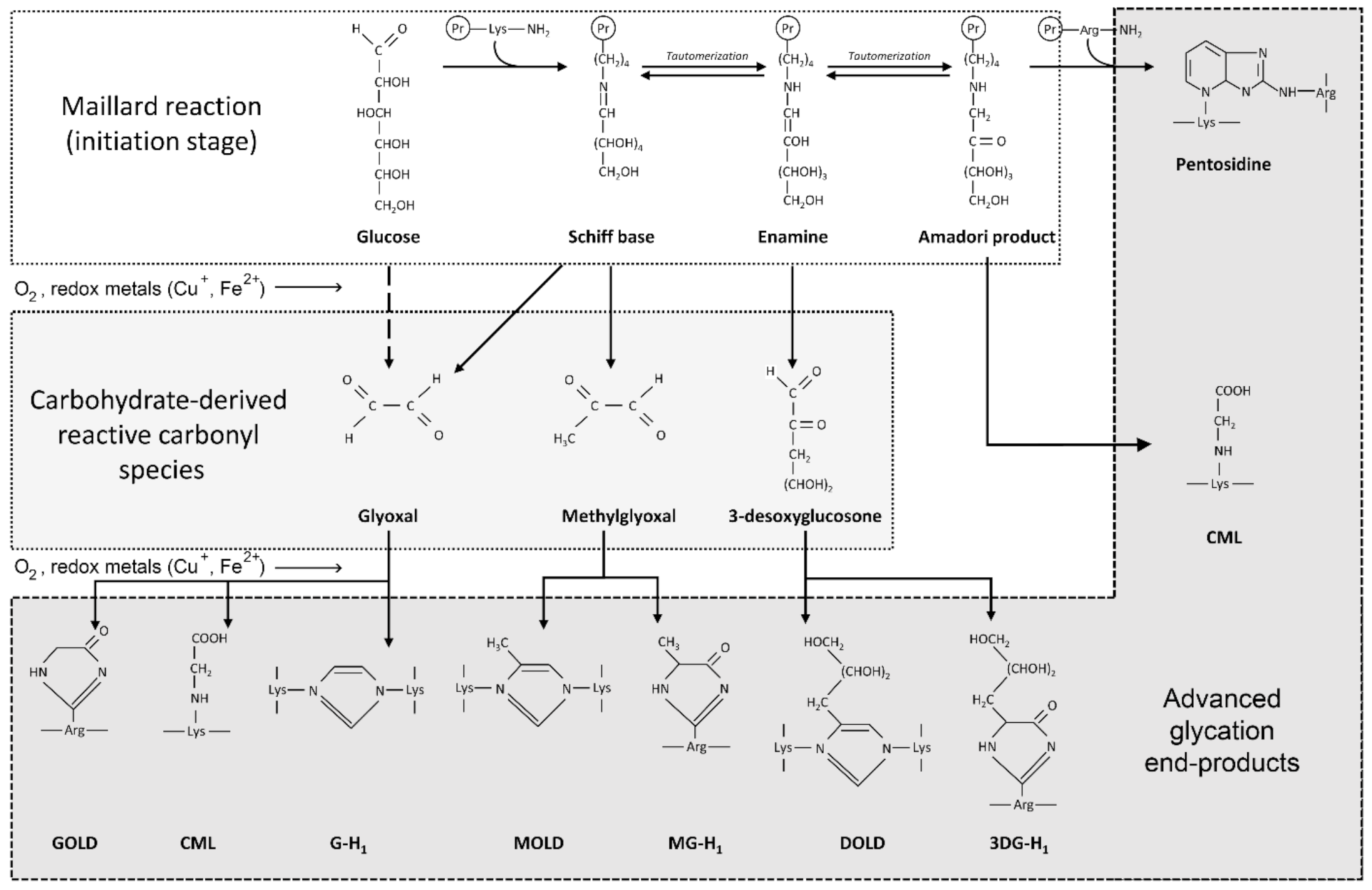{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. On Traditional Hypotheses for AD Etiology
2.1. Physiological APP Processing and Functions of Aβ
2.2. The Amyloid Cascade Hypothesis and the Immunotherapeutic Concept
2.3. New Insights into the Intracellular Metabolism of Aβ
2.4. Tau Hyperphosphorylation in AD
3. Hypothesis Involving Metabolic Syndrome and Glycation
3.1. The Hypothesis of a Role of Metabolic Syndrome and Dyslipidemia
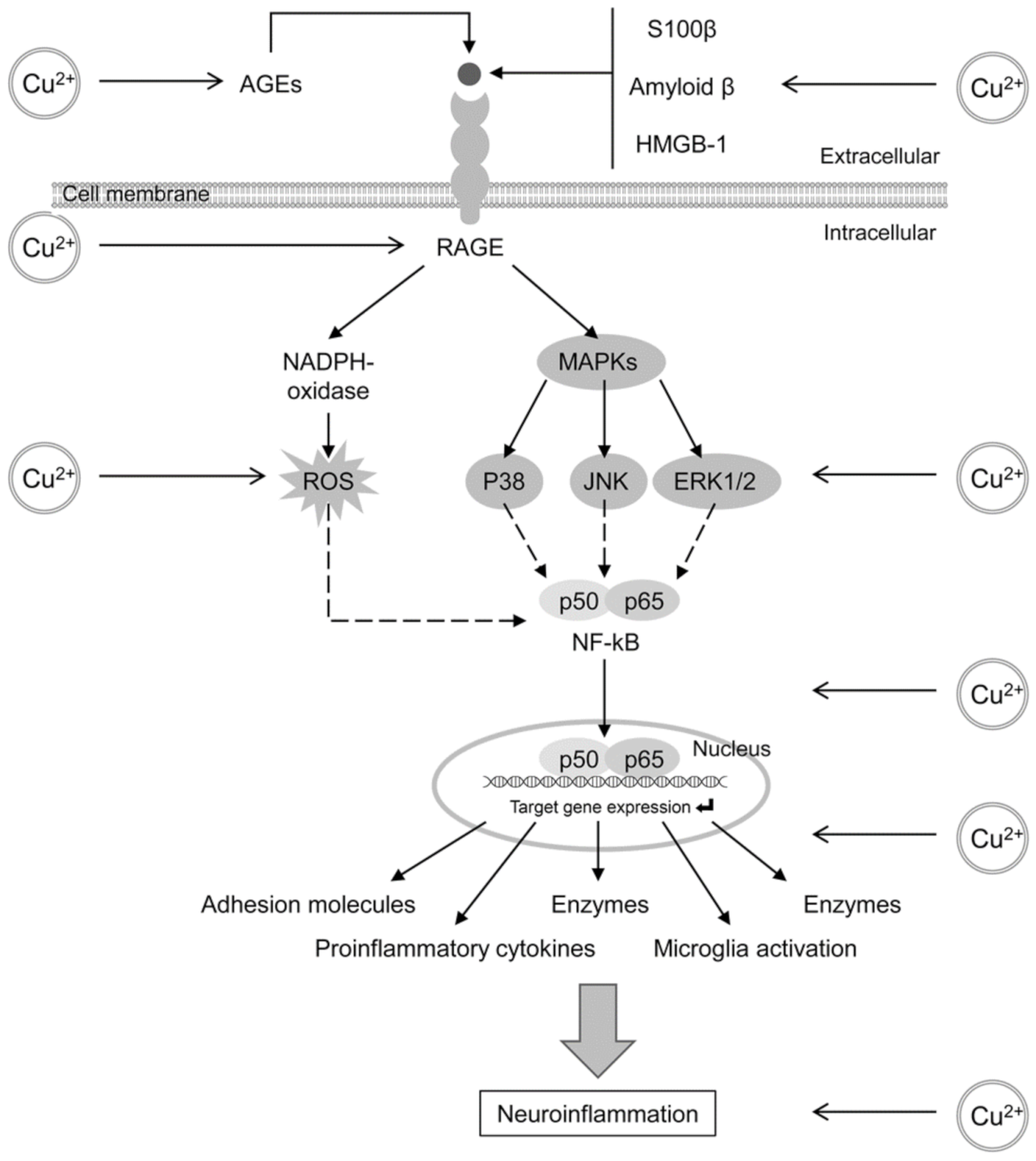
3.2. The Role of Glycation, AGE and RAGE in Alzheimer’s Disease
4. The Role of Copper (Cu) and Iron (Fe) in Alzheimer’s Disease
4.1. General Aspects
4.2. Glycation and AGE Toxicity in AD as Influenced by Cu
4.3. Glycation and AGE Toxicity in AD as Influenced by Fe
5. New Therapeutic Approaches to AD
5.1. Lipo-Glycemic Dysregulation—A Possible Therapeutic Target?
5.2. Metal Chelation—A Rational Strategy?
5.3. Selenium Compounds as Protective Agents
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Association. 2021 Alzheimers disease facts and figures. Alzheimers Dement. 2021, 17, 327–406. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Sadigh-Eteghad, S.; Talebi, M.; Farhoudi, M. Association of apolipoprotein E epsilon 4 allele with sporadic late onset Alzheimer’s disease. A meta-analysis. Neurosciences 2012, 17, 321–326. [Google Scholar] [PubMed]
- Jacobs, E.G.; Kroenke, C.; Lin, J.; Epel, E.S.; Kenna, H.A.; Blackburn, E.H.; Rasgon, N.L. Accelerated cell aging in female APOE-epsilon4 carriers: Implications for hormone therapy use. PLoS ONE 2013, 8, e54713. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Zerbinatti, C.V.; Zhang, J.; Hoe, H.S.; Wang, B.; Cole, S.L.; Herz, J.; Muglia, L.; Bu, G. Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron 2007, 56, 66–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweizer, U.; Bohleber, S.; Zhao, W.; Fradejas-Villar, N. The Neurobiology of Selenium: Looking Back and to the Future. Front. Neurosci 2021, 15, 652099. [Google Scholar] [CrossRef]
- Deane, R.; Bell, R.D.; Sagare, A.; Zlokovic, B.V. Clearance of amyloid-beta peptide across the blood-brain barrier: Implication for therapies in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2009, 8, 16–30. [Google Scholar] [CrossRef]
- Bachmeier, C.; Paris, D.; Beaulieu-Abdelahad, D.; Mouzon, B.; Mullan, M.; Crawford, F. A multifaceted role for apoE in the clearance of beta-amyloid across the blood-brain barrier. Neurodegener. Dis. 2013, 11, 13–21. [Google Scholar] [CrossRef]
- Drachman, D.A. The amyloid hypothesis, time to move on: Amyloid is the downstream result, not cause, of Alzheimer’s disease. Alzheimers Dement. 2014, 10, 372–380. [Google Scholar] [CrossRef]
- Jahn, H. Memory loss in Alzheimer’s disease. Dialogues Clin. Neurosci. 2013, 15, 445–454. [Google Scholar] [PubMed]
- Lim, H.K.; Jung, W.S.; Ahn, K.J.; Won, W.Y.; Hahn, C.; Lee, S.Y.; Kim, I.; Lee, C.U. Relationships between hippocampal shape and cognitive performances in drug-naive patients with Alzheimer’s disease. Neurosci. Lett. 2012, 516, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Konishi, K.; Hori, K.; Tani, M.; Tomioka, H.; Kitajima, Y.; Akashi, N.; Inamoto, A.; Kurosawa, K.; Yuda, H.; Hanashi, T.; et al. Hypothesis of Endogenous Anticholinergic Activity in Alzheimer’s Disease. Neurodegener. Dis. 2015, 15, 149–156. [Google Scholar] [CrossRef]
- Wallace, T.L.; Bertrand, D. Importance of the nicotinic acetylcholine receptor system in the prefrontal cortex. Biochem. Pharm. 2013, 85, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Parsons, C.G.; Stoffler, A.; Danysz, W. Memantine: A NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system—Too little activation is bad, too much is even worse. Neuropharmacology 2007, 53, 699–723. [Google Scholar] [CrossRef] [PubMed]
- Surguchov, A. Caveolin: A New Link Between Diabetes and AD. Cell Mol. Neurobiol. 2020, 40, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Pugazhenthi, S.; Qin, L.; Reddy, P.H. Common neurodegenerative pathways in obesity, diabetes, and Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1037–1045. [Google Scholar] [CrossRef]
- Rojas-Gutierrez, E.; Munoz-Arenas, G.; Trevino, S.; Espinosa, B.; Chavez, R.; Rojas, K.; Flores, G.; Diaz, A.; Guevara, J. Alzheimer’s disease and metabolic syndrome: A link from oxidative stress and inflammation to neurodegeneration. Synapse 2017, 71, e21990. [Google Scholar] [CrossRef]
- Rani, V.; Deep, G.; Singh, R.K.; Palle, K.; Yadav, U.C. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016, 148, 183–193. [Google Scholar] [CrossRef]
