
Dark Side of Cancer Therapy: Cancer Treatment-Induced Cardiopulmonary Inflammation, Fibrosis, and Immune Modulation

{kind=link}
{kind=link}
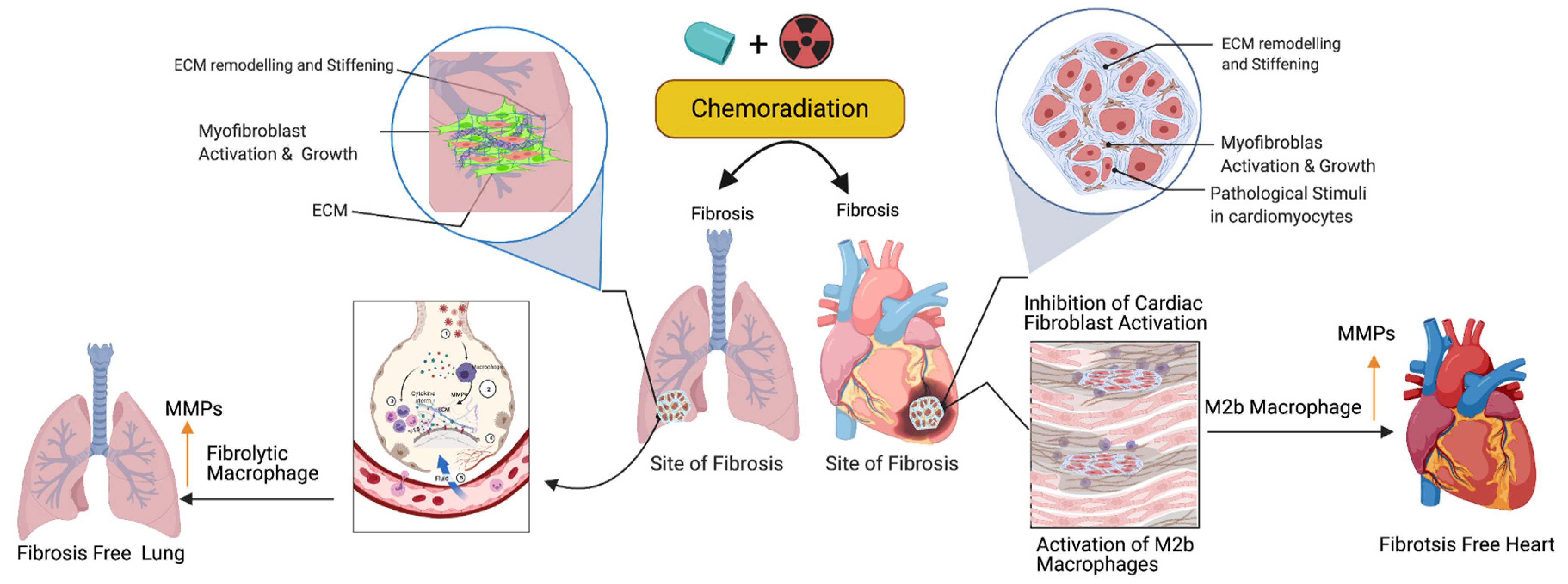{kind=link}
{kind=link}
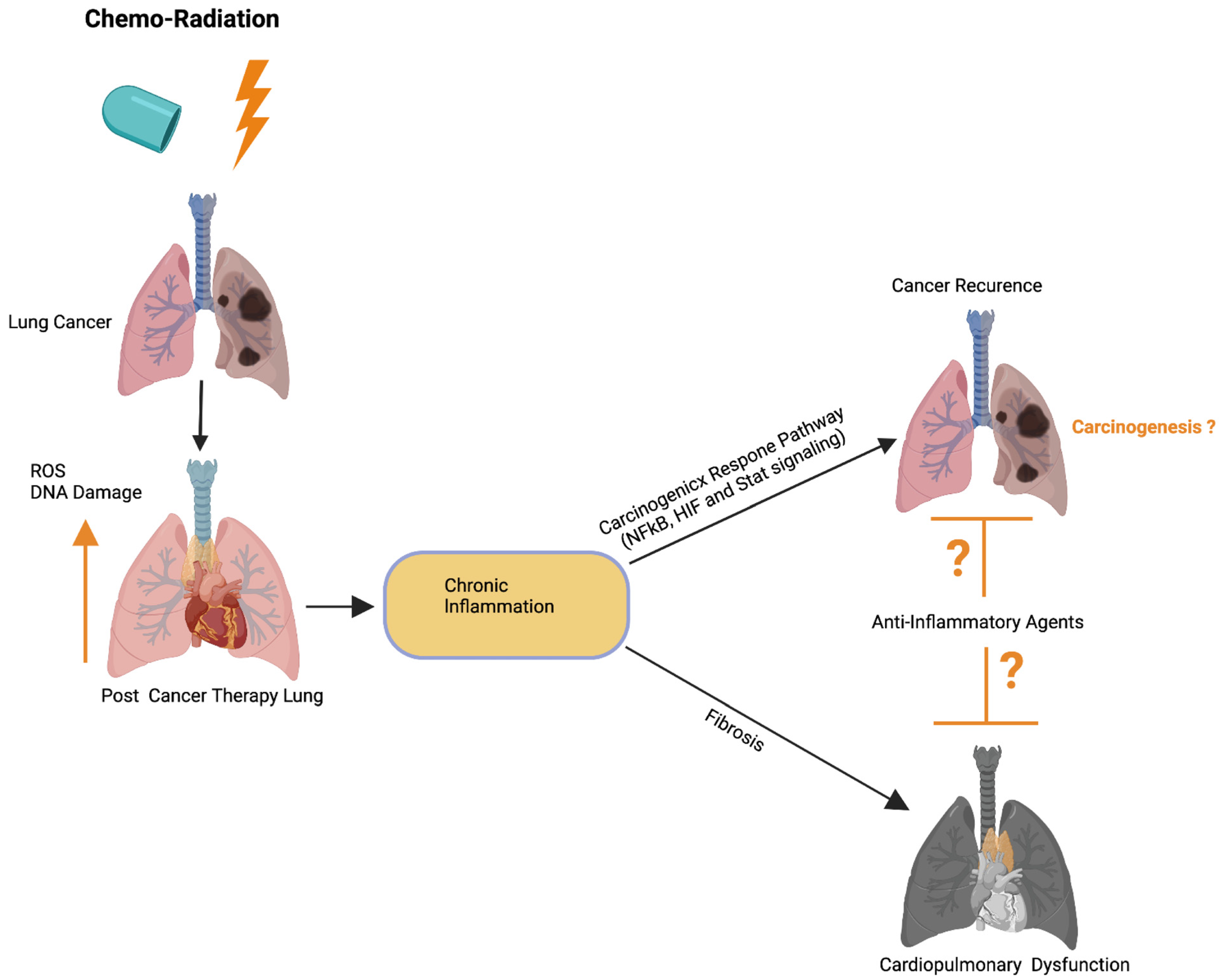{kind=link}
Abstract
:1. Introduction
2. Cancer Therapy-Induced Inflammation and Cardiopulmonary Fibrosis
Cancer Therapy Response Factors Promote Cardiopulmonary Toxicity via NFκB-Signaling
3. Cancer Therapy (Radiation)-Induced Immune Modulation
3.1. Fibroblast Activation and Cardiopulmonary Inflammation in Response to Cancer Therapy
3.2. Activation of M2 Macrophages, Maintenance of Tissue Archistructure, and Extracellular Matrix Remodeling (ECM)
3.3. Biomarker Signatures in Cardiopulmonary Toxicity Following Cancer Therapy
3.4. Targeted Therapy-Induced Cardiac Toxicities
3.5. Possible Antioxidant Treatments and Prevention of Cancer Therapy-Induced Toxicity
3.6. Cell Fat Function and Clinical Biomarker Determination
3.7. Role of Cancer Therapy-Induced Inflammation in Lung Cancer
3.8. Mechanisms of Inflammation-Induced Carcinogenesis and Cancer Therapy-Induced Tumor Recurrence
3.9. Role of the Inflammatory Microenvironment in Oncogenesis and Metastasis in Lung Cancer
4. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ROS | Reactive Oxygen Species |
| PD-L1 | Reactive Oxygen Species |
| NSCLC | Non-Small Cell Lung Cancer |
| TGF-β | Transforming Growth Factor Beta |
| CTRCPD | Cancer Therapy-Related Cardiopulmonary Dysfunction |
| SMAD3 | Mothers Against Decapendtaplegic Homolog 3 |
References
- Oser, M.G.; Niederst, M.J.; Sequist, L.V.; Engelman, J.A. Transformation from non-small-cell lung cancer to small-cell lung cancer: Molecular drivers and cells of origin. Lancet Oncol. 2015, 16, e165–e172. [Google Scholar] [CrossRef] [Green Version]
- Gago, J.; Câmara, G.; Dionísio, J.; Opinião, A. Pulmonary metastasis as sole manifestation of relapse in previously treated localised prostate cancer: Three exceptional case reports. Ecancermedicalscience 2016, 10, 645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Piao, H.-L.; Kim, B.-J.; Yao, F.; Han, Z.; Wang, Y.; Xiao, Z.; Siverly, A.N.; Lawhon, S.E.; Ton, B.N.; et al. Long noncoding RNA MALAT1 suppresses breast cancer metastasis. Nat. Genet. 2018, 50, 1705–1715. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.; Tashiro, K.; Dixit, A.; Soni, A.; Vogel, K.; Hall, B.; Shafqat, I.; Slaughter, J.; Param, N.; Le, A.; et al. Loss of HIF1A From Pancreatic Cancer Cells Increases Expression of PPP1R1B and Degradation of p53 to Promote Invasion and Metastasis. Gastroenterology 2020, 159, 1882–1897.e5. [Google Scholar] [CrossRef]
- Trott, K.R.; Herrmann, T.; Kasper, M. Target cells in radiation pneumopathy. Int. J. Radiat. Oncol. 2004, 58, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Wirsdörfer, F.; Jendrossek, V. The Role of Lymphocytes in Radiotherapy-Induced Adverse Late Effects in the Lung. Front. Immunol. 2016, 7, 591. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, H. Impact of neoadjuvant chemotherapy and chemoradiotherapy on postoperative cardiopulmonary complications in patients with esophageal cancer. Dis. Esophagus 2017, 30, 1–7. [Google Scholar] [CrossRef]
- Frey, B.; Rückert, M.; Deloch, L.; Rühle, P.F.; Derer, A.; Fietkau, R.; Gaipl, U.S. Immunomodulation by ionizing radiation-impact for design of radio-immunotherapies and for treatment of inflammatory diseases. Immunol. Rev. 2017, 280, 231–248. [Google Scholar] [CrossRef]
- Kim, S.-K.; Kwon, D.-A.; Lee, H.S.; Kim, H.K.; Kim, W.-K. Preventive Effect of the Herbal Preparation, HemoHIM, on Cisplatin-Induced Immune Suppression. Evidence-Based Complement. Altern. Med. 2019, 2019, 1–8. [Google Scholar]
- Wang, W.; Chapman, N.M.; Zhang, B.; Li, M.; Fan, M.; Laribee, R.N.; Zaidi, M.R.; Pfeffer, L.M.; Chi, H.; Wu, Z.-H. Upregulation of PD-L1 via HMGB1-Activated IRF3 and NF-κB Contributes to UV Radiation-Induced Immune Suppression. Cancer Res. 2019, 79, 2909–2922. [Google Scholar] [CrossRef]
- Fluorouracil and cardiotoxicity. Med. J. Aust. 1994, 160, 445. [CrossRef] [PubMed]
- Afsar, T.; Razak, S.; Almajwal, A.; Khan, M.R. Acacia hydaspica R. Parker ameliorates cisplatin induced oxidative stress, DNA damage and morphological alterations in rat pulmonary tissue. BMC Complement. Altern. Med. 2018, 18, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amraotkar, A.R.; Pachika, A.; Grubb, K.J.; DeFilippis, A.P. Rapid Extracorporeal Membrane Oxygenation Overcomes Fulminant Myocarditis Induced by 5-Fluorouracil. Tex. Heart Inst. J. 2016, 43, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Kanaoka, K.; Ikebe, S.; Ihara, S.; Tsuji, H.; Yasuoka, H.; Minami, S. Durvalumab-Induced Diffuse Alveolar Hemorrhage: An Autopsy Case Report. Case Rep. Oncol. 2020, 13, 696–701. [Google Scholar] [CrossRef]
