
Eosinophils as Drivers of Severe Eosinophilic Asthma: Endotypes or Plasticity?


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Eosinophils as Evolving “Usual Suspects” in Asthma
3. Refining the Role of Eosinophils in Asthma through Eosinophil-Targeting Biological Therapies
4. Eosinophil Endotypes or Local Plasticity as Potential Effectors in SEA
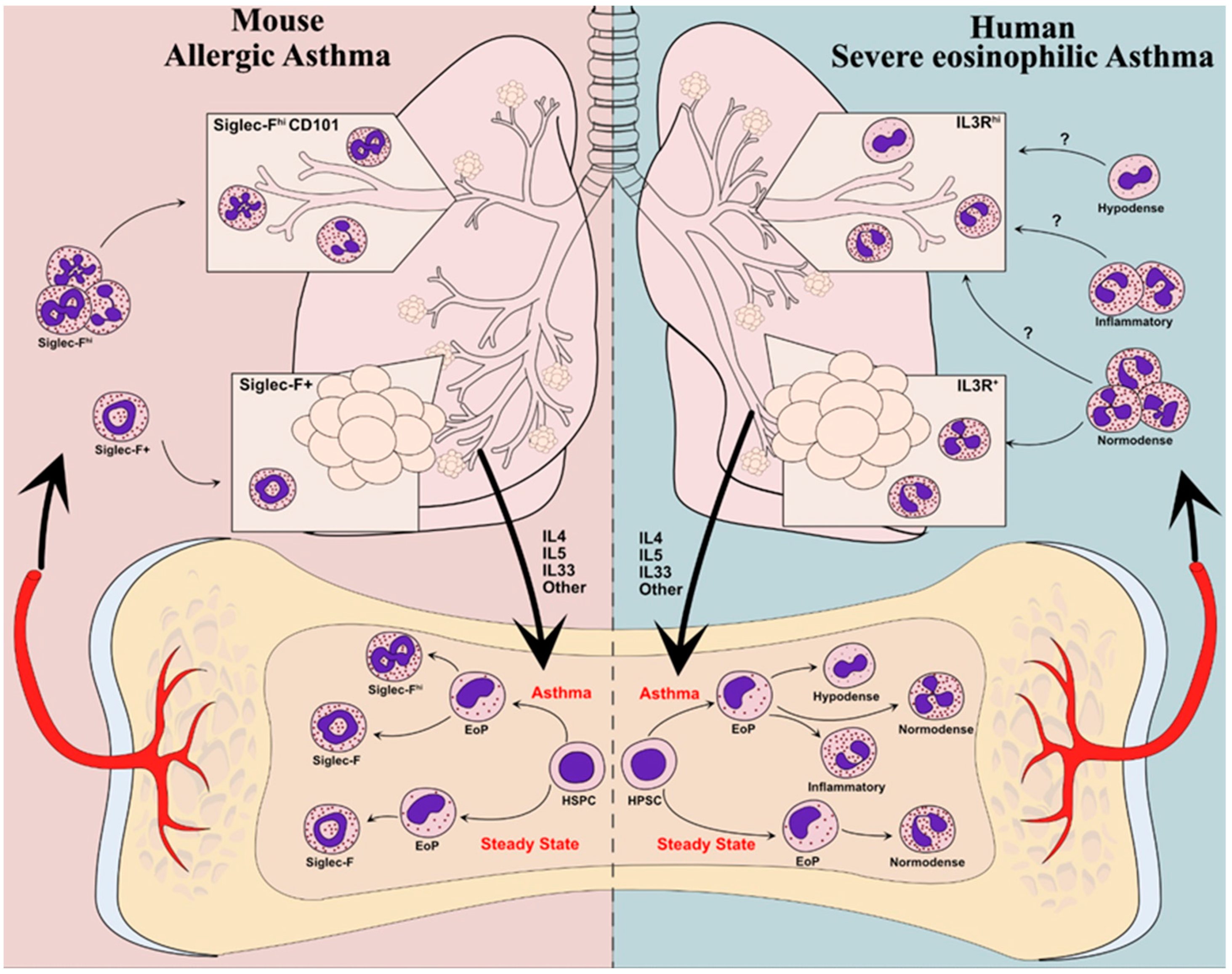
4.1. Emerging Evidence for Eosinophil “Endotypes”
4.2. Local Eosinophil Plasticity
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Soriano, J.B.; Abajobir, A.A.; Abate, K.H.; Abera, S.F.; Agrawal, A.; Ahmed, M.B.; Aichour, A.N.; Aichour, I.; Aichour, M.T.E.; Alam, K.; et al. Global, regional, and national deaths, prevalence, disability-adjusted life years, and years lived with disability for chronic obstructive pulmonary disease and asthma, 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet Respir. Med. 2017, 5, 691–706. [Google Scholar] [CrossRef] [Green Version]
- Reddel, H.K.; Bateman, E.D.; Becker, A.; Boulet, L.-P.; Cruz, A.A.; Drazen, J.M.; Haahtela, T.; Hurd, S.S.; Inoue, H.; de Jongste, J.C.; et al. A summary of the new GINA strategy: A roadmap to asthma control. Eur. Respir. J. 2015, 46, 622–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo, J.R.; Peters, S.P.; Busse, W.W. Asthma Exacerbations: Pathogenesis, Prevention, and Treatment. J. Allergy Clin. Immunol. Pract. 2017, 5, 918–927. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Brightling, C.; Pedersen, S.E.; Reddel, H.K. Asthma. Lancet 2018, 391, 783–800. [Google Scholar] [CrossRef]
- Chung, K.F.; Wenzel, S.E.; Brozek, J.L.; Bush, A.; Castro, M.; Sterk, P.J.; Adcock, I.M.; Bateman, E.D.; Bel, E.H.; Bleecker, E.R.; et al. International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. Eur. Respir. J. 2014, 43, 343–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Côté, A.; Godbout, K.; Boulet, L.-P. The management of severe asthma in 2020. Biochem. Pharmacol. 2020, 179, 114112. [Google Scholar] [CrossRef]
- Kaur, R.; Chupp, G. Phenotypes and endotypes of adult asthma: Moving toward precision medicine. J. Allergy Clin. Immunol. 2019, 144, 1–12. [Google Scholar] [CrossRef]
- Chung, K.F. Precision medicine in asthma: Linking phenotypes to targeted treatments. Curr. Opin. Pulmon. Med. 2018, 24, 1. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, P.G.; Modrek, B.; Choy, D.F.; Jia, G.; Abbas, A.R.; Ellwanger, A.; Arron, J.R.; Koth, L.L.; Fahy, V.J. T-helper Type 2–driven Inflammation Defines Major Subphenotypes of Asthma. Am. J. Respir. Crit. Care. Med. 2009, 180, 388–395. [Google Scholar] [CrossRef]
- Swedin, L.; Saarne, T.; Rehnberg, M.; Glader, P.; Niedzielska, M.; Johansson, G.; Hazon, P.; Catley, M.C. Patient stratification and the unmet need in asthma. Pharmacol. Ther. 2017, 169, 13–34. [Google Scholar] [CrossRef]
- Licona-Limon, P.; Kim, L.K.; Palm, N.W.; Flavell, R.A. TH2, allergy and group 2 innate lymphoid cells. Nat. Immunol. 2013, 14, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Hammad, H.; Lambrecht, B.N. The basic immunology of asthma. Cell 2021, 184, 2521–2522. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.L.; Scott, R.; Boyle, M.J.; Gibson, P.G. Inflammatory subtypes in asthma: Assessment and identification using induced sputum. Respirology 2006, 11, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Schleich, F.N.; Manise, M.; Sele, J.; Henket, M.; Seidel, L.; Louis, R. Distribution of sputum cellular phenotype in a large asthma cohort: Predicting factors for eosinophilic vs neutrophilic inflammation. BMC Pulm. Med. 2013, 13, 11. [Google Scholar] [CrossRef] [Green Version]
- Lambrecht, B.N.; Hammad, H. The immunology of asthma. Nat. Immunol. 2014, 16, 45–56. [Google Scholar] [CrossRef]