- Prohaska, J.R.; Gybina, A.A. Intracellular copper transport in mammals. J. Nutr. 2004, 134, 1003–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, U.C.; Deller, T.; Korte, M. Not just amyloid: Physiological functions of the amyloid precursor protein family. Nat. Rev. Neurosci. 2017, 18, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Mockett, B.G.; Richter, M.; Abraham, W.C.; Muller, U.C. Therapeutic Potential of Secreted Amyloid Precursor Protein APPsalpha. Front. Mol. Neurosci. 2017, 10, 30. [Google Scholar] [CrossRef] [Green Version]
- Guo, Q.; Wang, Z.; Li, H.; Wiese, M.; Zheng, H. APP physiological and pathophysiological functions: Insights from animal models. Cell Res. 2012, 22, 78–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, M.C.; Ludewig, S.; Winschel, A.; Abel, T.; Bold, C.; Salzburger, L.R.; Klein, S.; Han, K.; Weyer, S.W.; Fritz, A.K.; et al. Distinct in vivo roles of secreted APP ectodomain variants APPsalpha and APPsbeta in regulation of spine density, synaptic plasticity, and cognition. EMBO J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Morley, J.E.; Farr, S.A.; Nguyen, A.D.; Xu, F. Editorial: What is the Physiological Function of Amyloid-Beta Protein? J. Nutr. Health Aging 2019, 23, 225–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, S.A.; Spires-Jones, T.L.; Durrant, C.S. The physiological roles of tau and Abeta: Implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathol. 2020, 140, 417–447. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.L.; Serpell, L.C. Membrane and surface interactions of Alzheimer’s Abeta peptide—Insights into the mechanism of cytotoxicity. FEBS J. 2011, 278, 3905–3917. [Google Scholar] [CrossRef] [PubMed]
- Warmlander, S.; Tiiman, A.; Abelein, A.; Luo, J.; Jarvet, J.; Soderberg, K.L.; Danielsson, J.; Graslund, A. Biophysical studies of the amyloid beta-peptide: Interactions with metal ions and small molecules. ChemBioChem 2013, 14, 1692–1704. [Google Scholar] [CrossRef] [PubMed]
- Gilman, S.; Koller, M.; Black, R.S.; Jenkins, L.; Griffith, S.G.; Fox, N.C.; Eisner, L.; Kirby, L.; Rovira, M.B.; Forette, F.; et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 2005, 64, 1553–1562. [Google Scholar] [CrossRef]
- Panza, F.; Solfrizzi, V.; Imbimbo, B.P.; Tortelli, R.; Santamato, A.; Logroscino, G. Amyloid-based immunotherapy for Alzheimer’s disease in the time of prevention trials: The way forward. Expert Rev. Clin. Immunol. 2014, 10, 405–419. [Google Scholar] [CrossRef]
- Gold, M. Phase II clinical trials of anti-amyloid beta antibodies: When is enough, enough? Alzheimers Dement. N. Y. 2017, 3, 402–409. [Google Scholar] [CrossRef]
- Perez-Garmendia, R.; Gevorkian, G. Pyroglutamate-Modified Amyloid Beta Peptides: Emerging Targets for Alzheimer’s Disease Immunotherapy. Curr. Neuropharmacol. 2013, 11, 491–498. [Google Scholar] [CrossRef] [Green Version]
- Andreeva, T.V.; Lukiw, W.J.; Rogaev, E.I. Biological Basis for Amyloidogenesis in Alzheimer’s Disease. Biochemistry 2017, 82, 122–139. [Google Scholar] [CrossRef]
- Ghosal, K.; Vogt, D.L.; Liang, M.; Shen, Y.; Lamb, B.T.; Pimplikar, S.W. Alzheimer’s disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proc. Natl. Acad. Sci. USA 2009, 106, 18367–18372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakono, M.; Zako, T. Amyloid oligomers: Formation and toxicity of Abeta oligomers. FEBS J. 2010, 277, 1348–1358. [Google Scholar] [CrossRef]
- Alsunusi, S.; Kumosani, T.A.; Glabe, C.G.; Huwait, E.A.; Moselhy, S.S. In vitro study of the mechanism of intraneuronal beta-amyloid aggregation in Alzheimer’s disease. Arch. Physiol. Biochem. 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Shang, F.; Taylor, A. Ubiquitin-proteasome pathway and cellular responses to oxidative stress. Free Radic. Biol. Med. 2011, 51, 5–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nassif, N.D.; Cambray, S.E.; Kraut, D.A. Slipping up: Partial substrate degradation by ATP-dependent proteases. IUBMB Life 2014, 66, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Saez, I.; Vilchez, D. The Mechanistic Links Between Proteasome Activity, Aging and Age-related Diseases. Curr. Genom. 2014, 15, 38–51. [Google Scholar] [CrossRef] [Green Version]
- Keck, S.; Nitsch, R.; Grune, T.; Ullrich, O. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer’s disease. J. Neurochem. 2003, 85, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Dickey, C.A.; Koren, J.; Zhang, Y.J.; Xu, Y.F.; Jinwal, U.K.; Birnbaum, M.J.; Monks, B.; Sun, M.; Cheng, J.Q.; Patterson, C.; et al. Akt and CHIP coregulate tau degradation through coordinated interactions. Proc. Natl. Acad. Sci. USA 2008, 105, 3622–3627. [Google Scholar] [CrossRef] [Green Version]
- Tseng, B.P.; Green, K.N.; Chan, J.L.; Blurton-Jones, M.; LaFerla, F.M. Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol. Aging 2008, 29, 1607–1618. [Google Scholar] [CrossRef] [Green Version]