- Mosseri, M.; Fingert, H.J.; Varticovski, L.; Chokshi, S.; Isner, J.M. In vitro evidence that myocardial ischemia resulting from 5-fluorouracil chemotherapy is due to protein kinase C-mediated vasoconstriction of vascular smooth muscle. Cancer Res. 1993, 53, 3028–3033. [Google Scholar] [PubMed]
- Torre-Bouscoulet, L.; Muñoz-Montaño, W.R.; Martinez-Briseno, D.; Lozano-Ruiz, F.J.; Fernandez-Plata, R.; Beck-Magaña, J.A.; García-Sancho, C.; Guzmán-Barragán, A.; Vergara, E.; Blake-Cerda, M.; et al. Abnormal pulmonary function tests predict the development of radiation-induced pneumonitis in advanced non-small cell lung Cancer. Respir. Res. 2018, 19, 72. [Google Scholar] [CrossRef] [Green Version]
- Turner, R.R.; Layfield, L.J.; Bishop, B.C.; Epstein, A.L.; Parker, J.W. Flow cytometric measurements of proliferation-associated nuclear antigen p105 and DNA content in non-Hodgkin’s lymphomas. Arch. Pathol. Lab. Med. 1989, 113, 907–911. [Google Scholar]
- Varga, Z.; Ferdinandy, P.; Liaudet, L.; Pacher, P. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am. J. Physiol. Circ. Physiol. 2015, 309, H1453–H1467. [Google Scholar] [CrossRef] [Green Version]
- Yimit, A.; Adebali, O.; Sancar, A.; Jiang, Y. Differential damage and repair of DNA-adducts induced by anti-cancer drug cisplatin across mouse organs. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Shin, Y.K.; Zheng, Z.; Zhu, L.; Lee, I.J. Risk of radiation-induced pneumonitis after helical and static-port tomotherapy in lung cancer patients and experimental rats. Radiat. Oncol. 2015, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zucali, P.A.; De Pas, T.; Palmieri, G.; Favaretto, A.; Chella, A.; Tiseo, M.; Caruso, M.; Simonelli, M.; Perrino, M.; De Vincenzo, F.; et al. Phase II Study of Everolimus in Patients With Thymoma and Thymic Carcinoma Previously Treated With Cisplatin-Based Chemotherapy. J. Clin. Oncol. 2018, 36, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Wijesinghe, N. A case of late-onset severe cardiotoxicity from 5-fluorouracil therapy resulting in death. N. Z. Med. J. 2007, 120, 2836. [Google Scholar]
- Albini, A.; Pennesi, G.; Donatelli, F.; Cammarota, R.; De Flora, S.; Noonan, D. Cardiotoxicity of Anticancer Drugs: The Need for Cardio-Oncology and Cardio-Oncological Prevention. J. Natl. Cancer Inst. 2010, 102, 14–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geyer, C.E.; Forster, J.; Lindquist, D.; Chan, S.; Romieu, C.G.; Pienkowski, T.; Jagiello-Gruszfeld, A.; Crown, J.; Chan, A.; Kaufman, B.; et al. Lapatinib plus Capecitabine for HER2-Positive Advanced Breast Cancer. N. Engl. J. Med. 2006, 355, 2733–2743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinzerling, L.; Ott, P.A.; Hodi, F.S.; Husain, A.N.; Tajmir-Riahi, A.; Tawbi, H.; Pauschinger, M.; Gajewski, T.F.; Lipson, E.J.; Luke, J.J. Cardiotoxicity associated with CTLA4 and PD1 blocking immunotherapy. J. Immunother. Cancer 2016, 4, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oncel, C.R.; Ovey, I.S. The role of selenium in bevacizumab induced cardiotoxicity. Bratisl. Lek. List. 2019, 120, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Provencio, M.; Bonilla, F.; La Casta, A.; Espana, P. Cerebral infarction after cisplatin-based chemotherapy. Postgrad. Med. J. 1994, 70, 525–526. [Google Scholar] [CrossRef] [Green Version]
- Abid, S.H.; Malhotra, V.; Perry, M.C. Radiation-induced and chemotherapy-induced pulmonary injury. Curr. Opin. Oncol. 2001, 13, 242–248. [Google Scholar] [CrossRef]
- Hanania, A.N.; Mainwaring, W.; Ghebre, Y.T.; Hanania, N.A.; Ludwig, M. Radiation-Induced Lung Injury. Chest 2019, 156, 150–162. [Google Scholar] [CrossRef]
- Lierova, A.; Jelicova, M.; Nemcova, M.; Proksova, M.; Pejchal, J.; Zarybnicka, L.; Sinkorova, Z. Cytokines and radiation-induced pulmonary injuries. J. Radiat. Res. 2018, 59, 709–753. [Google Scholar] [CrossRef]
- Giuranno, L.; Ient, J.; De Ruysscher, D.; Vooijs, M.A. Radiation-Induced Lung Injury (RILI). Front. Oncol. 2019, 9, 877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terasaki, Y.; Ohsawa, I.; Terasaki, M.; Takahashi, M.; Kunugi, S.; Dedong, K.; Urushiyama, H.; Amenomori, S.; Kaneko-Togashi, M.; Kuwahara, N.; et al. Hydrogen therapy attenuates irradiation-induced lung damage by reducing oxidative stress. Am. J. Physiol. Cell. Mol. Physiol. 2011, 301, L415–L426. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Chen, Z. The pathophysiological role of mitochondrial oxidative stress in lung diseases. J. Transl. Med. 2017, 15, 1–13. [Google Scholar] [CrossRef]
- Grazioli, S.; Pugin, J. Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front. Immunol. 2018, 9, 832. [Google Scholar] [CrossRef]
- Ryter, S.W.; Kim, H.P.; Hoetzel, A.; Park, J.W.; Nakahira, K.; Wang, X.; Choi, A.M. Mechanisms of Cell Death in Oxidative Stress. Antioxid. Redox Signal. 2007, 9, 49–89. [Google Scholar] [CrossRef]
- Darby, I.A.; Hewitson, T.D. Fibroblast Differentiation in Wound Healing and Fibrosis. Adv. Appl. Microbiol. 2007, 257, 143–179. [Google Scholar]