- Godar, M.; Blanchetot, C.; de Haard, H.; Lambrecht, B.N.; Brusselle, G. Personalized medicine with biologics for severe type 2 asthma: Current status and future prospects. mAbs 2018, 10, 34–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, R.; Bureau, F.; Desmet, C.J. Advances toward precision medicine for asthma. Biochem. Pharmacol. 2020, 179, 114081. [Google Scholar] [CrossRef]
- Kroes, J.A.; Zielhuis, S.W.; van Roon, E.N.; ten Brinke, A. Prediction of response to biological treatment with monoclonal antibodies in severe asthma. Biochem. Pharmacol. 2020, 179, 113978. [Google Scholar] [CrossRef]
- Schleich, F.N.; Chevremont, A.; Paulus, V.; Henket, M.; Manise, M.; Seidel, L.; Louis, R. Importance of concomitant local and systemic eosinophilia in uncontrolled asthma. Eur. Respir. J. 2014, 44, 97–108. [Google Scholar] [CrossRef]
- Roufosse, F. Targeting the Interleukin-5 Pathway for Treatment of Eosinophilic Conditions Other than Asthma. Front. Med. 2018, 5, 49. [Google Scholar] [CrossRef] [Green Version]
- Jacobsen, E.A.; Jackson, D.J.; Heffler, E.; Mathur, S.K.; Bredenoord, A.J.; Pavord, I.D.; Akuthota, P.; Roufosse, F.; Rothenberg, M.E. Eosinophil Knockout Humans: Uncovering the Role of Eosinophils Through Eosinophil-Directed Biological Therapies. Annu. Rev. Immunol. 2021, 39, 1. [Google Scholar] [CrossRef]
- Pavord, I.D.; Korn, S.; Howarth, P.; Bleecker, E.R.; Buhl, R.; Keene, O.N.; Ortega, H.; Chanez, P. Mepolizumab for severe eosinophilic asthma (DREAM): A multicentre, double-blind, placebo-controlled trial. Lancet 2012, 380, 651–659. [Google Scholar] [CrossRef]
- Castro, M.; Zangrilli, J.; Wechsler, M.E.; Bateman, E.D.; Brusselle, G.G.; Bardin, P.; Murphy, K.; Maspero, J.F.; O’Brien, C.; Korn, S. Reslizumab for inadequately controlled asthma with elevated blood eosinophil counts: Results from two multicentre, parallel, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet Respir. Med. 2015, 3, 355–366. [Google Scholar] [CrossRef]
- Menzies-Gow, A.; Flood-Page, P.; Sehmi, R.; Burman, J.; Hamid, Q.; Robinson, D.S.; Kay, A.B.; Denburg, J. Anti-IL-5 (mepolizumab) therapy induces bone marrow eosinophil maturational arrest and decreases eosinophil progenitors in the bronchial mucosa of atopic asthmatics. J. Allergy Clin. Immunol. 2003, 111, 714–719. [Google Scholar] [CrossRef]
- FitzGerald, J.M.; Bleecker, E.R.; Nair, P.; Korn, S.; Ohta, K.; Lommatzsch, M.; Ferguson, G.T.; Busse, W.W.; Barker, P.; Sproule, S.; et al. Benralizumab, an anti-interleukin-5 receptor α monoclonal antibody, as add-on treatment for patients with severe, uncontrolled, eosinophilic asthma (CALIMA): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2016, 388, 2128–2141. [Google Scholar] [CrossRef]
- Laviolette, M.; Gossage, D.L.; Gauvreau, G.; Leigh, R.; Olivenstein, R.; Katial, R.; Busse, W.W.; Wenzel, S.; Wu, Y.; Datta, V.; et al. Effects of benralizumab on airway eosinophils in asthmatic patients with sputum eosinophilia. J. Allergy Clin. Immunol. 2013, 132, 1086–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flood-Page, P.; Swenson, C.; Faiferman, I.; Matthews, J.; Williams, M.; Brannick, L.; Robinson, D.; Wenzel, S.; Busse, W.; Hansel, T.T.; et al. A Study to Evaluate Safety and Efficacy of Mepolizumab in Patients with Moderate Persistent Asthma. Am. J. Respir. Crit. Care Med. 2007, 176, 1062–1071. [Google Scholar] [CrossRef]
- Brusselle, G.; Germinaro, M.; Weiss, S.; Zangrilli, J. Reslizumab in patients with inadequately controlled late-onset asthma and elevated blood eosinophils. Pulm. Pharmacol. Ther. 2017, 43, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Harrison, T.W.; Chanez, P.; Menzella, F.; Canonica, G.W.; Louis, R.; Cosio, B.G.; Lugogo, N.L.; Mohan, A.; Burden, A.; McDermott, L.; et al. Onset of effect and impact on health-related quality of life, exacerbation rate, lung function, and nasal polyposis symptoms for patients with severe eosinophilic asthma treated with benralizumab (ANDHI): A randomised, controlled, phase 3b trial. Lancet Respir. Med. 2021, 9, 260–274. [Google Scholar] [CrossRef]