- Bjorklund, G.; Aaseth, J.; Dadar, M.; Chirumbolo, S. Molecular Targets in Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 7032–7044. [Google Scholar] [CrossRef] [PubMed]
- Reeg, S.; Grune, T. Protein Oxidation in Aging: Does It Play a Role in Aging Progression? Antioxid. Redox Signal. 2015, 23, 239–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aaseth, J.; Alexander, J.; Bjorklund, G.; Hestad, K.; Dusek, P.; Roos, P.M.; Alehagen, U. Treatment strategies in Alzheimer’s disease: A review with focus on selenium supplementation. Biometals 2016, 29, 827–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreyev, A.Y.; Kushnareva, Y.E.; Murphy, A.N.; Starkov, A.A. Mitochondrial ROS Metabolism: 10 Years Later. Biochemistry 2015, 80, 517–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, P.; Loganathan, K.; Sekiguchi, M.; Matsuba, Y.; Hui, K.; Tsubuki, S.; Tanaka, M.; Iwata, N.; Saito, T.; Saido, T.C. Abeta secretion and plaque formation depend on autophagy. Cell Rep. 2013, 5, 61–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leal, N.S.; Martins, L.M. Mind the Gap: Mitochondria and the Endoplasmic Reticulum in Neurodegenerative Diseases. Biomedicines 2021, 9, 227. [Google Scholar] [CrossRef]
- Fang, E.F. Mitophagy and NAD+ inhibit Alzheimer disease. Autophagy 2019, 15, 1112–1114. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.Z.; Liu, F. Microtubule-associated protein tau in development, degeneration and protection of neurons. Prog. Neurobiol. 2008, 85, 148–175. [Google Scholar] [CrossRef]
- Aaseth, J.; Buha, A.; Wallace, D.R.; Bjorklund, G. Xenobiotics, Trace Metals and Genetics in the Pathogenesis of Tauopathies. Int. J. Environ. Res. Public Health 2020, 17, 1269. [Google Scholar] [CrossRef] [Green Version]
- Shukla, V.; Skuntz, S.; Pant, H.C. Deregulated Cdk5 activity is involved in inducing Alzheimer’s disease. Arch. Med. Res. 2012, 43, 655–662. [Google Scholar] [CrossRef] [Green Version]
- Mushtaq, G.; Greig, N.H.; Anwar, F.; Al-Abbasi, F.A.; Zamzami, M.A.; Al-Talhi, H.A.; Kamal, M.A. Neuroprotective Mechanisms Mediated by CDK5 Inhibition. Curr. Pharm. Des. 2016, 22, 527–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kivipelto, M.; Mangialasche, F.; Ngandu, T. Lifestyle interventions to prevent cognitive impairment, dementia and Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 653–666. [Google Scholar] [CrossRef]
- Schalkwijk, C.G.; Stehouwer, C.D.A. Methylglyoxal, a Highly Reactive Dicarbonyl Compound, in Diabetes, Its Vascular Complications, and Other Age-Related Diseases. Physiol. Rev. 2020, 100, 407–461. [Google Scholar] [CrossRef]
- Grossberg, G.T.; Tong, G.; Burke, A.D.; Tariot, P.N. Present Algorithms and Future Treatments for Alzheimer’s Disease. J. Alzheimers Dis. 2019, 67, 1157–1171. [Google Scholar] [CrossRef] [PubMed]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef]
- Chaudhuri, J.; Bains, Y.; Guha, S.; Kahn, A.; Hall, D.; Bose, N.; Gugliucci, A.; Kapahi, P. The Role of Advanced Glycation End Products in Aging and Metabolic Diseases: Bridging Association and Causality. Cell Metab. 2018, 28, 337–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brings, S.; Fleming, T.; Freichel, M.; Muckenthaler, M.U.; Herzig, S.; Nawroth, P.P. Dicarbonyls and Advanced Glycation End-Products in the Development of Diabetic Complications and Targets for Intervention. Int. J. Mol. Sci. 2017, 18, 984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rungratanawanich, W.; Qu, Y.; Wang, X.; Essa, M.M.; Song, B.J. Advanced glycation end products (AGEs) and other adducts in aging-related diseases and alcohol-mediated tissue injury. Exp. Mol. Med. 2021, 53, 168–188. [Google Scholar] [CrossRef] [PubMed]
- Alghamdi, A.; Forbes, S.; Birch, D.J.S.; Vyshemirsky, V.; Rolinski, O.J. Detecting beta-amyloid glycation by intrinsic fluorescence—Understanding the link between diabetes and Alzheimer’s disease. Arch. Biochem. Biophys. 2021, 704, 108886. [Google Scholar] [CrossRef]
- Li, X.H.; Du, L.L.; Cheng, X.S.; Jiang, X.; Zhang, Y.; Lv, B.L.; Liu, R.; Wang, J.Z.; Zhou, X.W. Glycation exacerbates the neuronal toxicity of beta-amyloid. Cell Death Dis. 2013, 4, e673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, S.Y.; Lin, Y.P.; Lin, Y.S.; Chang, S.S. Advanced glycation end products enhance amyloid precursor protein expression by inducing reactive oxygen species. Free Radic. Biol. Med. 2010, 49, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Fawver, J.N.; Schall, H.E.; Petrofes Chapa, R.D.; Zhu, X.; Murray, I.V. Amyloid-beta metabolite sensing: Biochemical linking of glycation modification and misfolding. J. Alzheimer’s Dis. 2012, 30, 63–73. [Google Scholar] [CrossRef]
