
Dynamic Crosstalk between Vascular Smooth Muscle Cells and the Aged Extracellular Matrix

 , ,
, , {kind=link}
{kind=link}
Abstract
:1. Introduction
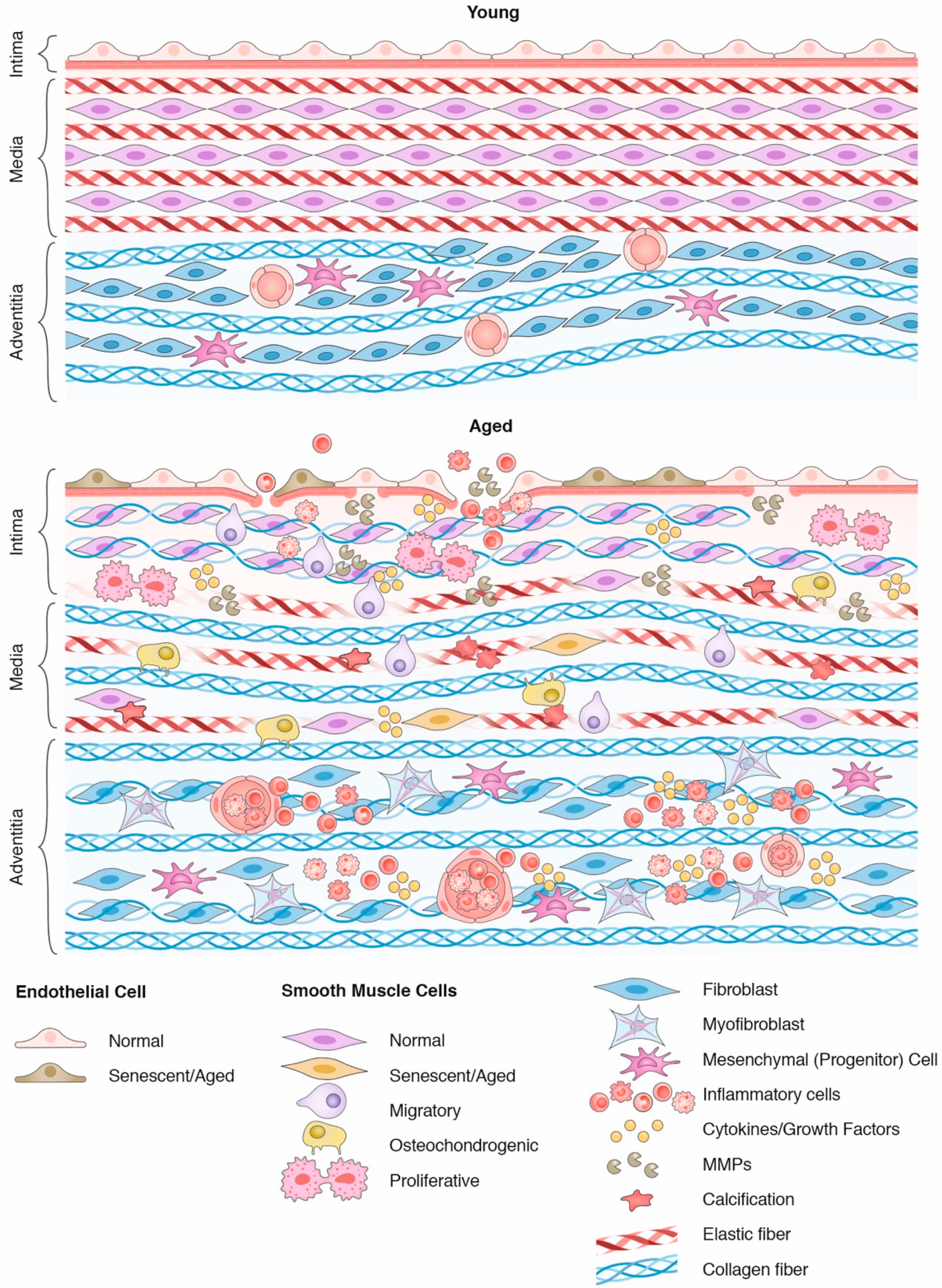
2. Aging and Vascular Extracellular Matrix Remodeling
2.1. Basic Organization of the Vascular Wall
2.2. Vascular Aging
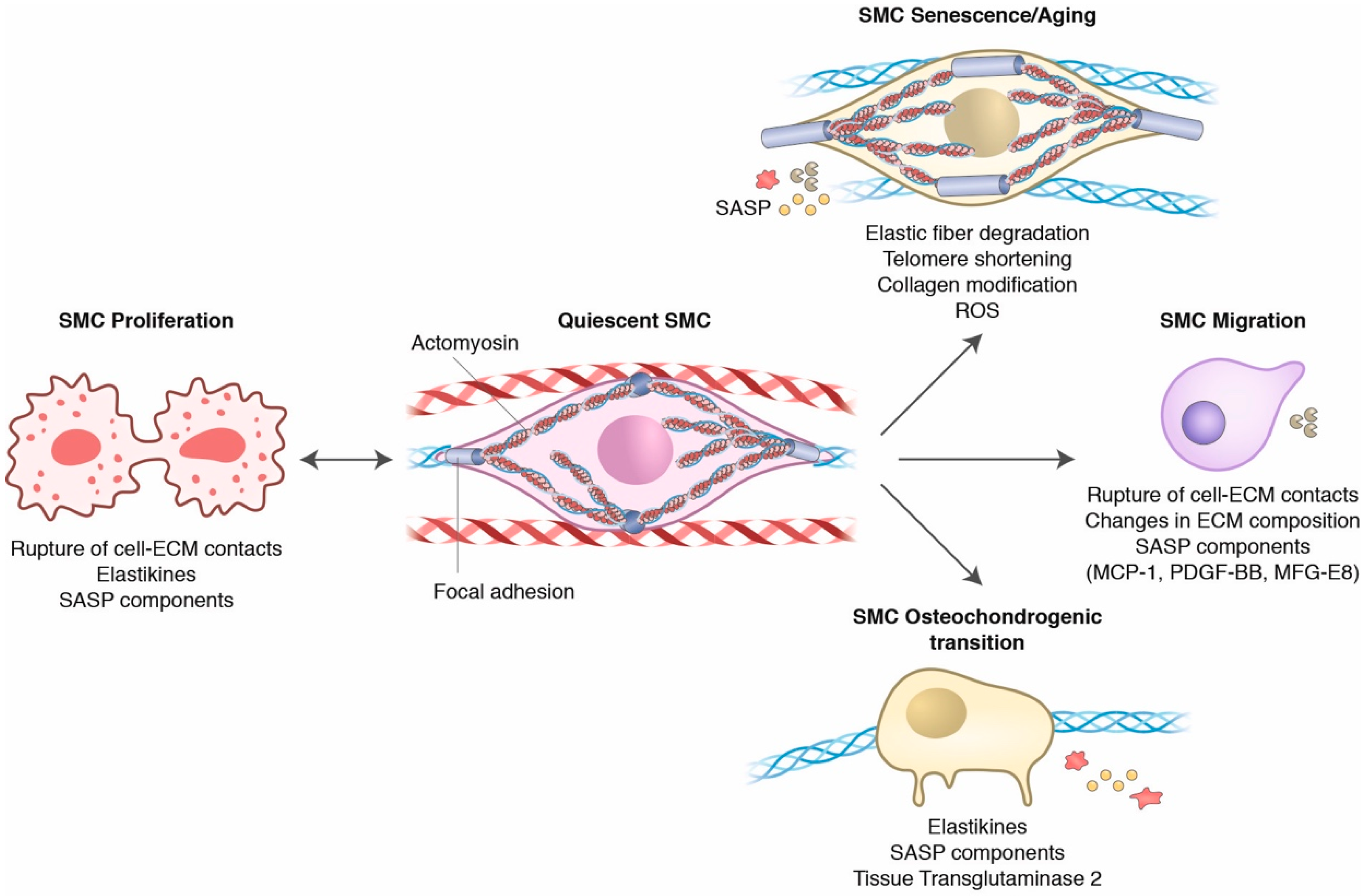
3. Dynamic Crosstalk between Smooth Muscle Cells and the Aged Extracellular Matrix
3.1. The Vascular Homeostatic Crosstalk of SMCs and the ECM
3.2. Aging Influences on SMC and ECM Crosstalk
3.3. Elastic Fiber Fragmentation and SMC Phenotypical Changes
3.4. ECM Changes and the Activation of TGF-β Signaling in SMCs
4. Cellular Senescence in Aging
4.1. Collagen in Promotion of SMC Senescence
4.2. The Senescence-Associated Secretory Phenotype (SASP)
5. Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mecham, R.P. Overview of Extracellular Matrix. Curr. Protoc. Cell Biol. 2012, 57, 10.1.1–10.1.14. [Google Scholar] [CrossRef]
- Hynes, R.O. The Extracellular Matrix: Not Just Pretty Fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [Green Version]
- Kang, L.S.; Kim, S.; Dominguez, J.M.; Sindler, A.L.; Dick, G.M.; Muller-Delp, J.M. Aging and muscle fiber type alter K + channel contributions to the myogenic response in skeletal muscle arterioles. J. Appl. Physiol. 2009, 107, 389–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, P.; Mora Solis, F.R.; Dominguez, J.M.; Spier, S.A.; Donato, A.J.; Delp, M.D.; Muller-Delp, J.M. Exercise training reverses aging-induced impairment of myogenic constriction in skeletal muscle arterioles. J. Appl. Physiol. 2015, 118, 904–911. [Google Scholar] [CrossRef] [Green Version]
- Gaballa, M.A.; Jacob, C.T.; Raya, T.E.; Liu, J.; Simon, B.; Goldman, S. Large Artery Remodeling During Aging. Hypertension 1998, 32, 437–443. [Google Scholar] [CrossRef]
- North, B.J.; Sinclair, D.A. The Intersection Between Aging and Cardiovascular Disease. Circ. Res. 2012, 110, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Fukazawa, R.; Ikegam, E.; Watanabe, M.; Hajikano, M.; Kamisago, M.; Katsube, Y.; Yamauchi, H.; Ochi, M.; Ogawa, S. Coronary Artery Aneurysm Induced by Kawasaki Disease in Children Show Features Typical Senescence. Circ. J. 2007, 71, 709–715. [Google Scholar] [CrossRef] [Green Version]
- Sueta, D.; Koibuchi, N.; Hasegawa, Y.; Toyama, K.; Uekawa, K.; Katayama, T.; Ma, M.; Nakagawa, T.; Waki, H.; Maeda, M.; et al. Blood pressure variability, impaired autonomic function and vascular senescence in aged spontaneously hypertensive rats are ameliorated by angiotensin blockade. Atherosclerosis 2014, 236, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.J.; Pham, S.; Wei, Y.; Mateo, D.; St-Pierre, M.; Fletcher, T.M.; Vazquez-Padron, R.I. Stress-induced senescence exaggerates postinjury neointimal formation in the old vasculature. Am. J. Physiol. Circ. Physiol. 2010, 298, H66–H74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, C.; Gorenne, I.; Scott, S.; Figg, N.; Kirkpatrick, P.; Ritchie, A.; Goddard, M.; Bennett, M. Vascular Smooth Muscle Cells Undergo Telomere-Based Senescence in Human Atherosclerosis. Circ. Res. 2006, 99, 156–164. [Google Scholar] [CrossRef] [Green Version]
- Brodsky, S.V.; Gealekman, O.; Chen, J.; Zhang, F.; Togashi, N.; Crabtree, M.; Gross, S.S.; Nasjletti, A.; Goligorsky, M.S. Prevention and Reversal of Premature Endothelial Cell Senescence and Vasculopathy in Obesity-Induced Diabetes by Ebselen. Circ. Res. 2004, 94, 377–384. [Google Scholar] [CrossRef] [Green Version]
- Nagy, N.; Freudenberger, T.; Melchior-Becker, A.; Röck, K.; ter Braak, M.; Jastrow, H.; Kinzig, M.; Lucke, S.; Suvorava, T.; Kojda, G.; et al. Inhibition of Hyaluronan Synthesis Accelerates Murine Atherosclerosis. Circulation 2010, 122, 2313–2322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dogné, S.; Flamion, B.; Caron, N. Endothelial glycocalyx as a shield against diabetic vascular complications: Involvement of hyaluronan and hyaluronidases. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1427–1439. [Google Scholar] [CrossRef]
- Wagner, D.D. Cell Biology of von Willebrand Factor. Annu. Rev. Cell Biol. 1990, 6, 217–242. [Google Scholar] [CrossRef] [PubMed]