- Bleecker, E.R.; Wechsler, M.E.; FitzGerald, J.M.; Menzies-Gow, A.; Wu, Y.; Hirsch, I.; Goldman, M.; Newbold, P.; Zangrilli, J.G. Baseline patient factors impact on the clinical efficacy of benralizumab for severe asthma. Eur. Respir. J. 2018, 52, 1800936. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.J.; Jacobsen, E.A.; Ochkur, S.I.; McGarry, M.P.; Condjella, R.M.; Doyle, A.D.; Luo, H.; Zellner, K.R.; Protheroe, C.A.; Willetts, L.; et al. Human versus mouse eosinophils: “that which we call an eosinophil, by any other name would stain as red”. J. Allergy Clin. Immunol. 2012, 130, 572–584. [Google Scholar] [CrossRef] [Green Version]
- Stacy, N.I.; Ackerman, S.J. A tribute to eosinophils from a comparative and evolutionary perspective. J. Allergy Clin. Immunol. 2021, 147, 1115–1116. [Google Scholar] [CrossRef]
- Klion, A.D.; Ackerman, S.J.; Bochner, B.S. Contributions of Eosinophils to Human Health and Disease. Annu. Rev. Pathol. 2020, 15, 179–209. [Google Scholar] [CrossRef] [Green Version]
- Shah, K.; Ignacio, A.; McCoy, K.D.; Harris, N.L. The emerging roles of eosinophils in mucosal homeostasis. Mucosal Immunology 2020, 13, 574–583. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.J.; Jacobsen, E.A.; McGarry, M.P.; Schleimer, R.P.; Lee, N.A. Eosinophils in health and disease: The LIAR hypothesis. Clin. Exp. Allergy 2010, 40, 563–575. [Google Scholar] [CrossRef] [Green Version]
- Kopf, M.; Brombacher, F.; Hodgkin, P.D.; Ramsay, A.J.; Milbourne, E.A.; Dai, W.J.; Ovington, K.S.; Behm, C.A.; Köhler, G.; Young, I.G.; et al. IL-5-Deficient Mice Have a Developmental Defect in CD5+ B-1 Cells and Lack Eosinophilia but Have Normal Antibody and Cytotoxic T Cell Responses. Immunity 1996, 4, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Khatri, S.; Moore, W.; Gibson, P.G.; Leigh, R.; Bourdin, A.; Maspero, J.; Barros, M.; Buhl, R.; Howarth, P.; Albers, F.C.; et al. Assessment of the long-term safety of mepolizumab and durability of clinical response in patients with severe eosinophilic asthma. J. Allergy Clin. Immunol. 2019, 143, 1742–1751.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, L.; Pekkarinen, P.T.; Lakshmikanth, T.; Tan, Z.; Consiglio, C.R.; Pou, C.; Chen, Y.; Mugabo, C.H.; Nguyen, N.A.; Nowlan, K.; et al. Systems-Level Immunomonitoring from Acute to Recovery Phase of Severe COVID-19. Cell Rep. Med. 2020, 1, 100078. [Google Scholar] [CrossRef] [PubMed]
- Poznanski, S.M.; Mukherjee, M.; Zhao, N.; Huang, C.; Radford, K.; Ashkar, A.A.; Nair, P. Asthma exacerbations on benralizumab are largely non-eosinophilic. Allergy 2021, 76, 375–379. [Google Scholar] [CrossRef]
- Bohrer, A.C.; Castro, E.; Hu, Z.; Queiroz, A.T.L.; Tocheny, C.E.; Assmann, M.; Sakai, S.; Nelson, C.; Baker, P.J.; Ma, H.; et al. Eosinophils are part of the granulocyte response in tuberculosis and promote host resistance in mice. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef]
- Marichal, T.; Mesnil, C.; Bureau, F. Homeostatic Eosinophils: Characteristics and Functions. Front. Med. 2017, 4, 101. [Google Scholar] [CrossRef] [Green Version]
- Abdala-Valencia, H.; Coden, M.E.; Chiarella, S.E.; Jacobsen, E.A.; Bochner, B.S.; Lee, J.J.; Berdnikovs, S. Shaping eosinophil identity in the tissue contexts of development, homeostasis, and disease. J. Leukoc. Biol. 2018, 104, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.E.; Schwartz, L.B.; Langmack, E.L.; Halliday, J.L.; Trudeau, J.B.; Gibbs, R.L.; Chu, H.W. Evidence that severe asthma can be divided pathologically into two inflammatory subtypes with distinct physiologic and clinical characteristics. Am. J. Respir. Crit. Care Med. 1999, 160, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Green, R.H.; Brightling, C.E.; McKenna, S.; Hargadon, B.; Parker, D.; Bradding, P.; Wardlaw, A.J.; Pavord, I.D. Asthma exacerbations and sputum eosinophil counts: A randomised controlled trial. Lancet 2002, 360, 1715–1721. [Google Scholar] [CrossRef]