- Batkulwar, K.; Godbole, R.; Banarjee, R.; Kassaar, O.; Williams, R.J.; Kulkarni, M.J. Advanced Glycation End Products Modulate Amyloidogenic APP Processing and Tau Phosphorylation: A Mechanistic Link between Glycation and the Development of Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 988–1000. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Park, J.H. Receptor for Advanced Glycation Endproducts (RAGE), Its Ligands, and Soluble RAGE: Potential Biomarkers for Diagnosis and Therapeutic Targets for Human Renal Diseases. Genom. Inf. 2013, 11, 224–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.; Liu, N.; Wang, C.; Qin, B.; Zhou, Y.; Xiao, M.; Chang, L.; Yan, L.J.; Zhao, B. Role of RAGE in Alzheimer’s Disease. Cell Mol. Neurobiol. 2016, 36, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Chen, H.; Li, Y. The potential mechanisms of Abeta-receptor for advanced glycation end-products interaction disrupting tight junctions of the blood-brain barrier in Alzheimer’s disease. Int. J. Neurosci. 2014, 124, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, F.; Du, Y.F.; Long, Y.; Reed, M.N.; Hu, M.; Suppiramaniam, V.; Hong, H.; Tang, S.S. Targeted inhibition of RAGE reduces amyloid-beta influx across the blood-brain barrier and improves cognitive deficits in db/db mice. Neuropharmacology 2018, 131, 143–153. [Google Scholar] [CrossRef]
- Zeng, F.; Liu, Y.; Huang, W.; Qing, H.; Kadowaki, T.; Kashiwazaki, H.; Ni, J.; Wu, Z. Receptor for advanced glycation end products up-regulation in cerebral endothelial cells mediates cerebrovascular-related amyloid beta accumulation after Porphyromonas gingivalis infection. J. Neurochem. 2020. [Google Scholar] [CrossRef]
- Barichello, T.; Generoso, J.S.; Giridharan, V.V.; Collodel, A.; Dominguini, D.; Petronilho, F.; Dal-Pizzol, F. Receptor for advanced glycation end products mediates meningitis-triggered amyloid-β accumulation and cognitive impairment. Alzheimers Dement. 2020, 16, e047199. [Google Scholar] [CrossRef]
- Ray, R.; Juranek, J.K.; Rai, V. RAGE axis in neuroinflammation, neurodegeneration and its emerging role in the pathogenesis of amyotrophic lateral sclerosis. Neurosci. Biobehav. Rev. 2016, 62, 48–55. [Google Scholar] [CrossRef]
- Tobon-Velasco, J.C.; Cuevas, E.; Torres-Ramos, M.A. Receptor for AGEs (RAGE) as mediator of NF-kB pathway activation in neuroinflammation and oxidative stress. CNS Neurol. Disord. Drug Targets 2014, 13, 1615–1626. [Google Scholar] [CrossRef]
- Bush, A.I. Copper, zinc, and the metallobiology of Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2003, 17, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Dexter, D.T.; Crichton, R.R. Neurodegenerative diseases and therapeutic strategies using iron chelators. J. Trace Elem. Med. Biol. 2015, 31, 267–273. [Google Scholar] [CrossRef] [Green Version]
- Torsdottir, G.; Kristinsson, J.; Snaedal, J.; Johannesson, T. Ceruloplasmin and iron proteins in the serum of patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Dis. Extra 2011, 1, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Squitti, R.; Ghidoni, R.; Simonelli, I.; Ivanova, I.D.; Colabufo, N.A.; Zuin, M.; Benussi, L.; Binetti, G.; Cassetta, E.; Rongioletti, M.; et al. Copper dyshomeostasis in Wilson disease and Alzheimer’s disease as shown by serum and urine copper indicators. J. Trace Elem. Med. Biol. 2018, 45, 181–188. [Google Scholar] [CrossRef]
- Kono, S. Aceruloplasminemia: An update. Int. Rev. Neurobiol. 2013, 110, 125–151. [Google Scholar] [CrossRef]
- Fica-Contreras, S.M.; Shuster, S.O.; Durfee, N.D.; Bowe, G.J.K.; Henning, N.J.; Hill, S.A.; Vrla, G.D.; Stillman, D.R.; Suralik, K.M.; Sandwick, R.K.; et al. Glycation of Lys-16 and Arg-5 in amyloid-beta and the presence of Cu2+ play a major role in the oxidative stress mechanism of Alzheimer’s disease. J. Biol. Inorg. Chem. 2017, 22, 1211–1222. [Google Scholar] [CrossRef] [PubMed]
- Sparks, D.L.; Schreurs, B.G. Trace amounts of copper in water induce beta-amyloid plaques and learning deficits in a rabbit model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 11065–11069. [Google Scholar] [CrossRef] [Green Version]
- Morris, M.C.; Evans, D.A.; Tangney, C.C.; Bienias, J.L.; Schneider, J.A.; Wilson, R.S.; Scherr, P.A. Dietary copper and high saturated and trans fat intakes associated with cognitive decline. Arch. Neurol. 2006, 63, 1085–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Squitti, R.; Ghidoni, R.; Siotto, M.; Ventriglia, M.; Benussi, L.; Paterlini, A.; Magri, M.; Binetti, G.; Cassetta, E.; Caprara, D.; et al. Value of serum nonceruloplasmin copper for prediction of mild cognitive impairment conversion to Alzheimer disease. Ann. Neurol. 2014, 75, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Meenakshi-Sundaram, S.; Mahadevan, A.; Taly, A.B.; Arunodaya, G.R.; Swamy, H.S.; Shankar, S.K. Wilson’s disease: A clinico-neuropathological autopsy study. J. Clin. Neurosci. 2008, 15, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Aigner, E.; Theurl, I.; Haufe, H.; Seifert, M.; Hohla, F.; Scharinger, L.; Stickel, F.; Mourlane, F.; Weiss, G.; Datz, C. Copper availability contributes to iron perturbations in human nonalcoholic fatty liver disease. Gastroenterology 2008, 135, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Ayton, S.; Faux, N.G.; Bush, A.I.; Alzheimer’s Disease Neuroimaging Initiative. Ferritin levels in the cerebrospinal fluid predict Alzheimer’s disease outcomes and are regulated by APOE. Nat. Commun. 2015, 6, 6760. [Google Scholar] [CrossRef] [Green Version]