- Davis, G.E.; Senger, D.R. Endothelial Extracellular Matrix. Circ. Res. 2005, 97, 1093–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauly, R.R.; Passaniti, A.; Crow, M.; Kinsella, J.L.; Papadopoulos, N.; Monticone, R.; Lakatta, E.G.; Martin, G.R. Experimental models that mimic the differentiation and dedifferentiation of vascular cells. Circulation 1992, 86, III68–III73. [Google Scholar]
- Dinardo, C.L.; Venturini, G.; Zhou, E.H.; Watanabe, I.S.; Campos, L.C.G.; Dariolli, R.; da Motta-Leal-Filho, J.M.; Carvalho, V.M.; Cardozo, K.H.M.; Krieger, J.E.; et al. Variation of mechanical properties and quantitative proteomics of VSMC along the arterial tree. Am. J. Physiol. Circ. Physiol. 2014, 306, H505–H516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacolley, P.; Regnault, V.; Segers, P.; Laurent, S. Vascular Smooth Muscle Cells and Arterial Stiffening: Relevance in Development, Aging, and Disease. Physiol. Rev. 2017, 97, 1555–1617. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.; Glomset, J.A. Atherosclerosis and the Arterial Smooth Muscle Cell. Science 1973, 180, 1332–1339. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, Z.; Torsney, E.; Afzal, A.R.; Davison, F.; Metzler, B.; Xu, Q. Abundant progenitor cells in the adventitia contribute to atheroscleroses of vein grafts in ApoE-deficient mice. J. Clin. Investig. 2004, 113, 1258–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karrer, H.E. An electron microscope study of the aorta in young and in aging mice. J. Ultrastruct. Res. 1961, 5, 1–27. [Google Scholar] [CrossRef]
- Majesky, M.W. Vascular Development. Arterioscler. Thromb. Vasc. Biol. 2018, 38, E17–E24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagai, Y.; Metter, E.J.; Earley, C.J.; Kemper, M.K.; Becker, L.C.; Lakatta, E.G.; Fleg, J.L. Increased Carotid Artery Intimal-Medial Thickness in Asymptomatic Older Subjects With Exercise-Induced Myocardial Ischemia. Circulation 1998, 98, 1504–1509. [Google Scholar] [CrossRef] [Green Version]
- Virmani, R.; Avolio, A.P.; Mergner, W.J.; Robinowitz, M.; Herderick, E.E.; Cornhill, J.F.; Guo, S.Y.; Liu, T.H.; Ou, D.Y.; O’Rourke, M. Effect of aging on aortic morphology in populations with high and low prevalence of hypertension and atherosclerosis. Comparison between occidental and Chinese communities. Am. J. Pathol. 1991, 139, 1119–1129. [Google Scholar]
- Li, Z.; Froehlich, J.; Galis, Z.S. Increased Expression of Matrix Metalloproteinase-2 in the Thickened Intima of Aged Rats. Hypertension 1999, 33, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Caro, C.G.; Fitz-Gerald, J.M.; Schroter, R.C. Arterial Wall Shear and Distribution of Early Atheroma in Man. Nature 1969, 223, 1159–1161. [Google Scholar] [CrossRef]
- Hahn, C.; Schwartz, M.A. Mechanotransduction in vascular physiology and atherogenesis. Nat. Rev. Mol. Cell Biol. 2009, 10, 53–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maleszewska, M.; Vanchin, B.; Harmsen, M.C.; Krenning, G. The decrease in histone methyltransferase EZH2 in response to fluid shear stress alters endothelial gene expression and promotes quiescence. Angiogenesis 2016, 19, 9–24. [Google Scholar] [CrossRef] [Green Version]
- Pedrigi, R.M.; Mehta, V.; Bovens, S.M.; Mohri, Z.; Poulsen, C.B.; Gsell, W.; Tremoleda, J.L.; Towhidi, L.; De Silva, R.; Petretto, E.; et al. Influence of shear stress magnitude and direction on atherosclerotic plaque composition. R. Soc. Open Sci. 2016, 3, 160588. [Google Scholar] [CrossRef] [Green Version]
- Zarins, C.K.; Giddens, D.P.; Bharadvaj, B.K.; Sottiurai, V.S.; Mabon, R.F.; Glagov, S. Carotid bifurcation atherosclerosis. Quantitative correlation of plaque localization with flow velocity profiles and wall shear stress. Circ. Res. 1983, 53, 502–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souilhol, C.; Serbanovic-Canic, J.; Fragiadaki, M.; Chico, T.J.; Ridger, V.; Roddie, H.; Evans, P.C. Endothelial responses to shear stress in atherosclerosis: A novel role for developmental genes. Nat. Rev. Cardiol. 2020, 17, 52–63. [Google Scholar] [CrossRef]