- Fleckenstein, K.; Zgonjanin, L.; Chen, L.; Rabbani, Z.; Jackson, I.L.; Thrasher, B.; Kirkpatrick, J.; Foster, W.M.; Vujaskovic, Z. Temporal Onset of Hypoxia and Oxidative Stress After Pulmonary Irradiation. Int. J. Radiat. Oncol. 2007, 68, 196–204. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Signal transduction to hypoxia-inducible factor 1. Biochem. Pharmacol. 2002, 64, 993–998. [Google Scholar] [CrossRef]
- Ma, H.; Jones, K.R.; Guo, R.; Xu, P.; Shen, Y.; Ren, J. Cisplatin compromises myocardial contractile function and mitochondrial ultrastructure: Role of endoplasmic reticulum stress. Clin. Exp. Pharmacol. Physiol. 2010, 37, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Rosa, G.M.; Gigli, L.; Tagliasacchi, M.I.; Di Iorio, C.; Carbone, F.; Nencioni, A.; Montecucco, F.; Brunelli, C. Update on cardiotoxicity of anti-cancer treatments. Eur. J. Clin. Investig. 2016, 46, 264–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fossella, F.V.; Lee, J.S.; Murphy, W.K.; Lippman, S.M.; Calayag, M.; Pang, A.; Chasen, M.; Shin, D.M.; Glisson, B.; Benner, S. Phase II study of docetaxel for recurrent or metastatic non-small-cell lung cancer. J. Clin. Oncol. 1994, 12, 1238–1244. [Google Scholar] [CrossRef]
- Oprea, A.D.; Russell, R.R.; Russell, K.S.; Abu-Khalaf, M. Chemotherapy Agents With Known Cardiovascular Side Effects and Their Anesthetic Implications. J. Cardiothorac. Vasc. Anesth. 2017, 31, 2206–2226. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Hou, J.; Yan, X.; Leng, J.; Li, R.; Zhang, J.; Xing, J.; Chen, C.; Wang, Z.; Li, W. Platycodon grandiflorum Saponins Ameliorate Cisplatin-Induced Acute Nephrotoxicity through the NF-κB-Mediated Inflammation and PI3K/Akt/Apoptosis Signaling Pathways. Nutrients 2018, 10, 1328. [Google Scholar] [CrossRef] [Green Version]
- Gordon, J.; Shaw, J.A.; Kirshenbaum, L.A. Multiple Facets of NF-κB in the Heart. Circ. Res. 2011, 108, 1122–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moding, E.J.; Kastan, M.B.; Kirsch, D.G. Strategies for optimizing the response of cancer and normal tissues to radiation. Nat. Rev. Drug Discov. 2013, 12, 526–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaffray, D.A. Image-guided radiotherapy: From current concept to future perspectives. Nat. Rev. Clin. Oncol. 2012, 9, 688–699. [Google Scholar] [CrossRef]
- Shigematsu, A.; Adachi, Y.; Koike-Kiriyama, N.; Suzuki, Y.; Iwasaki, M.; Koike, Y.; Nakano, K.; Mukaide, H.; Imamura, M.; Ikehara, S. Effects of Low-dose Irradiation on Enhancement of Immunity by Dendritic Cells. J. Radiat. Res. 2007, 48, 51–55. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.-S.; Liu, Z.-M.; Yu, X.-Y.; Song, A.-Q.; Liu, N.; Wang, H. Low-dose Radiation Induces Antitumor Effects and Erythrocyte System Hormesis. Asian Pac. J. Cancer Prev. 2013, 14, 4121–4126. [Google Scholar] [CrossRef] [Green Version]
- Gameiro, S.R.; Jammeh, M.L.; Wattenberg, M.M.; Tsang, K.Y.; Ferrone, S.; Hodge, J.W. Radiation-induced immunogenic modulation of tumor enhances antigen processing and calreticulin exposure, resulting in enhanced T-cell killing. Oncotarget 2014, 5, 403–416. [Google Scholar] [CrossRef] [Green Version]
- Reits, E.A.; Hodge, J.W.; Herberts, C.A.; Groothuis, T.; Chakraborty, M.; Wansley, E.; Camphausen, K.; Luiten, R.; De Ru, A.H.; Neijssen, J.; et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J. Exp. Med. 2006, 203, 1259–1271. [Google Scholar] [CrossRef]
- Chen, J.; Liu, X.; Zeng, Z.; Li, J.; Luo, Y.; Sun, W.; Gong, Y.; Zhang, J.; Wu, Q.; Xie, C. Immunomodulation of NK Cells by Ionizing Radiation. Front. Oncol. 2020, 10, 874. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.-D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunology 2014, 41, 843–852. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-Y.; Son, Y.; Park, S.-W.; Bae, J.-H.; Chung, J.S.; Kim, H.H.; Chung, B.-S.; Kim, S.-H.; Kang, C.-D. Increase of NKG2D ligands and sensitivity to NK cell-mediated cytotoxicity of tumor cells by heat shock and ionizing radiation. Exp. Mol. Med. 2006, 38, 474–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lugade, A.A.; Sorensen, E.; Gerber, S.A.; Moran, J.P.; Frelinger, J.G.; Lord, E.M. Radiation-Induced IFN-γ Production within the Tumor Microenvironment Influences Antitumor Immunity. J. Immunol. 2008, 180, 3132–3139. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, S.; Wang, B.; Kawashima, N.; Braunstein, S.; Badura, M.; Cameron, T.O.; Babb, J.; Schneider, R.; Formenti, S.C.; Dustin, M.; et al. Radiation-Induced CXCL16 Release by Breast Cancer Cells Attracts Effector T Cells. J. Immunol. 2008, 181, 3099–3107. [Google Scholar] [CrossRef] [PubMed]
- Kolesnick, R.; Fuks, Z. Radiation and ceramide-induced apoptosis. Oncogene 2003, 22, 5897–5906. [Google Scholar] [CrossRef] [Green Version]