- Ayton, S.; Faux, N.G.; Bush, A.I. Association of Cerebrospinal Fluid Ferritin Level with Preclinical Cognitive Decline in APOE-epsilon4 Carriers. JAMA Neurol. 2017, 74, 122–125. [Google Scholar] [CrossRef] [Green Version]
- Ahmadi, S.; Zhu, S.; Sharma, R.; Wilson, D.J.; Kraatz, H.B. Interaction of metal ions with tau protein. The case for a metal-mediated tau aggregation. J. Inorg. Biochem. 2019, 194, 44–51. [Google Scholar] [CrossRef]
- Bourassa, M.W.; Leskovjan, A.C.; Tappero, R.V.; Farquhar, E.R.; Colton, C.A.; Van Nostrand, W.E.; Miller, L.M. Elevated copper in the amyloid plaques and iron in the cortex are observed in mouse models of Alzheimer’s disease that exhibit neurodegeneration. Biomed. Spectrosc. Imaging 2013, 2, 129–139. [Google Scholar] [CrossRef] [Green Version]
- Masaldan, S.; Bush, A.I.; Devos, D.; Rolland, A.S.; Moreau, C. Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. Free Radic. Biol. Med. 2019, 133, 221–233. [Google Scholar] [CrossRef]
- Qian, M.; Liu, M.; Eaton, J.W. Transition metals bind to glycated proteins forming redox active “glycochelates”: Implications for the pathogenesis of certain diabetic complications. Biochem. Biophys. Res. Commun. 1998, 250, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Marques, C.M.S.; Nunes, E.A.; Lago, L.; Pedron, C.N.; Manieri, T.M.; Sato, R.H.; Oliveira, V.X.J.; Cerchiaro, G. Generation of Advanced Glycation End-Products (AGEs) by glycoxidation mediated by copper and ROS in a human serum albumin (HSA) model peptide: Reaction mechanism and damage in motor neuron cells. Mutat. Res. 2017, 824, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Siotto, M.; Squitti, R. Copper imbalance in Alzheimer’s disease: Overview of the exchangeable copper component in plasma and the intriguing role albumin plays. Coord. Chem. Rev. 2018, 371, 86–95. [Google Scholar] [CrossRef]
- Loske, C.; Gerdemann, A.; Schepl, W.; Wycislo, M.; Schinzel, R.; Palm, D.; Riederer, P.; Munch, G. Transition metal-mediated glycoxidation accelerates cross-linking of beta-amyloid peptide. Eur J. Biochem. 2000, 267, 4171–4178. [Google Scholar] [CrossRef] [PubMed]
- Ryan, T.M.; Kirby, N.; Mertens, H.D.; Roberts, B.; Barnham, K.J.; Cappai, R.; Pham Cle, L.; Masters, C.L.; Curtain, C.C. Small angle X-ray scattering analysis of Cu2+-induced oligomers of the Alzheimer’s amyloid beta peptide. Metallomics 2015, 7, 536–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahmadi, A.; Steiner, N.; Munch, G. Advanced glycation endproducts as gerontotoxins and biomarkers for carbonyl-based degenerative processes in Alzheimer’s disease. Clin. Chem. Lab. Med. 2011, 49, 385–391. [Google Scholar] [CrossRef]
- Xiao, H.; Cai, G.; Liu, M. Fe2+-catalyzed non-enzymatic glycosylation alters collagen conformation during AGE-collagen formation in vitro. Arch. Biochem. Biophys. 2007, 468, 183–192. [Google Scholar] [CrossRef]
- Mirlohi, M.S.; Yaghooti, H.; Shirali, S.; Aminasnafi, A.; Olapour, S. Increased levels of advanced glycation end products positively correlate with iron overload and oxidative stress markers in patients with beta-thalassemia major. Ann. Hematol. 2018, 97, 679–684. [Google Scholar] [CrossRef]
- Chen, S.H.; Yuan, K.C.; Lee, Y.C.; Shih, C.K.; Tseng, S.H.; Tinkov, A.A.; Skalny, A.V.; Chang, J.S. Iron and Advanced Glycation End Products: Emerging Role of Iron in Androgen Deficiency in Obesity. Antioxidants 2020, 9, 261. [Google Scholar] [CrossRef] [Green Version]
- Shahab, U.; Tabrez, S.; Khan, M.S.; Akhter, F.; Khan, M.S.; Saeed, M.; Ahmad, K.; Srivastava, A.K.; Ahmad, S. Immunogenicity of DNA-advanced glycation end product fashioned through glyoxal and arginine in the presence of Fe3+: Its potential role in prompt recognition of diabetes mellitus auto-antibodies. Chem. Biol. Interact. 2014, 219, 229–240. [Google Scholar] [CrossRef]
- Alagiakrishnan, K.; Sankaralingam, S.; Ghosh, M.; Mereu, L.; Senior, P. Antidiabetic drugs and their potential role in treating mild cognitive impairment and Alzheimer’s disease. Discov. Med. 2013, 16, 277–286. [Google Scholar] [PubMed]
- Kuan, Y.C.; Huang, K.W.; Lin, C.L.; Hu, C.J.; Kao, C.H. Effects of metformin exposure on neurodegenerative diseases in elderly patients with type 2 diabetes mellitus. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 79, 77–83. [Google Scholar] [CrossRef]