- Ajami, N.E.; Gupta, S.; Maurya, M.R.; Nguyen, P.; Li, J.Y.S.; Shyy, J.Y.J.; Chen, Z.; Chien, S.; Subramaniam, S. Systems biology analysis of longitudinal functional response of endothelial cells to shear stress. Proc. Natl. Acad. Sci. USA 2017, 114, 10990–10995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, K.; Joshi, D.; Timalsina, S.; Schwartz, M.A. Early events in endothelial flow sensing. Cytoskeleton 2021, cm.21652. [Google Scholar] [CrossRef]
- Levy, B.I.; Benessiano, J.; Poitevin, P.; Safar, M.E. Endothelium-dependent mechanical properties of the carotid artery in WKY and SHR. Role of angiotensin converting enzyme inhibition. Circ. Res. 1990, 66, 321–328. [Google Scholar] [CrossRef] [Green Version]
- Levy, B.I.; Michel, J.B.; Salzmann, J.L.; Azizi, M.; Poitevin, P.; Safar, M.; Camilleri, J.P. Effects of chronic inhibition of converting enzyme on mechanical and structural properties of arteries in rat renovascular hypertension. Circ. Res. 1988, 63, 227–239. [Google Scholar] [CrossRef] [Green Version]
- Harrison, D.G. Cellular and molecular mechanisms of endothelial cell dysfunction. J. Clin. Investig. 1997, 100, 2153–2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donato, A.J.; Gano, L.B.; Eskurza, I.; Silver, A.E.; Gates, P.E.; Jablonski, K.; Seals, D.R. Vascular endothelial dysfunction with aging: Endothelin-1 and endothelial nitric oxide synthase. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H425–H432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, Oxidants, and Aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Epstein, F.H.; Gibbons, G.H.; Dzau, V.J. The Emerging Concept of Vascular Remodeling. N. Engl. J. Med. 1994, 330, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- Schiffrin, E. Remodeling of resistance arteries in essential hypertension and effects of antihypertensive treatment. Am. J. Hypertens. 2004, 17, 1192–1200. [Google Scholar] [CrossRef] [Green Version]
- Meyer, T. De Studying telomeres in a longitudinal population based study. Front. Biosci. 2008, 13, 2960. [Google Scholar] [CrossRef] [Green Version]
- Matsui-Hirai, H.; Hayashi, T.; Yamamoto, S.; Ina, K.; Maeda, M.; Kotani, H.; Iguchi, A.; Ignarro, L.J.; Hattori, Y. Dose-dependent modulatory effects of insulin on glucose-induced endothelial senescence in vitro and in vivo: A relationship between telomeres and nitric oxide. J. Pharmacol. Exp. Ther. 2011, 337, 591–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donato, A.J.; Black, A.D.; Jablonski, K.L.; Gano, L.B.; Seals, D.R. Aging is associated with greater nuclear NFκB, reduced IκBα, and increased expression of proinflammatory cytokines in vascular endothelial cells of healthy humans. Aging Cell 2008, 7, 805–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donato, A.J.; Eskurza, I.; Silver, A.E.; Levy, A.S.; Pierce, G.L.; Gates, P.E.; Seals, D.R. Direct Evidence of Endothelial Oxidative Stress With Aging in Humans. Circ. Res. 2007, 100, 1659–1666. [Google Scholar] [CrossRef] [Green Version]
- Chang, F.; Flavahan, S.; Flavahan, N.A. Impaired activity of adherens junctions contributes to endothelial dilator dysfunction in ageing rat arteries. J. Physiol. 2017, 595, 5143–5158. [Google Scholar] [CrossRef] [PubMed]
- Krouwer, V.J.D.; Hekking, L.H.P.; Langelaar-Makkinje, M.; Regan-Klapisz, E.; Post, J.A. Endothelial cell senescence is associated with disrupted cell-cell junctions and increased monolayer permeability. Vasc. Cell 2012, 4, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesh, D.; Fredette, N.; Rostama, B.; Tang, Y.; Vary, C.P.H.; Liaw, L.; Urs, S. RhoA-mediated signaling in Notch-induced senescence-like growth arrest and endothelial barrier dysfunction. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 876–882. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Rha, G.B.; Chen, L.; Seelbach, M.J.; Zhang, B.; András, I.E.; Bruemmer, D.; Hennig, B.; Toborek, M. Inhibition of telomerase activity alters tight junction protein expression and induces transendothelial migration of HIV-1-infected cells. Am. J. Physiol. Circ. Physiol. 2010, 298, H1136–H1145. [Google Scholar] [CrossRef] [PubMed]