- Leuppi, J.D.; Salome, C.M.; Jenkins, C.R.; Anderson, S.D.; Xuan, W.E.I.; Marks, G.B.; Koskela, H.; Brannan, J.D.; Freed, R.; Andersson, M.; et al. Predictive Markers of Asthma Exacerbation during Stepwise Dose Reduction of Inhaled Corticosteroids. Am. J. Respir. Crit. Care Med. 2001, 163, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Deykin, A.; Lazarus, S.C.; Fahy, V.J.; Wechsler, M.E.; Boushey, H.A.; Chinchilli, V.M.; Craig, T.J.; Dimango, E.; Kraft, M.; Leone, F.; et al. Sputum eosinophil counts predict asthma control after discontinuation of inhaled corticosteroids. J. Allergy Clin. Immunol. 2005, 115, 720–727. [Google Scholar] [CrossRef]
- Fulkerson, P.C.; Rothenberg, M.E. Eosinophil development, disease involvement, and therapeutic suppression. Adv. Immunol. 2018, 138, 1–34. [Google Scholar]
- Jacobsen, S.E.W.; Nerlov, C. Haematopoiesis in the era of advanced single-cell technologies. Nat. Cell Biol. 2019, 21, 2–8. [Google Scholar] [CrossRef]
- Berdnikovs, S. The twilight zone: Plasticity and mixed ontogeny of neutrophil and eosinophil granulocyte subsets. Semin. Immunopathol. 2021, 43, 337–346. [Google Scholar] [CrossRef]
- Haldar, P.; Brightling, C.E.; Hargadon, B.; Gupta, S.; Monteiro, W.; Sousa, A.; Marshall, R.P.; Bradding, P.; Green, R.H.; Wardlaw, A.J.; et al. Mepolizumab and exacerbations of refractory eosinophilic asthma. N. Engl. J. Med. 2009, 360, 973–984. [Google Scholar] [CrossRef] [Green Version]
- Dunn, J.L.M.; Caldwell, J.M.; Ballaban, A.; Ben-Baruch Morgenstern, N.; Rochman, M.; Rothenberg, M.E. Bidirectional crosstalk between eosinophils and esophageal epithelial cells regulates inflammatory and remodeling processes. Mucosal Immunol. 2021, 14, 1133–1143. [Google Scholar] [CrossRef]
- Fettrelet, T.; Gigon, L.; Karaulov, A.; Yousefi, S.; Simon, H.U. The Enigma of Eosinophil Degranulation. Int. J. Mol. Sci. 2021, 22, 7091. [Google Scholar] [CrossRef]
- Melo, R.C.; Spencer, L.A.; Dvorak, A.M.; Weller, P.F. Mechanisms of eosinophil secretion: Large vesiculotubular carriers mediate transport and release of granule-derived cytokines and other proteins. J. Leukoc. Biol. 2008, 83, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Shamri, R.; Melo, R.C.; Young, K.M.; Bivas-Benita, M.; Xenakis, J.J.; Spencer, L.A.; Weller, P.F. CCL11 elicits secretion of RNases from mouse eosinophils and their cell-free granules. FASEB J. 2012, 26, 2084–2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persson, E.K.; Verstraete, K.; Heyndrickx, I.; Gevaert, E.; Aegerter, H.; Percier, J.M.; Deswarte, K.; Verschueren, K.H.G.; Dansercoer, A.; Gras, D.; et al. Protein crystallization promotes type 2 immunity and is reversible by antibody treatment. Science 2019, 364, aaw4295. [Google Scholar] [CrossRef] [PubMed]
- Ueki, S.; Konno, Y.; Takeda, M.; Moritoki, Y.; Hirokawa, M.; Matsuwaki, Y.; Honda, K.; Ohta, N.; Yamamoto, S.; Takagi, Y.; et al. Eosinophil extracellular trap cell death-derived DNA traps: Their presence in secretions and functional attributes. J. Allergy Clin. Immunol. 2016, 137, 258–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davoine, F.; Lacy, P. Eosinophil cytokines, chemokines, and growth factors: Emerging roles in immunity. Front. Immunol. 2014, 5, 570. [Google Scholar] [CrossRef] [Green Version]
- Pascual, R.M.; Peters, S.P. Airway remodeling contributes to the progressive loss of lung function in asthma: An overview. J. Allergy Clin. Immunol. 2005, 116, 477–486; quiz 487. [Google Scholar] [CrossRef]
- Redington, A.E.; Madden, J.; Frew, A.J.; Djukanovic, R.; Roche, W.R.; Holgate, S.T.; Howarth, P.H. Transforming growth factor-beta 1 in asthma. Measurement in bronchoalveolar lavage fluid. Am. J. Respir. Crit. Care Med. 1997, 156, 642–647. [Google Scholar] [CrossRef]
- Wong, D.T.; Elovic, A.; Matossian, K.; Nagura, N.; McBride, J.; Chou, M.Y.; Gordon, J.R.; Rand, T.H.; Galli, S.J.; Weller, P.F. Eosinophils from patients with blood eosinophilia express transforming growth factor beta 1. Blood 1991, 78, 2702–2707. [Google Scholar] [CrossRef] [Green Version]