- Feinkohl, I.; Janke, J.; Hadzidiakos, D.; Slooter, A.; Winterer, G.; Spies, C.; Pischon, T. Associations of the metabolic syndrome and its components with cognitive impairment in older adults. BMC Geriatr. 2019, 19, 77. [Google Scholar] [CrossRef] [Green Version]
- Lane-Donovan, C.; Herz, J. Is apolipoprotein e required for cognitive function in humans? Implications for Alzheimer drug development. JAMA Neurol. 2014, 71, 1213–1215. [Google Scholar] [CrossRef] [Green Version]
- Davis, K.A.S.; Bishara, D.; Perera, G.; Molokhia, M.; Rajendran, L.; Stewart, R.J. Benefits and Harms of Statins in People with Dementia: A Systematic Review and Meta-Analysis. J. Am. Geriatr. Soc. 2020, 68, 650–658. [Google Scholar] [CrossRef]
- Qin, L.; Chong, T.; Rodriguez, R.; Pugazhenthi, S. Glucagon-Like Peptide-1-Mediated Modulation of Inflammatory Pathways in the Diabetic Brain: Relevance to Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 1346–1355. [Google Scholar] [CrossRef] [PubMed]
- Wicinski, M.; Socha, M.; Malinowski, B.; Wodkiewicz, E.; Walczak, M.; Gorski, K.; Slupski, M.; Pawlak-Osinska, K. Liraglutide and its Neuroprotective Properties-Focus on Possible Biochemical Mechanisms in Alzheimer’s Disease and Cerebral Ischemic Events. Int. J. Mol. Sci. 2019, 20, 1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, K.T.; Wroolie, T.E.; Tong, G.; Foland-Ross, L.C.; Frangou, S.; Singh, M.; McIntyre, R.S.; Roat-Shumway, S.; Myoraku, A.; Reiss, A.L.; et al. Neural correlates of liraglutide effects in persons at risk for Alzheimer’s disease. Behav. Brain Res. 2019, 356, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Suliman, M.; Buckley, A.; Al Tikriti, A.; Tan, T.; le Roux, C.W.; Lessan, N.; Barakat, M. Routine clinical use of liraglutide 3 mg for the treatment of obesity: Outcomes in non-surgical and bariatric surgery patients. Diabetes Obes. Metab. 2019, 21, 1498–1501. [Google Scholar] [CrossRef]
- Diaz-Gerevini, G.T.; Repossi, G.; Dain, A.; Tarres, M.C.; Das, U.N.; Eynard, A.R. Beneficial action of resveratrol: How and why? Nutrition 2016, 32, 174–178. [Google Scholar] [CrossRef]
- Juillerat-Jeanneret, L. Dipeptidyl peptidase IV and its inhibitors: Therapeutics for type 2 diabetes and what else? J. Med. Chem. 2014, 57, 2197–2212. [Google Scholar] [CrossRef]
- Bernstein, H.G.; Dobrowolny, H.; Keilhoff, G.; Steiner, J. Dipeptidyl peptidase IV, which probably plays important roles in Alzheimer disease (AD) pathology, is upregulated in AD brain neurons and associates with amyloid plaques. Neurochem. Int. 2018, 114, 55–57. [Google Scholar] [CrossRef]
- D’Amico, M.; Di Filippo, C.; Marfella, R.; Abbatecola, A.M.; Ferraraccio, F.; Rossi, F.; Paolisso, G. Long-term inhibition of dipeptidyl peptidase-4 in Alzheimer’s prone mice. Exp. Gerontol. 2010, 45, 202–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLachlan, D.R.; Smith, W.L.; Kruck, T.P. Desferrioxamine and Alzheimer’s disease: Video home behavior assessment of clinical course and measures of brain aluminum. Ther. Drug Monit. 1993, 15, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.; Portbury, S.D.; Lago, L.; Bush, A.I.; Adlard, P.A. The Iron Chelator Deferiprone Improves the Phenotype in a Mouse Model of Tauopathy. J. Alzheimer’s Dis. 2020, 78, 1783. [Google Scholar] [CrossRef] [PubMed]
- Grinan-Ferre, C.; Bellver-Sanchis, A.; Izquierdo, V.; Corpas, R.; Roig-Soriano, J.; Chillon, M.; Andres-Lacueva, C.; Somogyvari, M.; Soti, C.; Sanfeliu, C.; et al. The pleiotropic neuroprotective effects of resveratrol in cognitive decline and Alzheimer’s disease pathology: From antioxidant to epigenetic therapy. Ageing Res. Rev. 2021, 67, 101271. [Google Scholar] [CrossRef]
- Gülçin, İ. Antioxidant properties of resveratrol: A structure-activity insight. Innov. Food Sci. Emerg. Technol. 2010, 11, 210–218. [Google Scholar] [CrossRef]
- Bush, A.I. Metal complexing agents as therapies for Alzheimer’s disease. Neurobiol. Aging 2002, 23, 1031–1038. [Google Scholar] [CrossRef]
- Faux, N.G.; Ritchie, C.W.; Gunn, A.; Rembach, A.; Tsatsanis, A.; Bedo, J.; Harrison, J.; Lannfelt, L.; Blennow, K.; Zetterberg, H.; et al. PBT2 rapidly improves cognition in Alzheimer’s Disease: Additional phase II analyses. J. Alzheimer’s Dis. 2010, 20, 509–516. [Google Scholar] [CrossRef] [Green Version]
- Galatti, L.; Giustini, S.E.; Sessa, A.; Polimeni, G.; Salvo, F.; Spina, E.; Caputi, A.P. Neuropsychiatric reactions to drugs: An analysis of spontaneous reports from general practitioners in Italy. Pharm. Res. 2005, 51, 211–216. [Google Scholar] [CrossRef]
- Burk, R.F.; Hill, K.E. Selenoprotein P-expression, functions, and roles in mammals. Biochim. Biophys. Acta 2009, 1790, 1441–1447. [Google Scholar] [CrossRef] [Green Version]