- Vestweber, D. Relevance of endothelial junctions in leukocyte extravasation and vascular permeability. Ann. N. Y. Acad. Sci. 2012, 1257, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Nourshargh, S.; Hordijk, P.L.; Sixt, M. Breaching multiple barriers: Leukocyte motility through venular walls and the interstitium. Nat. Rev. Mol. Cell Biol. 2010, 11, 366–378. [Google Scholar] [CrossRef]
- Wang, M.; Lakatta, E.G. Altered regulation of matrix metalloproteinase-2 in aortic remodeling during aging. Hypertension 2002, 39, 865–873. [Google Scholar] [CrossRef]
- Belz, G.G. Elastic properties and Windkessel function of the human aorta. Cardiovasc. Drugs Ther. 1995, 9, 73–83. [Google Scholar] [CrossRef]
- Boutouyrie, P.; Bussy, C.; Lacolley, P.; Girerd, X.; Laloux, B.; Laurent, S. Association Between Local Pulse Pressure, Mean Blood Pressure, and Large-Artery Remodeling. Circulation 1999, 100, 1387–1393. [Google Scholar] [CrossRef] [Green Version]
- Chow, M.J.; Turcotte, R.; Lin, C.P.; Zhang, Y. Arterial extracellular matrix: A mechanobiological study of the contributions and interactions of elastin and collagen. Biophys. J. 2014, 106, 2684–2692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolinsky, H.; Glagov, S. Nature of Species Differences in the Medial Distribution of Aortic Vasa Vasorum in Mammals. Circ. Res. 1967, 20, 409–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armentano, R.L.; Levenson, J.; Barra, J.G.; Fischer, E.I.C.; Breitbart, G.J.; Pichel, R.H.; Simon, A. Assessment of elastin and collagen contribution to aortic elasticity in conscious dogs. Am. J. Physiol. Heart Circ. Physiol. 1991, 260, H1870–H1877. [Google Scholar] [CrossRef]
- Fritze, O.; Romero, B.; Schleicher, M.; Jacob, M.P.; Oh, D.Y.; Starcher, B.; Schenke-Layland, K.; Bujan, J.; Stock, U.A. Age-related changes in the elastic tissue of the human aorta. J. Vasc. Res. 2011, 49, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Fleenor, B.S.; Marshall, K.D.; Durrant, J.R.; Lesniewski, L.A.; Seals, D.R. Arterial stiffening with ageing is associated with transforming growth factor-β1-related changes in adventitial collagen: Reversal by aerobic exercise. J. Physiol. 2010, 588, 3971–3982. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; O’Brien, J.E.; Fard, A.; Mannion, J.D.; Wang, D.; Zalewski, A. Adventitial myofibroblasts contribute to neointimal formation in injured porcine coronary arteries. Circulation 1996, 94, 1655–1664. [Google Scholar] [CrossRef]
- Scott, N.A.; Cipolla, G.D.; Ross, C.E.; Dunn, B.; Martin, F.H.; Simonet, L.; Wilcox, J.N. Identification of a potential role for the adventitia in vascular lesion formation after balloon overstretch injury of porcine coronary arteries. Circulation 1996, 93, 2178–2187. [Google Scholar] [CrossRef] [PubMed]
- Tieu, B.C.; Ju, X.; Lee, C.; Sun, H.; Lejeune, W.; Recinos, A.; Brasier, A.R.; Tilton, R.G. Aortic adventitial fibroblasts participate in angiotensin-induced vascular wall inflammation and remodeling. J. Vasc. Res. 2011, 48, 261–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef] [Green Version]
- Zaidel-Bar, R.; Itzkovitz, S.; Ma’ayan, A.; Iyengar, R.; Geiger, B. Functional atlas of the integrin adhesome. Nat. Cell Biol. 2007, 9, 858–867. [Google Scholar] [CrossRef] [PubMed]
- Horton, E.R.; Byron, A.; Askari, J.A.; Ng, D.H.J.; Millon-Frémillon, A.; Robertson, J.; Koper, E.J.; Paul, N.R.; Warwood, S.; Knight, D.; et al. Definition of a consensus integrin adhesome and its dynamics during adhesion complex assembly and disassembly. Nat. Cell Biol. 2015, 17, 1577–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingber, D.E. Cellular mechanotransduction: Putting all the pieces together again. FASEB J. 2006, 20, 811–827. [Google Scholar] [CrossRef] [PubMed]
- Turner, C.E.; Pietras, K.M.; Taylor, D.S.; Molloy, C.J. Angiotensin II stimulation of rapid paxillin tyrosine phosphorylation correlates with the formation of focal adhesions in rat aortic smooth muscle cells. J. Cell Sci. 1995, 108, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro-Silva, J.C.; Miyakawa, A.A.; Krieger, J.E. Focal adhesion signaling: Vascular smooth muscle cell contractility beyond calcium mechanisms. Clin. Sci. 2021, 135, 1189–1207. [Google Scholar] [CrossRef] [PubMed]