- Relling, D.P.; Hintz, K.K.; Ren, J. Acute exposure of ceramide enhances cardiac contractile function in isolated ventricular myocytes. Br. J. Pharmacol. 2003, 140, 1163–1168. [Google Scholar] [CrossRef] [Green Version]
- Bodas, M.; Pehote, G.; Silverberg, D.; Gulbins, E.; Vij, N. Autophagy augmentation alleviates cigarette smoke-induced CFTR-dysfunction, ceramide-accumulation and COPD-emphysema pathogenesis. Free. Radic. Biol. Med. 2019, 131, 81–97. [Google Scholar] [CrossRef]
- Suryadevara, V.; Fu, P.; Ebenezer, D.L.; Berdyshev, E.; Bronova, I.A.; Huang, L.S.; Harijith, A.; Natarajan, V. Sphingolipids in Ventilator Induced Lung Injury: Role of Sphingosine-1-Phosphate Lyase. Int. J. Mol. Sci. 2018, 19, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, H.; Gong, J.; Peterson, A.L.; Lu, X.; Zhang, P.; Dennery, P.A. Fatty Acid Oxidation Protects against Hyperoxia-induced Endothelial Cell Apoptosis and Lung Injury in Neonatal Mice. Am. J. Respir. Cell Mol. Biol. 2019, 60, 667–677. [Google Scholar] [CrossRef]
- Kang, J.; Jung, M.; Choudhury, M.; Leof, E.B. Transforming growth factor beta induces fibroblasts to express and release the immunomodulatory protein PD-L1 into extracellular vesicles. FASEB J. 2020, 34, 2213–2226. [Google Scholar] [CrossRef] [Green Version]
- Gabbiani, G. The myofibroblast in wound healing and fibrocontractive diseases. J. Pathol. 2003, 200, 500–503. [Google Scholar] [CrossRef]
- Feghali, C.A.; Wright, T.M. Cytokines in acute and chronic inflammation. Front. Biosci. 1997, 2, d12–d26. [Google Scholar]
- Gharaee-Kermani, M.; McCullumsmith, R.E.; Charo, I.F.; Kunkel, S.L.; Phan, S.H. CC-chemokine receptor 2 required for bleomycin-induced pulmonary fibrosis. Cytokine 2003, 24, 266–276. [Google Scholar] [CrossRef]
- Scotton, C.J.; Chambers, R.C. Molecular targets in pulmonary fibrosis: The myofibroblast in focus. Chest 2007, 132, 1311. [Google Scholar] [CrossRef] [PubMed]
- Nagineni, C.N.; William, A.; Cherukuri, A.; Samuel, W.; Hooks, J.J.; Detrick, B. Inflammatory cytokines regulate secretion of VEGF and chemokines by human conjunctival fibroblasts: Role in dysfunctional tear syndrome. Cytokine 2016, 78, 16–19. [Google Scholar] [CrossRef] [PubMed]
- Riedel, A.; Shorthouse, D.; Haas, L.; Hall, B.A.; Shields, J. Tumor-induced stromal reprogramming drives lymph node transformation. Nat. Immunol. 2016, 17, 1118–1127. [Google Scholar] [CrossRef]
- Sjöberg, E.; Meyrath, M.; Milde, L.; Herrera, M.; Lövrot, J.; Hägerstrand, D.; Frings, O.; Bartish, M.; Rolny, C.; Sonnhammer, E.; et al. A Novel ACKR2-Dependent Role of Fibroblast-Derived CXCL14 in Epithelial-to-Mesenchymal Transition and Metastasis of Breast Cancer. Clin. Cancer Res. 2019, 25, 3702–3717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, P.; Langevitz, P.; Alderdice, C.A.; Aubrey, M.; Baer, P.A.; Baron, M.; Buskila, D.; Dutz, J.P.; Khostanteen, I.; Piper, S. Mortality in systemic sclerosis (scleroderma). Q. J. Med. 1992, 82, 139–148. [Google Scholar] [PubMed]
- Taunk, N.; Haffty, B.G.; Kostis, J.B.; Egoyal, S. Radiation-Induced Heart Disease: Pathologic Abnormalities and Putative Mechanisms. Front. Oncol. 2015, 5, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madan, R.; Benson, R.; Sharma, D.; Julka, P.; Rath, G. Radiation induced heart disease: Pathogenesis, management and review literature. J. Egypt. Natl. Cancer Inst. 2015, 27, 187–193. [Google Scholar] [CrossRef]
- Camargo, R.D.O.; Abual’Anaz, B.; Rattan, S.G.; Filomeno, K.L.; Dixon, I.M.C. Novel factors that activate and deactivate cardiac fibroblasts: A new perspective for treatment of cardiac fibrosis. Wound Repair Regen. 2021, 29, 667–677. [Google Scholar] [CrossRef]
- Ulmasov, B.; Neuschwander-Tetri, B.A.; Lai, J.; Monastyrskiy, V.; Bhat, T.; Yates, M.P.; Oliva, J.; Prinsen, M.J.; Ruminski, P.G.; Griggs, D.W. Inhibitors of Arg-Gly-Asp-Binding Integrins Reduce Development of Pancreatic Fibrosis in Mice. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 499–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, G.A.; Masters, K.S.; Shah, D.N.; Anseth, K.S.; Leinwand, L.A. Valvular myofibroblast activation by transforming growth factor-beta: Implications for pathological extracellular matrix remodeling in heart valve disease. Circ. Res. 2004, 95, 253–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krippendorf, B.B.; Riley, D.A. Distinguishing unloading. Versus reloading-induced changes in rat soleus muscle. Muscle Nerve 1993, 16, 99–108. [Google Scholar] [CrossRef]
- Pierre, B.A.S.; Tidball, J.G. Differential response of macrophage subpopulations to soleus muscle reloading after rat hindlimb suspension. J. Appl. Physiol. 1994, 77, 290–297. [Google Scholar] [CrossRef]
- Tidball, J.G.; Berchenko, E.; Frenette, J. Macrophage invasion does not contribute to muscle membrane injury during inflammation. J. Leukoc. Biol. 1999, 65, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Newby, A.C. Metalloproteinase production from macrophages—A perfect storm leading to atherosclerotic plaque rupture and myocardial infarction. Exp. Physiol. 2016, 101, 1327–1337. [Google Scholar] [CrossRef] [Green Version]