- Schweizer, U.; Brauer, A.U.; Kohrle, J.; Nitsch, R.; Savaskan, N.E. Selenium and brain function: A poorly recognized liaison. Brain Res. Brain Res. Rev. 2004, 45, 164–178. [Google Scholar] [CrossRef]
- Mitozo, P.A.; de Souza, L.F.; Loch-Neckel, G.; Flesch, S.; Maris, A.F.; Figueiredo, C.P.; Dos Santos, A.R.; Farina, M.; Dafre, A.L. A study of the relative importance of the peroxiredoxin-, catalase-, and glutathione-dependent systems in neural peroxide metabolism. Free Radic. Biol. Med. 2011, 51, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Steinbrenner, H.; Sies, H. Selenium homeostasis and antioxidant selenoproteins in brain: Implications for disorders in the central nervous system. Arch. Biochem. Biophys. 2013, 536, 152–157. [Google Scholar] [CrossRef]
- Nakayama, A.; Hill, K.E.; Austin, L.M.; Motley, A.K.; Burk, R.F. All regions of mouse brain are dependent on selenoprotein P for maintenance of selenium. J. Nutr. 2007, 137, 690–693. [Google Scholar] [CrossRef]
- Caito, S.W.; Milatovic, D.; Hill, K.E.; Aschner, M.; Burk, R.F.; Valentine, W.M. Progression of neurodegeneration and morphologic changes in the brains of juvenile mice with selenoprotein P deleted. Brain Res. 2011, 1398, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahar, A.; Patel, K.V.; Semba, R.D.; Bandinelli, S.; Shahar, D.R.; Ferrucci, L.; Guralnik, J.M. Plasma selenium is positively related to performance in neurological tasks assessing coordination and motor speed. Mov. Disord. 2010, 25, 1909–1915. [Google Scholar] [CrossRef] [Green Version]
- Berr, C.; Balansard, B.; Arnaud, J.; Roussel, A.M.; Alperovitch, A. Cognitive decline is associated with systemic oxidative stress: The EVA study. Etude du Vieillissement Arteriel. J. Am. Geriatr. Soc. 2000, 48, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Dominguez, R.; Garcia-Barrera, T.; Gomez-Ariza, J.L. Homeostasis of metals in the progression of Alzheimer’s disease. Biometals 2014, 27, 539–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, B.R.; Roberts, B.R.; Bush, A.I.; Hare, D.J. Selenium, selenoproteins and neurodegenerative diseases. Metallomics 2015, 7, 1213–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kryscio, R.J.; Abner, E.L.; Caban-Holt, A.; Lovell, M.; Goodman, P.; Darke, A.K.; Yee, M.; Crowley, J.; Schmitt, F.A. Association of Antioxidant Supplement Use and Dementia in the Prevention of Alzheimer’s Disease by Vitamin E and Selenium Trial (PREADViSE). JAMA Neurol. 2017, 74, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Varikasuvu, S.R.; Prasad, V.S.; Kothapalli, J.; Manne, M. Brain Selenium in Alzheimer’s Disease (BRAIN SEAD Study): A Systematic Review and Meta-Analysis. Biol. Trace Elem. Res. 2019, 189, 361–369. [Google Scholar] [CrossRef]
- Alehagen, U.; Aaseth, J.; Alexander, J.; Johansson, P.; Larsson, A. Supplemental selenium and coenzyme Q10 reduce glycation along with cardiovascular mortality in an elderly population with low selenium status—A four-year, prospective, randomised, double-blind placebo-controlled trial. J. Trace Elem. Med. Biol. 2020, 61, 126541. [Google Scholar] [CrossRef]
- Alehagen, U.; Alexander, J.; Aaseth, J.; Larsson, A. Decrease in inflammatory biomarker concentration by intervention with selenium and coenzyme Q10: A subanalysis of osteopontin, osteoprotergerin, TNFr1, TNFr2 and TWEAK. J. Inflamm. 2019, 16, 5. [Google Scholar] [CrossRef] [Green Version]
- Gwon, A.R.; Park, J.S.; Park, J.H.; Baik, S.H.; Jeong, H.Y.; Hyun, D.H.; Park, K.W.; Jo, D.G. Selenium attenuates A beta production and A beta-induced neuronal death. Neurosci. Lett. 2010, 469, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Song, G.L.; Chen, C.; Wu, Q.Y.; Zhang, Z.H.; Zheng, R.; Chen, Y.; Jia, S.Z.; Ni, J.Z. Selenium-enriched yeast inhibited beta-amyloid production and modulated autophagy in a triple transgenic mouse model of Alzheimer’s disease. Metallomics 2018, 10, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Zurdo, D.; Romero-Sanchez, I.; Rosales-Conrado, N.; Leon-Gonzalez, M.E.; Madrid, Y. Ability of selenium species to inhibit metal-induced Abeta aggregation involved in the development of Alzheimer’s disease. Anal. Bioanal. Chem. 2020, 412, 6485–6497. [Google Scholar] [CrossRef]
- Li, G.Z.; Liu, F.; Xu, C.; Li, J.Y.; Xu, Y.J. Selenium and Zinc against Abeta25-35-Induced Cytotoxicity and Tau Phosphorylation in PC12 Cells and Inhibits gamma-cleavage of APP. Biol. Trace Elem. Res. 2018, 184, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, B.R.; Roberts, B.R.; Malpas, C.B.; Vivash, L.; Genc, S.; Saling, M.M.; Desmond, P.; Steward, C.; Hicks, R.J.; Callahan, J.; et al. Supranutritional Sodium Selenate Supplementation Delivers Selenium to the Central Nervous System: Results from a Randomized Controlled Pilot Trial in Alzheimer’s Disease. Neurotherapeutics 2019, 16, 192–202. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.L.; Wright, D.K.; Liu, S.; Hovens, C.; O’Brien, T.J.; Shultz, S.R. Sodium selenate, a protein phosphatase 2A activator, mitigates hyperphosphorylated tau and improves repeated mild traumatic brain injury outcomes. Neuropharmacology 2016, 108, 382–393. [Google Scholar] [CrossRef]