- Kirby, T.J.; Lammerding, J. Emerging views of the nucleus as a cellular mechanosensor. Nat. Cell Biol. 2018, 20, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Pawłowski, R.; Rajakylä, E.K.; Vartiainen, M.K.; Treisman, R. An actin-regulated importin α/β-dependent extended bipartite NLS directs nuclear import of MRTF-A. EMBO J. 2010, 29, 3448–3458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.; Azzolin, L.; Di Biagio, D.; Zanconato, F.; Battilana, G.; Lucon Xiccato, R.; Aragona, M.; Giulitti, S.; Panciera, T.; Gandin, A.; et al. The SWI/SNF complex is a mechanoregulated inhibitor of YAP and TAZ. Nature 2018, 563, 265–269. [Google Scholar] [CrossRef]
- Kuwahara, K.; Barrientos, T.; Pipes, G.C.T.; Li, S.; Olson, E.N. Muscle-Specific Signaling Mechanism That Links Actin Dynamics to Serum Response Factor. Mol. Cell. Biol. 2005, 25, 3173–3181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambillara, V.; Thacher, T.; Silacci, P.; Stergiopulos, N. Effects of Reduced Cyclic Stretch on Vascular Smooth Muscle Cell Function of Pig Carotids Perfused Ex Vivo. Am. J. Hypertens. 2008, 21, 425–431. [Google Scholar] [CrossRef] [Green Version]
- Leung, D.; Glagov, S.; Mathews, M. Cyclic stretching stimulates synthesis of matrix components by arterial smooth muscle cells in vitro. Science 2003, 191, 475–477. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, J.D.; Dufresne, E.R.; Schwartz, M.A. Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 2014, 15, 802–812. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, J.D.; Schwartz, M.A.; Tellides, G.; Milewicz, D.M. Role of mechanotransduction in vascular biology: Focus on thoracic aortic aneurysms and dissections. Circ. Res. 2015, 116, 1448–1461. [Google Scholar] [CrossRef] [Green Version]
- Li, D.Y.; Brooke, B.; Davis, E.C.; Mecham, R.P.; Sorensen, L.K.; Boak, B.B.; Eichwald, E.; Keating, M.T. Elastin is an essential determinant of arterial morphogenesis. Nature 1998, 393, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Karnik, S.K.; Wythe, J.D.; Sorensen, L.; Brooke, B.S.; Urness, L.D.; Li, D.Y. Elastin induces myofibrillogenesis via a specific domain, VGVAPG. Matrix Biol. 2003, 22, 409–425. [Google Scholar] [CrossRef]
- Hinek, A.; Boyle, J.; Rabinovitch, M. Vascular smooth muscle cell detachment from elastin and migration through elastic laminae is promoted by chondroitin sulfate-induced “shedding” of the 67-kDa cell surface elastin binding protein. Exp. Cell Res. 1992, 203, 344–353. [Google Scholar] [CrossRef]
- Wheeler, J.B.; Mukherjee, R.; Stroud, R.E.; Jones, J.A.; Ikonomidis, J.S. Relation of Murine Thoracic Aortic Structural and Cellular Changes With Aging to Passive and Active Mechanical Properties. J. Am. Heart Assoc. 2015, 4, e001744. [Google Scholar] [CrossRef] [Green Version]
- Toda, T.; Tsuda, N.; Nishimori, I.; Leszczynski, D.E.; Kummerow, F.A. Morphometrical analysis of the aging process in human arteries and aorta. Acta Anat. 1980, 106, 35–44. [Google Scholar] [CrossRef]
- Ramirez, F.; Pereira, L. The fibrillins. Int. J. Biochem. Cell Biol. 1999, 31, 255–259. [Google Scholar] [CrossRef]
- Wolinsky, H.; Glagov, S. A Lamellar Unit of Aortic Medial Structure and Function in Mammals. Circ. Res. 1967, 20, 99–111. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, M.F.; Hashimoto, J. Mechanical Factors in Arterial Aging. J. Am. Coll. Cardiol. 2007, 50, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Elliott, R.J.; McGrath, L.T. Calcification of the human thoracic aorta during aging. Calcif. Tissue Int. 1994, 54, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Duca, L.; Blaise, S.; Romier, B.; Laffargue, M.; Gayral, S.; El Btaouri, H.; Kawecki, C.; Guillot, A.; Martiny, L.; Debelle, L.; et al. Matrix ageing and vascular impacts: Focus on elastin fragmentation. Cardiovasc. Res. 2016, 110, 298–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robins, S.P. Biochemistry and functional significance of collagen cross-linking. Biochem. Soc. Trans. 2007, 35, 849–852. [Google Scholar] [CrossRef] [Green Version]