- Patole, P.S.; Schubert, S.; Hildinger, K.; Khandoga, S.; Khandoga, A.; Segerer, S.; Henger, A.; Kretzler, M.; Werner, M.; Krombach, F.; et al. Toll-like receptor-4: Renal cells and bone marrow cells signal for neutrophil recruitment during pyelonephritis. Kidney Int. 2005, 68, 2582–2587. [Google Scholar] [CrossRef] [Green Version]
- Yue, Y.; Huang, S.; Wang, L.; Wu, Z.; Liang, M.; Li, H.; Lv, L.; Li, W.; Wu, Z. M2b Macrophages Regulate Cardiac Fibroblast Activation and Alleviate Cardiac Fibrosis After Reperfusion Injury. Circ. J. 2020, 84, 626–635. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, L.; Su, H.; Liu, Q.; Shen, J.; Dai, H.; Zheng, W.; Lu, Y.; Zhang, W.; Bei, Y.; et al. Chimeric antigen receptor macrophage therapy for breast tumours mediated by targeting the tumour extracellular matrix. Br. J. Cancer 2019, 121, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Asmis, R.; Hawkins, G.A.; Howard, T.D.; Cox, L.A. The Need for Multi-Omics Biomarker Signatures in Precision Medicine. Int. J. Mol. Sci. 2019, 20, 4781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.G.; Chen, W.S.; Li, H.; Foye, A.; Zhang, M.; Sjöström, M.; Aggarwal, R.; Playdle, D.; Liao, A.; Alumkal, J.J.; et al. The DNA methylation landscape of advanced prostate cancer. Nat. Genet. 2020, 52, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Brun, A.; Magallanes, M.E.; Del Rio, C.M.; Barrett-Wilt, G.A.; Karasov, W.H.; Caviedes-Vidal, E. A Fast and Accurate Method to Identify and Quantify Enzymes in Brush-Border Membranes: In Situ Hydrolysis Followed by Nano LC-MS/MS. Methods Protoc. 2020, 3, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacMullan, M.; Dunn, Z.S.; Graham, N.; Yang, L.; Wang, P. Quantitative Proteomics and Metabolomics Reveal Biomarkers of Disease as Potential Immunotherapy Targets and Indicators of Therapeutic Efficacy. Theranostics 2019, 9, 7872–7888. [Google Scholar] [CrossRef]
- Hristova, V.A.; Chan, D.W. Cancer biomarker discovery and translation: Proteomics and beyond. Expert Rev. Proteom. 2019, 16, 93–103. [Google Scholar] [CrossRef]
- Chu, H.-W.; Chang, K.-P.; Hsu, C.-W.; Chang, I.Y.-F.; Liu, H.-P.; Chen, Y.-T.; Wu, C.-C. Identification of Salivary Biomarkers for Oral Cancer Detection with Untargeted and Targeted Quantitative Proteomics Approaches. Mol. Cell. Proteom. 2019, 18, 1796–1806. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Shin, Y.; Kim, T.H.; Kim, D.-H.; Lee, A. Plasma metabolites as possible biomarkers for diagnosis of breast cancer. PLoS ONE 2019, 14, e0225129. [Google Scholar] [CrossRef]
- Sabet, N.S.; Atashbar, S.; Khanlou, E.M.; Kahrizi, F.; Salimi, A. Curcumin attenuates bevacizumab-induced toxicity via suppressing oxidative stress and preventing mitochondrial dysfunction in heart mitochondria. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2020, 393, 1447–1457. [Google Scholar] [CrossRef]
- Zaborowska-Szmit, M.; Krzakowski, M.; Kowalski, D.M.; Szmit, S. Cardiovascular Complications of Systemic Therapy in Non-Small-Cell Lung Cancer. J. Clin. Med. 2020, 9, 1268. [Google Scholar] [CrossRef]
- Economopoulou, P.; Kentepozidis, N.; Kotsakis, A.; Kapiris, I. Cancer therapy and cardiovascular risk: Focus on bevacizumab. Cancer Manag. Res. 2015, 7, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Pereira, G.M.; Miller, J.F.; Shevach, E.M. Mechanism of action of cyclosporine A in vivo. II. T cell priming in vivo to alloantigen can be mediated by an IL-2-independent cyclosporine A-resistant pathway. J. Immunol. 1990, 144, 2109–2116. [Google Scholar]
- Boyiadzis, M.; Foon, K.A. Approved monoclonal antibodies for cancer therapy. Expert Opin. Biol. Ther. 2008, 8, 1151–1158. [Google Scholar] [CrossRef]
- Cammisotto, V.; Nocella, C.; Bartimoccia, S.; Sanguigni, V.; Francomano, D.; Sciarretta, S.; Pastori, D.; Peruzzi, M.; Cavarretta, E.; D’Amico, A.; et al. The Role of Antioxidants Supplementation in Clinical Practice: Focus on Cardiovascular Risk Factors. Antioxidants 2021, 10, 146. [Google Scholar] [CrossRef] [PubMed]
- Lubrano, S.B.V.; Balzan, S. Enzymatic antioxidant system in vascular inflammation and coronary artery disease. World J. Exp. Med. 2015, 5, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Nocella, C.; Cammisotto, V.; Pigozzi, F.; Borrione, P.; Fossati, C.; D’Amico, A.; Cangemi, R.; Peruzzi, M.; Gobbi, G.; Ettorre, E.; et al. Impairment between Oxidant and Antioxidant Systems: Short- and Long-term Implications for Athletes’ Health. Nutrients 2019, 11, 1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aboul-Enein, H.Y.; Kruk, I.; Kładna, A.; Lichszteld, K.; Michalska, T. Scavenging effects of phenolic compounds on reactive oxygen species. Biopolymers 2007, 86, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, R.; Loffredo, L.; Pignatelli, P.; Nocella, C.; Bartimoccia, S.; Di Santo, S.; Martino, F.; Catasca, E.; Perri, L.; Violi, F. Dark chocolate inhibits platelet isoprostanes via NOX2 down-regulation in smokers. J. Thromb. Haemost. 2012, 10, 125–132. [Google Scholar] [CrossRef]