- Januskevicius, A.; Vaitkiene, S.; Gosens, R.; Janulaityte, I.; Hoppenot, D.; Sakalauskas, R.; Malakauskas, K. Eosinophils enhance WNT-5a and TGF-beta1 genes expression in airway smooth muscle cells and promote their proliferation by increased extracellular matrix proteins production in asthma. BMC Pulm. Med. 2016, 16, 94. [Google Scholar] [CrossRef] [Green Version]
- Flood-Page, P.T.; Menzies-Gow, A.N.; Kay, A.B.; Robinson, D.S. Eosinophil’s role remains uncertain as anti-interleukin-5 only partially depletes numbers in asthmatic airway. Am. J. Respir. Crit. Care Med. 2003, 167, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.J.; McGarry, M.P.; Farmer, S.C.; Denzler, K.L.; Larson, K.A.; Carrigan, P.E.; Brenneise, I.E.; Horton, M.A.; Haczku, A.; Gelfand, E.W.; et al. Interleukin-5 expression in the lung epithelium of transgenic mice leads to pulmonary changes pathognomonic of asthma. J. Exp. Med. 1997, 185, 2143–2156. [Google Scholar] [CrossRef]
- Kariyawasam, H.H.; Robinson, D.S. The role of eosinophils in airway tissue remodelling in asthma. Curr. Opin. Immunol. 2007, 19, 681–686. [Google Scholar] [CrossRef]
- Pavord, I.D.; Menzies-Gow, A.; Buhl, R.; Chanez, P.; Dransfield, M.; Lugogo, N.; Keene, O.N.; Bradford, E.S.; Yancey, S.W. Clinical Development of Mepolizumab for the Treatment of Severe Eosinophilic Asthma: On the Path to Personalized Medicine. J. Allergy Clin. Immunol. Pract. 2020, 9, 1121–1132.e7. [Google Scholar] [CrossRef]
- Leckie, M.J.; Brinke, T.A.; Khan, J.; Diamant, Z.; O’Connor, B.J.; Walls, C.M.; Mathur, A.K.; Cowley, H.C.; Chung, K.F.; Djukanovic, R.; et al. Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyper-responsìveness, and the late asthmatic response. Lancet 2000, 356, 2144–2148. [Google Scholar] [CrossRef]
- Bleecker, E.R.; FitzGerald, J.M.; Chanez, P.; Papi, A.; Weinstein, S.F.; Barker, P.; Sproule, S.; Gilmartin, G.; Aurivillius, M.; Werkström, V.; et al. Efficacy and safety of benralizumab for patients with severe asthma uncontrolled with high-dosage inhaled corticosteroids and long-acting β2-agonists (SIROCCO): A randomised, multicentre, placebo-controlled phase 3 trial. Lancet 2016, 388, 2115–2127. [Google Scholar] [CrossRef]
- Bjermer, L.; Lemiere, C.; Maspero, J.; Weiss, S.; Zangrilli, J.; Germinaro, M. Reslizumab for Inadequately Controlled Asthma With Elevated Blood Eosinophil Levels: A Randomized Phase 3 Study. Chest 2016, 150, 789–798. [Google Scholar] [CrossRef] [Green Version]
- Chipps, B.E.; Hirsch, I.; Trudo, F.; Alacqua, M.; Zangrilli, J.G. Benralizumab efficacy for patients with fixed airflow obstruction and severe, uncontrolled eosinophilic asthma. Ann. Allergy Asthma Immunol. 2020, 124, 79–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corren, J.; Weinstein, S.; Janka, L.; Zangrilli, J.; Garin, M. Phase 3 Study of Reslizumab in Patients With Poorly Controlled Asthma: Effects Across a Broad Range of Eosinophil Counts. Chest 2016, 150, 799–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega, H.G.; Yancey, S.W.; Mayer, B.; Gunsoy, N.B.; Keene, O.N.; Bleecker, E.R.; Brightling, C.E.; Pavord, I.D. Severe eosinophilic asthma treated with mepolizumab stratified by baseline eosinophil thresholds: A secondary analysis of the DREAM and MENSA studies. Lancet Respir. Med. 2016, 4, 549–556. [Google Scholar] [CrossRef]
- Albers, F.C.; Licskai, C.; Chanez, P.; Bratton, D.J.; Bradford, E.S.; Yancey, S.W.; Kwon, N.; Quirce, S. Baseline blood eosinophil count as a predictor of treatment response to the licensed dose of mepolizumab in severe eosinophilic asthma. Respir. Med. 2019, 159, 105806. [Google Scholar] [CrossRef]
- Shrimanker, R.; Keene, O.; Hynes, G.; Wenzel, S.; Yancey, S.; Pavord, I.D. Prognostic and Predictive Value of Blood Eosinophil Count, Fractional Exhaled Nitric Oxide, and Their Combination in Severe Asthma: A Post Hoc Analysis. Am. J. Respir. Crit. Care Med. 2019, 200, 1308–1312. [Google Scholar] [CrossRef] [PubMed]