- Panwar, P.; Butler, G.S.; Jamroz, A.; Azizi, P.; Overall, C.M.; Brömme, D. Aging-associated modifications of collagen affect its degradation by matrix metalloproteinases. Matrix Biol. 2018, 65, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Monnier, V.M.; Mustata, G.T.; Biemel, K.L.; Reihl, O.; Lederer, M.O.; Zhenyu, D.; Sell, D.R. Cross-Linking of the Extracellular Matrix by the Maillard Reaction in Aging and Diabetes: An Update on “a Puzzle Nearing Resolution”. Ann. N. Y. Acad. Sci. 2005, 1043, 533–544. [Google Scholar] [CrossRef]
- Atherton, P.; Stutchbury, B.; Wang, D.-Y.; Jethwa, D.; Tsang, R.; Meiler-Rodriguez, E.; Wang, P.; Bate, N.; Zent, R.; Barsukov, I.L.; et al. Vinculin controls talin engagement with the actomyosin machinery. Nat. Commun. 2015, 6, 10038. [Google Scholar] [CrossRef] [Green Version]
- Qiu, H.; Zhu, Y.; Sun, Z.; Trzeciakowski, J.P.; Gansner, M.; Depre, C.; Resuello, R.R.G.; Natividad, F.F.; Hunter, W.C.; Genin, G.M.; et al. Short Communication: Vascular Smooth Muscle Cell Stiffness As a Mechanism for Increased Aortic Stiffness with Aging. Circ. Res. 2010, 107, 615–619. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Qiu, H.; Trzeciakowski, J.P.; Sun, Z.; Li, Z.; Hong, Z.; Hill, M.A.; Hunter, W.C.; Vatner, D.E.; Vatner, S.F.; et al. Temporal analysis of vascular smooth muscle cell elasticity and adhesion reveals oscillation waveforms that differ with aging. Aging Cell 2012, 11, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Seawright, J.W.; Sreenivasappa, H.; Gibbs, H.C.; Padgham, S.; Shin, S.Y.; Chaponnier, C.; Yeh, A.T.; Trzeciakowski, J.P.; Woodman, C.R.; Trache, A. Vascular Smooth Muscle Contractile Function Declines With Age in Skeletal Muscle Feed Arteries. Front. Physiol. 2018, 9, 856. [Google Scholar] [CrossRef]
- Nicholson, C.J.; Singh, K.; Saphirstein, R.J.; Gao, Y.Z.; Li, Q.; Chiu, J.G.; Leavis, P.; Verwoert, G.C.; Mitchell, G.F.; Porter, T.; et al. Reversal of Aging-Induced Increases in Aortic Stiffness by Targeting Cytoskeletal Protein-Protein Interfaces. J. Am. Heart Assoc. 2018, 7, e008926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daamen, W.F.; Quaglino, D. Signaling pathways in elastic tissues. Cell. Signal. 2019, 63, 109364. [Google Scholar] [CrossRef] [PubMed]
- Le Page, A.; Khalil, A.; Vermette, P.; Frost, E.H.; Larbi, A.; Witkowski, J.M.; Fulop, T. The role of elastin-derived peptides in human physiology and diseases. Matrix Biol. 2019, 84, 81–96. [Google Scholar] [CrossRef] [PubMed]
- Scandolera, A.; Odoul, L.; Salesse, S.; Guillot, A.; Blaise, S.; Kawecki, C.; Maurice, P.; El Btaouri, H.; Romier-Crouzet, B.; Martiny, L.; et al. The Elastin Receptor Complex: A Unique Matricellular Receptor with High Anti-tumoral Potential. Front. Pharmacol. 2016, 7, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mochizuki, S.; Brassart, B.; Hinek, A. Signaling Pathways Transduced through the Elastin Receptor Facilitate Proliferation of Arterial Smooth Muscle Cells*. J. Biol. Chem. 2002, 277, 44854–44863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular Regulation of Vascular Smooth Muscle Cell Differentiation in Development and Disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef] [PubMed]
- Haudenschild, C.C.; Prescott, M.F.; Chobanian, A. V Aortic endothelial and subendothelial cells in experimental hypertension and aging. Hypertension 1981, 3, 148–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simionescu, A.; Philips, K.; Vyavahare, N. Elastin-derived peptides and TGF-β1 induce osteogenic responses in smooth muscle cells. Biochem. Biophys. Res. Commun. 2005, 334, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Bobryshev, Y.V. Transdifferentiation of smooth muscle cells into chondrocytes in atherosclerotic arteries in situ: Implications for diffuse intimal calcification. J. Pathol. 