- Li, Y.; Cao, Z.; Zhu, H. Upregulation of endogenous antioxidants and phase 2 enzymes by the red wine polyphenol, resveratrol in cultured aortic smooth muscle cells leads to cytoprotection against oxidative and electrophilic stress. Pharmacol. Res. 2005, 53, 6–15. [Google Scholar] [CrossRef]
- de Jager, T.L.; Cockrell, A.E.; Du Plessis, S.S. Ultraviolet Light Induced Generation of Reactive Oxygen Species. Adv. Exp. Med. Biol. 2017, 996, 15–23. [Google Scholar]
- Klebelsberg, D. Psychological characteristics of motorization development. Hefte Unf. 1978, 1978, 348–355. [Google Scholar]
- Li, Q.; Liang, X.; Yang, Y.; Zeng, X.; Zhong, X.; Huang, C. Panax notoginsengsaponins ameliorate cisplatin-induced mitochondrial injury via the HIF-1α/mitochondria/ROS pathway. FEBS Open Bio 2020, 10, 118–126. [Google Scholar] [CrossRef]
- Meng, X.-M.; Ren, G.-L.; Gao, L.; Yang, Q.; Li, H.-D.; Wu, W.-F.; Huang, C.; Zhang, L.; Lv, X.-W.; Li, J. NADPH oxidase 4 promotes cisplatin-induced acute kidney injury via ROS-mediated programmed cell death and inflammation. Lab. Investig. 2018, 98, 63–78. [Google Scholar] [CrossRef] [Green Version]
- Vincent, D.T.; Ibrahim, Y.F.; Espey, M.G.; Suzuki, Y.J. The role of antioxidants in the era of cardio-oncology. Cancer Chemother. Pharmacol. 2013, 72, 1157–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghareeb, A.E.; Moawed, F.S.M.; Ghareeb, D.A.; Kandil, E.I. Potential Prophylactic Effect of Berberine against Rat Colon Carcinoma Induce by 1,2-Dimethyl Hydrazine. Asian Pac. J. Cancer Prev. 2018, 19, 1685–1690. [Google Scholar]
- Koukourakis, M.I. Radiation damage and radioprotectants: New concepts in the era of molecular medicine. Br. J. Radiol. 2012, 85, 313–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebmann, J.; DeLuca, A.M.; Epstein, A.; Steinberg, S.M.; Morstyn, G.; Mitchell, J.B. Protection from Lethal Irradiation by the Combination of Stem Cell Factor and Tempol. Radiat. Res. 1994, 137, 400. [Google Scholar] [CrossRef]
- Wang, J.; He, D.; Zhang, Q.; Han, Y.; Jin, S.; Qi, F. Resveratrol Protects Against Cisplatin-Induced Cardiotoxicity by Alleviating Oxidative Damage. Cancer Biother. Radiopharm. 2009, 24, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Fung, C.; Dinh, P.; Ardeshir-Rouhani-Fard, S.; Schaffer, K.; Fossa, S.D.; Travis, L.B. Toxicities Associated with Cisplatin-Based Chemotherapy and Radiotherapy in Long-Term Testicular Cancer Survivors. Adv. Urol. 2018, 2018, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Kerns, S.L.; Fung, C.; Fossa, S.D.; Dinh, P.C.; Monahan, P.; Sesso, H.D.; Frisina, R.D.; Feldman, D.R.; Hamilton, R.J.; Vaughn, D.; et al. Relationship of Cisplatin-Related Adverse Health Outcomes With Disability and Unemployment Among Testicular Cancer Survivors. JNCI Cancer Spectr. 2020, 4, pkaa022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabinowits, M.; Souhami, L.; Gil, R.A.; Andrade, C.A.V.; Paiva, H.C. Increased Pulmonary Toxicity with Bleomycin and Cisplatin Chemotherapy Combinations. Am. J. Clin. Oncol. 1990, 13, 132–138. [Google Scholar] [CrossRef] [PubMed]
- El-Awady, E.-S.E.; Moustafa, Y.M.; Abo-Elmatty, D.; Radwan, A. Cisplatin-induced cardiotoxicity: Mechanisms and cardioprotective strategies. Eur. J. Pharmacol. 2011, 650, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.D.; Tsuji, P.A.; Milner, J.A. Selenoproteins and Cancer Prevention. Annu. Rev. Nutr. 2012, 32, 73–95. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Kliment, C.R.; Suliman, H.B.; Tobolewski, J.M.; Reynolds, C.M.; Day, B.J.; Zhu, X.; McTiernan, C.F.; McGaffin, K.R.; Piantadosi, C.A.; Oury, T.D. Extracellular superoxide dismutase regulates cardiac function and fibrosis. J. Mol. Cell. Cardiol. 2009, 47, 730–742. [Google Scholar] [CrossRef] [Green Version]
- Adhami, V.M.; Mukhtar, H. Anti-oxidants from green tea and pomegranate for chemoprevention of prostate cancer. Mol. Biotechnol. 2007, 37, 52–57. [Google Scholar] [CrossRef]
- Mániková, D.; Šestáková, Z.; Rendeková, J.; Vlasáková, D.; Lukáčová, P.; Paegle, E.; Arsenyan, P.; Chovanec, M. Resveratrol-Inspired Benzo[b]selenophenes Act as Anti-Oxidants in Yeast. Molecules 2018, 23, 507. [Google Scholar] [CrossRef] [Green Version]
- Milisav, I.; Ribarič, S.; Poljsak, B. Antioxidant vitamins and ageing. In Subcellular Biochemistry; Springer: New York, NY, USA, 2018; Volume 90, pp. 1–23. [Google Scholar]
- Tripathi, S.; Kumari, U.; Mazumder, P.M. Ameliorative effects of apple cider vinegar on neurological complications via regulation of oxidative stress markers. J. Food Biochem. 2020, 44, e13504. [Google Scholar] [CrossRef]