- Nair, P.; Bardin, P.; Humbert, M.; Murphy, K.R.; Hickey, L.; Garin, M.; Vanlandingham, R.; Chanez, P. Efficacy of Intravenous Reslizumab in Oral Corticosteroid–Dependent Asthma. J. Allergy Clin. Immunol. Pract. 2020, 8, 555–564. [Google Scholar] [CrossRef] [PubMed]
- FitzGerald, J.M.; Bleecker, E.R.; Menzies-Gow, A.; Zangrilli, J.G.; Hirsch, I.; Metcalfe, P.; Newbold, P.; Goldman, M. Predictors of enhanced response with benralizumab for patients with severe asthma: Pooled analysis of the SIROCCO and CALIMA studies. Lancet Respir. Med. 2018, 6, 51–64. [Google Scholar] [CrossRef]
- Chipps, B.E.; Newbold, P.; Hirsch, I.; Trudo, F.; Goldman, M. Benralizumab efficacy by atopy status and serum immunoglobulin E for patients with severe, uncontrolled asthma. Ann. Allergy Asthma Immunol. 2018, 120, 504–511.e4. [Google Scholar] [CrossRef] [Green Version]
- Humbert, M.; Albers, F.C.; Bratton, D.J.; Yancey, S.W.; Liu, M.C.; Hozawa, S.; Llanos, J.-P.; Kwon, N. Effect of mepolizumab in severe eosinophilic asthma according to omalizumab eligibility. Respir. Med. 2019, 154, 69–75. [Google Scholar] [CrossRef]
- Dhariwal, J.; Hearn, A.P.; Kavanagh, J.E.; d’Ancona, G.; Green, L.; Fernandes, M.; Thomson, L.; Roxas, C.; Kent, B.D.; Nanzer, A.M.; et al. Real-World Effectiveness of Anti–IL-5/5R Therapy in Severe Atopic Eosinophilic Asthma with Fungal Sensitization. J. Allergy Clin. Immunol. Pract. 2021, 9, 2315–2320. [Google Scholar] [CrossRef]
- Prazma, C.M.; Idzko, M.; Douglass, J.A.; Bourdin, A.; Mallett, S.; Albers, F.C.; Yancey, S.W. Response to Mepolizumab Treatment in Patients with Severe Eosinophilic Asthma and Atopic Phenotypes. J. Asthma Allergy 2021, 14, 675–683. [Google Scholar] [CrossRef]
- Albers, F.C.; Papi, A.; Taillé, C.; Bratton, D.J.; Bradford, E.S.; Yancey, S.W.; Kwon, N. Mepolizumab reduces exacerbations in patients with severe eosinophilic asthma, irrespective of body weight/body mass index: Meta-analysis of MENSA and MUSCA. Respir. Res. 2019, 20, 169. [Google Scholar] [CrossRef] [Green Version]
- McDowell, P.J.; Diver, S.; Yang, F.; Borg, C.; Busby, J.; Brown, V.; Shrimanker, R.; Cox, C.; Brightling, C.E.; Chaudhuri, R.; et al. The inflammatory profile of exacerbations in patients with severe refractory eosinophilic asthma receiving mepolizumab (the MEX study): A prospective observational study. Lancet Respir. Med. 2021. [Google Scholar] [CrossRef]
- Kelly, E.A.; Esnault, S.; Liu, L.Y.; Evans, M.D.; Johansson, M.W.; Mathur, S.; Mosher, D.F.; Denlinger, L.C.; Jarjour, N.N. Mepolizumab Attenuates Airway Eosinophil Numbers, but Not Their Functional Phenotype, in Asthma. Am. J. Respir. Crit. Care Med. 2017, 196, 1385–1395. [Google Scholar] [CrossRef]
- Van Hulst, G.; Jorssen, J.; Jacobs, N.; Henket, M.; Louis, R.; Schleich, F.; Bureau, F.; Desmet, C.J. Anti-IL-5 mepolizumab minimally influences residual blood eosinophils in severe asthma. Eur. Respir. J. 2021, in press. [Google Scholar] [CrossRef]
- Kraft, M.; Brusselle, G.; Mark FitzGerald, J.; Pavord, I.D.; Keith, M.; Fagerås, M.; Garcia Gil, E.; Hirsch, I.; Goldman, M.; Colice, G. Patient characteristics, biomarkers, and exacerbation risk in severe, uncontrolled asthma. Eur. Respir. J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Zeiger, R.S.; Schatz, M.; Zhang, F.; Crawford, W.W.; Kaplan, M.S.; Roth, R.M.; Chen, W. Elevated exhaled nitric oxide is a clinical indicator of future uncontrolled asthma in asthmatic patients on inhaled corticosteroids. J. Allergy Clin. Immunol. 2011, 128, 412–414. [Google Scholar] [CrossRef] [PubMed]