2005, 205, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Speer, M.Y.; Yang, H.Y.; Brabb, T.; Leaf, E.; Look, A.; Lin, W.L.; Frutkin, A.; Dichek, D.; Giachelli, C.M. Smooth muscle cells give rise to osteochondrogenic precursors and chondrocytes in calcifying arteries. Circ. Res. 2009, 104, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Shanahan, C.M.; Cary, N.R.B.; Salisbury, J.R.; Proudfoot, D.; Weissberg, P.L.; Edmonds, M.E. Medial localization of mineralization-regulating proteins in association with Monckeberg’s sclerosis: Evidence for smooth muscle cell-mediated vascular calcification. Circulation 1999, 100, 2168–2176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyson, K.L.; Reynolds, J.L.; McNair, R.; Zhang, Q.; Weissberg, P.L.; Shanahan, C.M. Osteo/chondrocytic transcription factors and their target genes exhibit distinct patterns of expression in human arterial calcification. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 489–494. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Zhang, J.; Monticone, R.E.; Telljohann, R.; Wu, J.; Wang, M.; Lakatta, E.G. Calpain-1 regulation of matrix metalloproteinase 2 activity in vascular smooth muscle cells facilitates age-associated aortic wall calcification and fibrosis. Hypertension 2012, 60, 1192–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steppan, J.; Sikka, G.; Jandu, S.; Barodka, V.; Halushka, M.K.; Flavahan, N.A.; Belkin, A.M.; Nyhan, D.; Butlin, M.; Avolio, A.; et al. Exercise, vascular stiffness, and tissue transglutaminase. J. Am. Heart Assoc. 2014, 3, e000599. [Google Scholar] [CrossRef] [Green Version]
- Santhanam, L.; Tuday, E.C.; Webb, A.K.; Dowzicky, P.; Kim, J.H.; Oh, Y.J.; Sikka, G.; Kuo, M.; Halushka, M.K.; MacGregor, A.M.; et al. Decreased S-nitrosylation of tissue transglutaminase contributes to age-related increases in vascular stiffness. Circ. Res. 2010, 107, 117–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, K.A.; Polewski, M.; Terkeltaub, R.A. Transglutaminase 2 is central to induction of the arterial calcification program by smooth muscle cells. Circ. Res. 2008, 102, 529–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, S.M.; Jandu, S.; Steppan, J.; Belkin, A.; An, S.S.; Pak, A.; Choi, E.Y.; Nyhan, D.; Butlin, M.; Viegas, K.; et al. Increased tissue transglutaminase activity contributes to central vascular stiffness in eNOS knockout mice. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H803. [Google Scholar] [CrossRef] [Green Version]
- Kielty, C.M. Elastic fibres in health and disease. Expert Rev. Mol. Med. 2006, 8, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Berk, D.R.; Bentley, D.D.; Bayliss, S.J.; Lind, A.; Urban, Z. Cutis laxa: A review. J. Am. Acad. Dermatol. 2012, 66, 842.e1–842.e17. [Google Scholar] [CrossRef] [PubMed]
- Gheduzzi, D.; Guerra, D.; Bochicchio, B.; Pepe, A.; Tamburro, A.M.; Quaglino, D.; Mithieux, S.; Weiss, A.S.; Ronchetti, I.P. Heparan sulphate interacts with tropoelastin, with some tropoelastin peptides and is present in human dermis elastic fibers. Matrix Biol. 2005, 24, 15–25. [Google Scholar] [CrossRef]
- Baccarani-Contri, M.; Vincenzi, D.; Cicchetti, F.; Mori, G.; Pasquali-Ronchetti, I. Immunocytochemical localization of proteoglycans within normal elastin fibers. Eur. J. Cell Biol. 1990, 53, 305–312. [Google Scholar]
- Wang, M.; Zhao, D.; Spinetti, G.; Zhang, J.; Jiang, L.Q.; Pintus, G.; Monticone, R.; Lakatta, E.G. Matrix metalloproteinase 2 activation of Transforming Growth Factor-β1 (TGF-β1) and TGF-β1-type II receptor signaling within the aged arterial wall. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1503–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douillet, C.D.; Velarde, V.; Christopher, J.T.; Mayfield, R.K.; Trojanowska, M.E.; Jaffa, A.A. Mechanisms by which bradykinin promotes fibrosis in vascular smooth muscle cells: Role of TGF-β and MAPK. Am. J. Physiol. Circ. Physiol. 2000, 279, H2829–H2837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Callaghan, C.J.; Williams, B. Mechanical strain-induced extracellular matrix production by human vascular smooth muscle cells: Role of TGF-β1. Hypertension 2000, 36, 319–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeshita, K.; Yamamoto, K.; Ito, M.; Kondo, T.; Matsushita, T.; Hirai, M.; Kojima, T.; Nishimura, M.; Nabeshima, Y.; Loskutoff, D.J.; et al. Increased expression of plasminogen activator inhibitor-1 with fibrin deposition in a murine model of aging, “Klotho” mouse. Semin. Thromb. Hemost. 2002, 28, 545–553. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Kobayashi, A.; Yamazaki, N.; Sugawara, Y.; Takada, Y.; Takada, A. Relationship between age and plasma t-PA, PA-inhibitor, and PA activity. Thromb. Res. 1987, 46, 625–633. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Takeshita, K.; Saito, H. Plasminogen activator inhibitor-1 in aging. Semin. Thromb. Hemost. 2014, 40, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Björkerud, S. Effects of transforming growth factor-beta 1 on human arterial smooth muscle cells in vitro. Arterioscler. Thromb. A J. Vasc. Biol. 1991, 11, 892–902. [Google Scholar] [CrossRef] [Green Version]