- Chakraborthy, A.; Ramani, P.; Sherlin, H.; Premkumar, P.; Natesan, A. Antioxidant and pro-oxidant activity of Vitamin C in oral environment. Indian J. Dent. Res. 2014, 25, 499. [Google Scholar] [CrossRef]
- Njus, D.; Kelley, P.M.; Tu, Y.-J.; Schlegel, H.B. Ascorbic acid: The chemistry underlying its antioxidant properties. Free. Radic. Biol. Med. 2020, 159, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Sorice, A.; Guerriero, E.; Capone, F.; Colonna, G.; Castello, G.; Costantini, S. Ascorbic Acid: Its Role in Immune System and Chronic Inflammation Diseases. Mini-Rev. Med. Chem. 2014, 14, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Bresciani, G.; da Cruz, I.B.M.; González-Gallego, J. Manganese Superoxide Dismutase and Oxidative Stress Modulation. Adv. Virus Res. 2015, 68, 87–130. [Google Scholar]
- Wang, Y.; Branicky, R.; Noe, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Ronaldson-Bouchard, K.; Ma, S.P.; Yeager, K.; Chen, T.; Song, L.; Sirabella, D.; Morikawa, K.; Teles, D.; Yazawa, M.; Vunjak-Novakovic, G. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 2018, 556, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Franks, T.J.; Colby, T.V.; Travis, W.D.; Tuder, R.M.; Reynolds, H.Y.; Brody, A.R.; Cardoso, W.V.; Crystal, R.G.; Drake, C.J.; Engelhardt, J.; et al. Resident Cellular Components of the Human Lung: Current Knowledge and Goals for Research on Cell Phenotyping and Function. Proc. Am. Thorac. Soc. 2008, 5, 763–766. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Molinaro, C.; Martoriati, A.; Cailliau, K. Proteins from the DNA Damage Response: Regulation, Dysfunction, and Anticancer Strategies. Cancers 2021, 13, 3819. [Google Scholar] [CrossRef] [PubMed]
- Antwih, D.A.; Gabbara, K.M.; Lancaster, W.D.; Ruden, D.M.; Zielske, S.P. Radiation-induced epigenetic DNA methylation modification of radiation-response pathways. Epigenetics 2013, 8, 839–848. [Google Scholar] [CrossRef]
- Deng, J.-S.; Jiang, W.-P.; Chen, C.-C.; Lee, L.-Y.; Li, P.-Y.; Huang, W.-C.; Liao, J.-C.; Chen, H.-Y.; Huang, S.-S.; Huang, G.-J. Cordyceps cicadae Mycelia Ameliorate Cisplatin-Induced Acute Kidney Injury by Suppressing the TLR4/NF-κB/MAPK and Activating the HO-1/Nrf2 and Sirt-1/AMPK Pathways in Mice. Oxid. Med. Cell. Longev. 2020, 2020, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Citrin, D.E.; Prasanna, P.G.S.; Walker, A.J.; Freeman, M.L.; Eke, I.; Barcellos-Hoff, M.H.; Arankalayil, M.J.; Cohen, E.P.; Wilkins, R.; Ahmed, M.M.; et al. Radiation-Induced Fibrosis: Mechanisms and Opportunities to Mitigate. Report of an NCI Workshop, September 19, 2016. Radiat. Res. 2017, 188, 1–20. [Google Scholar] [CrossRef]
- Johnston, C.J.; Hernady, E.; Reed, C.; Thurston, S.W.; Finkelstein, J.N.; Williams, J.P. Early Alterations in Cytokine Expression in Adult Compared to Developing Lung in Mice after Radiation Exposure. Radiat. Res. 2010, 173, 522–535. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, E.L.; Walser, T.C.; Krysan, K.; Liclican, E.L.; Grant, J.L.; Rodriguez, N.L.; Dubinett, S.M. The Inflammatory Tumor Microenvironment, Epithelial Mesenchymal Transition and Lung Carcinogenesis. Cancer Microenviron. 2012, 5, 5–18. [Google Scholar] [CrossRef] [Green Version]
- Willis, B.C.; Borok, Z. TGF-β-induced EMT: Mechanisms and implications for fibrotic lung disease. Am. J. Physiol. Cell. Mol. Physiol. 2007, 293, L525–L534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Huang, Y.-J.; Liu, C.; Yang, Y.-Y.; Liu, H.; Cui, J.-G.; Cheng, Y.; Gao, F.; Cai, J.-M.; Li, B.-L. Inhibition of TBK1 attenuates radiation-induced epithelial–mesenchymal transition of A549 human lung cancer cells via activation of GSK-3β and repression of ZEB1. Lab. Investig. 2014, 94, 362–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Zhong, Y.; Chen, J.; Lin, X.; Lin, Z.; Wang, N.; Lin, S. Radiation Enhances the Epithelial– Mesenchymal Transition of A549 Cells via miR3591-5p/USP33/PPM1A. Cell. Physiol. Biochem. 2018, 50, 721–733. [Google Scholar] [CrossRef]
- Dennis, J.M.; Witting, P.K. Protective role for antioxidants in acute kidney disease. Nutrients 2017, 9, 718. [Google Scholar] [CrossRef] [Green Version]
- Bonifant, C.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef] [PubMed]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and Toxicity Management of 19-28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2014, 6, 224ra25. [Google Scholar] [CrossRef] [Green Version]
- Gomes, M.; Teixeira, A.L.; Coelho, A.; Araújo, A.; Medeiros, R. The Role of Inflammation in Lung Cancer. Adv. Exp. Med. Biol. 2014, 816, 1–23. [Google Scholar]
- Kamp, D.W.; Shacter, E.; Weitzman, S.A. Chronic inflammation and cancer: The role of the mitochondria. Oncology 2011, 25, 400. [Google Scholar]