- Tolonen, M.; Halme, M.; Sarna, S. Vitamin E and selenium supplementation in geriatric patients: A double-blind preliminary clinical trial. Biol. Trace Elem. Res. 1985, 7, 161–168. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Wen, L.; Wu, Q.Y.; Chen, C.; Zheng, R.; Liu, Q.; Ni, J.Z.; Song, G.L. Long-Term Dietary Supplementation with Selenium-Enriched Yeast Improves Cognitive Impairment, Reverses Synaptic Deficits, and Mitigates Tau Pathology in a Triple Transgenic Mouse Model of Alzheimer’s Disease. J. Agric. Food Chem. 2017, 65, 4970–4979. [Google Scholar] [CrossRef]
- van Eersel, J.; Ke, Y.D.; Liu, X.; Delerue, F.; Kril, J.J.; Gotz, J.; Ittner, L.M. Sodium selenate mitigates tau pathology, neurodegeneration, and functional deficits in Alzheimer’s disease models. Proc. Natl. Acad. Sci. USA 2010, 107, 13888–13893. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Tan, Y.; Zheng, Y.; Du, X.; Liu, Q. Ebselen ameliorates beta-amyloid pathology, tau pathology, and cognitive impairment in triple-transgenic Alzheimer’s disease mice. J. Biol. Inorg. Chem. 2017, 22, 851–865. [Google Scholar] [CrossRef] [PubMed]
- Godoi, G.L.; de Oliveira Porciuncula, L.; Schulz, J.F.; Kaufmann, F.N.; da Rocha, J.B.; de Souza, D.O.; Ghisleni, G.; de Almeida, H.L., Jr. Selenium compounds prevent amyloid beta-peptide neurotoxicity in rat primary hippocampal neurons. Neurochem. Res. 2013, 38, 2359–2363. [Google Scholar] [CrossRef] [PubMed]
- Van der Jeugd, A.; Parra-Damas, A.; Baeta-Corral, R.; Soto-Faguas, C.M.; Ahmed, T.; LaFerla, F.M.; Gimenez-Llort, L.; D’Hooge, R.; Saura, C.A. Reversal of memory and neuropsychiatric symptoms and reduced tau pathology by selenium in 3xTg-AD mice. Sci. Rep. 2018, 8, 6431. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Zhang, W.; Liu, W.; Zhu, W.; Guo, R.; Wang, Y.; Zhang, D.; Wang, J. The inhibitory effect of selenium nanoparticles on protein glycation in vitro. Nanotechnology 2015, 26, 145703. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.B.; Han, J.Y.; Jiang, W.; Wang, J. Selenium inhibits high glucose-induced cyclooxygenase-2 and P-selectin expression in vascular endothelial cells. Mol. Biol. Rep. 2011, 38, 2301–2306. [Google Scholar] [CrossRef]
- Pillai, S.S.; Sugathan, J.K.; Indira, M. Selenium downregulates RAGE and NFkappaB expression in diabetic rats. Biol. Trace Elem. Res. 2012, 149, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.T.; Bayse, C.A.; Ramoutar, R.R.; Brumaghim, J.L. Sulfur and selenium antioxidants: Challenging radical scavenging mechanisms and developing structure-activity relationships based on metal binding. J. Inorg. Biochem. 2015, 145, 30–40. [Google Scholar] [CrossRef]
- Battin, E.E.; Perron, N.R.; Brumaghim, J.L. The central role of metal coordination in selenium antioxidant activity. Inorg. Chem. 2006, 45, 499–501. [Google Scholar] [CrossRef]
- Du, X.; Wang, Z.; Zheng, Y.; Li, H.; Ni, J.; Liu, Q. Inhibitory effect of selenoprotein P on Cu+/Cu2+-induced Abeta42 aggregation and toxicity. Inorg. Chem. 2014, 53, 1672–1678. [Google Scholar] [CrossRef]
- Du, X.; Zheng, Y.; Wang, Z.; Chen, Y.; Zhou, R.; Song, G.; Ni, J.; Liu, Q. Inhibitory act of selenoprotein P on Cu+/Cu2+-induced tau aggregation and neurotoxicity. Inorg. Chem. 2014, 53, 11221–11230. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aaseth, J.; Skalny, A.V.; Roos, P.M.; Alexander, J.; Aschner, M.; Tinkov, A.A. Copper, Iron, Selenium and Lipo-Glycemic Dysmetabolism in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 9461. https://doi.org/10.3390/ijms22179461
Aaseth J, Skalny AV, Roos PM, Alexander J, Aschner M, Tinkov AA. Copper, Iron, Selenium and Lipo-Glycemic Dysmetabolism in Alzheimer’s Disease. International Journal of Molecular Sciences. 2021; 22(17):9461. https://doi.org/10.3390/ijms22179461
Chicago/Turabian StyleAaseth, Jan, Anatoly V. Skalny, Per M. Roos, Jan Alexander, Michael Aschner, and Alexey A. Tinkov. 2021. "Copper, Iron, Selenium and Lipo-Glycemic Dysmetabolism in Alzheimer’s Disease" International Journal of Molecular Sciences 22, no. 17: 9461. https://doi.org/10.3390/ijms22179461
APA StyleAaseth, J., Skalny, A. V., Roos, P. M., Alexander, J., Aschner, M., & Tinkov, A. A. (2021). Copper, Iron, Selenium and Lipo-Glycemic Dysmetabolism in Alzheimer’s Disease. International Journal of Molecular Sciences, 22(17), 9461. https://doi.org/10.3390/ijms22179461