- Reyes, M.; Filbin, M.R.; Bhattacharyya, R.P.; Sonny, A.; Mehta, A.; Billman, K.; Kays, K.R.; Pinilla-Vera, M.; Benson, M.E.; Cosimi, L.A.; et al. Plasma from patients with bacterial sepsis or severe COVID-19 induces suppressive myeloid cell production from hematopoietic progenitors in vitro. Sci. Transl. Med. 2021, 13, eabe9599. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Hashimoto, A.; Dweep, H.; Sanseviero, E.; De Leo, A.; Tcyganov, E.; Kossenkov, A.; Mulligan, C.; Nam, B.; Masters, G.; et al. Analysis of classical neutrophils and polymorphonuclear myeloid-derived suppressor cells in cancer patients and tumor-bearing mice. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, H.; Mizuno, S.-i.; Mayfield, R.; Shigematsu, H.; Arinobu, Y.; Seed, B.; Gurish, M.F.; Takatsu, K.; Akashi, K. Identification of eosinophil lineage–committed progenitors in the murine bone marrow. J. Exp. Med. 2005, 201, 1891–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, Y.; Iwasaki, H.; Kohno, K.; Yoshimoto, G.; Kikushige, Y.; Okeda, A.; Uike, N.; Niiro, H.; Takenaka, K.; Nagafuji, K.; et al. Identification of the human eosinophil lineage-committed progenitor: Revision of phenotypic definition of the human common myeloid progenitor. J. Exp. Med. 2009, 206, 183–193. [Google Scholar] [CrossRef]
- Drissen, R.; Buza-Vidas, N.; Woll, P.; Thongjuea, S.; Gambardella, A.; Giustacchini, A.; Mancini, E.; Zriwil, A.; Lutteropp, M.; Grover, A.; et al. Distinct myeloid progenitor-differentiation pathways identified through single-cell RNA sequencing. Nat. Immunol. 2016, 17, 666–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drissen, R.; Thongjuea, S.; Theilgaard-Mönch, K.; Nerlov, C. Identification of two distinct pathways of human myelopoiesis. Sci. Immunol. 2019, 4, eaau7148. [Google Scholar] [CrossRef]
- Tusi, B.K.; Wolock, S.L.; Weinreb, C.; Hwang, Y.; Hidalgo, D.; Zilionis, R.; Waisman, A.; Huh, J.R.; Klein, A.M.; Socolovsky, M. Population snapshots predict early haematopoietic and erythroid hierarchies. Nature 2018, 555, 54. [Google Scholar] [CrossRef]
- Weinreb, C.; Rodriguez-Fraticelli, A.; Camargo, F.D.; Klein, A.M. Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science 2020, 367, eaaw3381. [Google Scholar] [CrossRef]
- Jeong, B.M.; Walker, M.T.; Rodriguez, R.; Coden, M.E.; Nagasaka, R.; Doan, T.C.; Politanska, Y.; Abdala-Valencia, H.; Berdnikovs, S. More than neutrophils: Lin(+)Ly6G(+)IL-5Rα(+) multipotent myeloid cells (MMCs) are dominant in normal murine bone marrow and retain capacity to differentiate into eosinophils and monocytes. J. Leukoc. Biol. 2021. [Google Scholar] [CrossRef]
- Mesnil, C.; Raulier, S.; Paulissen, G.; Xiao, X.; Birrell, M.A.; Pirottin, D.; Janss, T.; Starkl, P.; Ramery, E.; Henket, M.; et al. Lung-resident eosinophils represent a distinct regulatory eosinophil subset. J. Clin. Investig. 2016, 126, 3279–3295. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Weng, Q.-Y.; Zhou, L.-R.; Cao, C.; Li, F.; Wu, Y.-F.; Wu, Y.-P.; Li, M.; Hu, Y.; Shen, J.-X.; et al. Homeostatic and early-recruited CD101− eosinophils suppress endotoxin-induced acute lung injury. Eur. Respir. J. 2020, 56, 1902354. [Google Scholar] [CrossRef] [PubMed]
- Venkateshaiah, S.U.; Mishra, A.; Manohar, M.; Verma, A.K.; Rajavelu, P.; Niranjan, R.; Wild, L.G.; Parada, N.A.; Blecker, U.; Lasky, J.A.; et al. A critical role for IL-18 in transformation and maturation of naive eosinophils to pathogenic eosinophils. J. Allergy Clin. Immunol. 2018, 142, 301–305. [Google Scholar] [CrossRef] [Green Version]
- Fairfax, K.A.; Bolden, J.E.; Robinson, A.J.; Lucas, E.C.; Baldwin, T.M.; Ramsay, K.A.; Cole, R.; Hilton, D.J.; de Graaf, C.A. Transcriptional profiling of eosinophil subsets in interleukin-5 transgenic mice. J. Leukoc. Biol. 2018, 104, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Dunnette, S.L.; Reed, C.E.; Ackerman, S.J.; Peters, M.S.; Gleich, G.J. Increased numbers of hypodense eosinophils in the blood of patients with bronchial asthma. Am. Rev. Respir. Dis. 1985, 132, 981–985. [Google Scholar] [CrossRef] [PubMed]