- Multhoff, G.; Molls, M.; Radons, J. Chronic Inflammation in Cancer Development. Front. Immunol. 2012, 2, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Callaghan, D.S.; O’Donnell, D.; O’Connell, F.; O’Byrne, K.J. The Role of Inflammation in the Pathogenesis of Non-small Cell Lung Cancer. J. Thorac. Oncol. 2010, 5, 2024–2036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindu, S.; Mazumder, S.; Bandyopadhyay, U. Non-steroidal anti-inflammatory drugs (NSAIDs) and organ damage: A current perspective. Biochem. Pharmacol. 2020, 180, 114147. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Gu, D.-N.; Dai, J.-J.; Huang, Q.; Tian, L. Dark Side of Cytotoxic Therapy: Chemoradiation-Induced Cell Death and Tumor Repopulation. Trends Cancer 2020, 6, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Patidar, A.; Selvaraj, S.; Sarode, A.; Chauhan, P.; Chattopadhyay, D.; Saha, B. DAMP-TLR-cytokine axis dictates the fate of tumor. Cytokine 2018, 104, 114–123. [Google Scholar] [CrossRef]
- Fang, H.; Ang, B.; Xu, X.; Huang, X.; Wu, Y.; Sun, Y.; Wang, W.; Li, N.; Cao, X.; Wan, T. TLR4 is essential for dendritic cell activation and anti-tumor T-cell response enhancement by DAMPs released from chemically stressed cancer cells. Cell. Mol. Immunol. 2014, 11, 150–159. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, Y.; Steller, H. Programmed Cell Death in Animal Development and Disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef] [Green Version]
- Lito, P.; Pratilas, C.A.; Joseph, E.W.; Tadi, M.; Halilovic, E.; Zubrowski, M.; Huang, A.; Wong, W.L.; Callahan, M.K.; Merghoub, T.; et al. Relief of Profound Feedback Inhibition of Mitogenic Signaling by RAF Inhibitors Attenuates Their Activity in BRAFV600E Melanomas. Cancer Cell 2012, 22, 668–682. [Google Scholar] [CrossRef] [Green Version]
- Lito, P.; Rosen, N.; Solit, D.B. Tumor adaptation and resistance to RAF inhibitors. Nat. Med. 2013, 19, 1401–1409. [Google Scholar] [CrossRef] [PubMed]
- Obenauf, A.C.; Zou, Y.; Ji, A.L.; Vanharanta, S.; Shu, W.; Shi, H.; Kong, X.; Bosenberg, M.C.; Wiesner, T.; Rosen, N.; et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature 2015, 520, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Crusz, S.M.; Balkwill, F. Inflammation and cancer: Advances and new agents. Nat. Rev. Clin. Oncol. 2015, 12, 584–596. [Google Scholar] [CrossRef]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Strang, R.R. Preliminary report on a new anti-Parkinson drug, UCB 1549. Acta Neurol. Psychiatr. Belg. 1966, 66, 771–776. [Google Scholar] [PubMed]
- Blackhall, F.; Faivre-Finn, C. Treatment of limited small cell lung cancer: An old or new challenge? Curr. Opin. Oncol. 2011, 23, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Yeung, K.T.; Yang, J. Epithelial-mesenchymal transition in tumor metastasis. Mol. Oncol. 2017, 11, 28–39. [Google Scholar] [CrossRef] [Green Version]
- Bracun, V.; Aboumsallem, J.P.; Van Der Meer, P.; De Boer, R.A. Cardiac Biomarkers in Patients with Cancer: Considerations, Clinical Implications, and Future Avenues. Curr. Oncol. Rep. 2020, 22, 1–11. [Google Scholar] [CrossRef]
- Aghajanian, H.; Kimura, T.; Rurik, J.; Hancock, A.S.; Leibowitz, M.S.; Linares, R.; Scholler, J.; Monslow, J.; Lo, A.; Han, W.; et al. Targeting cardiac fibrosis with engineered T cells. Nature 2019, 573, 430–433. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fang, Y.; Chen, X.; Wang, Z.; Liang, X.; Zhang, T.; Liu, M.; Zhou, N.; Lv, J.; Tang, K.; et al. Gasdermin E–mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci. Immunol. 2020, 5, eaax7969. [Google Scholar] [CrossRef] [PubMed]
- Benderitter, M.; Caviggioli, F.; Chapel, A.; Coppes, R.P.; Guha, C.; Klinger, M.; Malard, O.; Stewart, F.; Tamarat, R.; Van Luijk, P.; et al. Stem Cell Therapies for the Treatment of Radiation-Induced Normal Tissue Side Effects. Antioxid. Redox Signal 2014, 21, 338–355. [Google Scholar] [CrossRef]
- Hawkins, F.; Kotton, D.N. Embryonic and Induced Pluripotent Stem Cells for Lung Regeneration. Ann. Am. Thorac. Soc. 2015, 12, S50–S53. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boopathi, E.; Thangavel, C. Dark Side of Cancer Therapy: Cancer Treatment-Induced Cardiopulmonary Inflammation, Fibrosis, and Immune Modulation. Int. J. Mol. Sci. 2021, 22, 10126. https://doi.org/10.3390/ijms221810126
Boopathi E, Thangavel C. Dark Side of Cancer Therapy: Cancer Treatment-Induced Cardiopulmonary Inflammation, Fibrosis, and Immune Modulation. International Journal of Molecular Sciences. 2021; 22(18):10126. https://doi.org/10.3390/ijms221810126
Chicago/Turabian StyleBoopathi, Ettickan, and Chellappagounder Thangavel. 2021. "Dark Side of Cancer Therapy: Cancer Treatment-Induced Cardiopulmonary Inflammation, Fibrosis, and Immune Modulation" International Journal of Molecular Sciences 22, no. 18: 10126. https://doi.org/10.3390/ijms221810126
APA StyleBoopathi, E., & Thangavel, C. (2021). Dark Side of Cancer Therapy: Cancer Treatment-Induced Cardiopulmonary Inflammation, Fibrosis, and Immune Modulation. International Journal of Molecular Sciences, 22(18), 10126. https://doi.org/10.3390/ijms221810126