- Fulkerson, P.C.; Schollaert, K.L.; Bouffi, C.; Rothenberg, M.E. IL-5 Triggers a Cooperative Cytokine Network That Promotes Eosinophil Precursor Maturation. J. Immunol. 2014, 193, 4043–4052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, M.S.; Gleich, G.J.; Dunnette, S.L.; Fukuda, T. Ultrastructural study of eosinophils from patients with the hypereosinophilic syndrome: A morphological basis of hypodense eosinophils. Blood 1988, 71, 780–785. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, T.; Gleich, G. Heterogeneity of human eosinophils. J. Allergy Clin. Immunol. 1989, 83, 369–373. [Google Scholar] [CrossRef]
- Garcia-Romo, G.S.; Caielli, S.; Vega, B.; Connolly, J.; Allantaz, F.; Xu, Z.; Punaro, M.; Baisch, J.; Guiducci, C.; Coffman, R.L.; et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 2011, 3, 73ra20. [Google Scholar] [CrossRef] [Green Version]
- Chatfield, S.M.; Thieblemont, N.; Witko-Sarsat, V. Expanding Neutrophil Horizons: New Concepts in Inflammation. J. Innate. Immun. 2018, 10, 422–431. [Google Scholar] [CrossRef]
- Van Hulst, G.; Batugedara, H.M.; Jorssen, J.; Louis, R.; Bureau, F.; Desmet, C.J. Eosinophil diversity in asthma. Biochem. Pharmacol. 2020, 179, 113963. [Google Scholar] [CrossRef] [PubMed]
- Barnig, C.; Alsaleh, G.; Jung, N.; Dembélé, D.; Paul, N.; Poirot, A.; Uring-Lambert, B.; Georgel, P.; de Blay, F.; Bahram, S. Circulating Human Eosinophils Share a Similar Transcriptional Profile in Asthma and Other Hypereosinophilic Disorders. PLoS ONE 2015, 10, e0141740. [Google Scholar] [CrossRef] [PubMed]
- Abdala Valencia, H.; Loffredo, L.F.; Misharin, V.A.; Berdnikovs, S. Phenotypic plasticity and targeting of Siglec-FhighCD11clow eosinophils to the airway in a murine model of asthma. Allergy 2016, 71, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Svedberg, F.R. Does tissue imprinting restrict macrophage plasticity? Nat. Immunol. 2021, 22, 118–127. [Google Scholar] [CrossRef]
- Prin, L.; Charon, J.; Capron, M.; Gosset, P.; Taelman, H.; Tonnel, A.B.; Capron, A. Heterogeneity of human eosinophils. II. Variability of respiratory burst activity related to cell density. Clin. Exp. Immunol. 1984, 57, 735–742. [Google Scholar]
- Kroegel, C.; Liu, M.; Hubbard, W.; Lichtenstein, L.; Bochner, B. Blood and bronchoalveolar eosinophils in allergic subjects after segmental antigen challenge: Surface phenotype, density heterogeneity, and prostanoid production. J. Allergy Clin. Immunol. 1994, 93, 725–734. [Google Scholar] [CrossRef]
- Drake, M.G.; Lebold, K.M.; Roth-Carter, Q.R.; Pincus, A.B.; Blum, E.D.; Proskocil, B.J.; Jacoby, D.B.; Fryer, A.D.; Nie, Z. Eosinophil and airway nerve interactions in asthma. J. Leukoc. Biol. 2018, 104, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Drake, M.G.; Scott, G.D.; Blum, E.D.; Lebold, K.M.; Nie, Z.; Lee, J.J.; Fryer, A.D.; Costello, R.W.; Jacoby, D.B. Eosinophils increase airway sensory nerve density in mice and in human asthma. Sci. Transl. Med. 2018, 10, eaar8477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebold, K.M.; Drake, M.G.; Hales-Beck, L.B.; Fryer, A.D.; Jacoby, D.B. IL-5 Exposure In Utero Increases Lung Nerve Density and Airway Reactivity in Adult Offspring. Am. J. Respir. Cell Mol. Biol. 2019, 62, 493–502. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Hulst, G.; Bureau, F.; Desmet, C.J. Eosinophils as Drivers of Severe Eosinophilic Asthma: Endotypes or Plasticity? Int. J. Mol. Sci. 2021, 22, 10150. https://doi.org/10.3390/ijms221810150
Van Hulst G, Bureau F, Desmet CJ. Eosinophils as Drivers of Severe Eosinophilic Asthma: Endotypes or Plasticity? International Journal of Molecular Sciences. 2021; 22(18):10150. https://doi.org/10.3390/ijms221810150
Chicago/Turabian StyleVan Hulst, Glenn, Fabrice Bureau, and Christophe J. Desmet. 2021. "Eosinophils as Drivers of Severe Eosinophilic Asthma: Endotypes or Plasticity?" International Journal of Molecular Sciences 22, no. 18: 10150. https://doi.org/10.3390/ijms221810150
APA StyleVan Hulst, G., Bureau, F., & Desmet, C. J. (2021). Eosinophils as Drivers of Severe Eosinophilic Asthma: Endotypes or Plasticity? International Journal of Molecular Sciences, 22(18), 10150. https://doi.org/10.3390/ijms221810150