- Reddy, K.B.; Howe, P.H. Transforming growth factor β1-mediated inhibition of smooth muscle cell proliferation is associated with a late G1 cell cycle arrest. J. Cell. Physiol. 1993, 156, 48–55. [Google Scholar] [CrossRef] [PubMed]
- McCaffrey, T.A.; Consigli, S.; Du, B.; Falcone, D.J.; Sanborn, T.A.; Spokojny, A.M.; Bush, H.L. Decreased type II/type I TGF-β receptor ratio in cells derived from human atherosclerotic lesions: Conversion from an antiproliferative to profibrotic response to TGF-β1. J. Clin. Investig. 1995, 96, 2667–2675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCaffrey, T.A.; Falcone, D.J. Evidence for an age-related dysfunction in the antiproliferative response to transforming growth factor-β in vascular smooth muscle cells. Mol. Biol. Cell 1993, 4, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Torella, D.; Leosco, D.; Indolfi, C.; Curcio, A.; Coppola, C.; Ellison, G.M.; Russo, V.G.; Torella, M.; Li Volti, G.; Rengo, F.; et al. Aging exacerbates negative remodeling and impairs endothelial regeneration after balloon injury. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2850–H2860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Peng, X.; Lassance-Soares, R.M.; Najafi, A.H.; Alderman, L.O.; Sood, S.; Xue, Z.; Chan, R.; Faber, J.E.; Epstein, S.E.; et al. Aging-induced collateral dysfunction: Impaired responsiveness of collaterals and susceptibility to apoptosis via dysfunctional eNOS signaling. J. Cardiovasc. Transl. Res. 2011, 4, 779–789. [Google Scholar] [CrossRef] [Green Version]
- Fornieri, C.; Quaglino, D.; Mori, G. Role of the extracellular matrix in age-related modifications of the rat aorta—Ultrastmctural, morphometric, and enzymatic evaluations. Arterioscler. Thromb. Vasc. Biol. 1992, 12, 1008–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbert, K.E.; Mistry, Y.; Hastings, R.; Poolman, T.; Niklason, L.; Williams, B. Angiotensin II-mediated oxidative DNA damage accelerates cellular senescence in cultured human vascular smooth muscle cells via telomere-dependent and independent pathways. Circ. Res. 2008, 102, 201–208. [Google Scholar] [CrossRef] [PubMed]
- McCrann, D.J.; Yang, D.; Chen, H.; Carroll, S.; Ravid, K. Upregulation of Nox4 in the aging vasculature and its association with smooth muscle cell polyploidy. Cell Cycle 2009, 8, 902–908. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; McCrann, D.J.; Nguyen, H.; Hilaire, C.S.; DePinho, R.A.; Jones, M.R.; Ravid, K. Increased polyploidy in aortic vascular smooth muscle cells during aging is marked by cellular senescence. Aging Cell 2007, 6, 257–260. [Google Scholar] [CrossRef] [Green Version]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Herbig, U.; Jobling, W.A.; Chen, B.P.C.; Chen, D.J.; Sedivy, J.M. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21CIP1, but not p16INK4a. Mol. Cell 2004, 14, 501–513. [Google Scholar] [CrossRef]
- Blom, N.; Gammeltoft, S.; Brunak, S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 1999, 294, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Torres, A.; Lozano, R.; Melón, J.; Carraro, R. Age-Dependent Decline of in Vitro Migration (Basal and Stimulated by IGF-1 or Insulin) of Human Vascular Smooth Muscle Cells. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2003, 58, 1074–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vafaie, F.; Yin, H.; O’Neil, C.; Nong, Z.; Watson, A.; Arpino, J.M.; Chu, M.W.A.; Wayne Holdsworth, D.; Gros, R.; Pickering, J.G. Collagenase-resistant collagen promotes mouse aging and vascular cell senescence. Aging Cell 2014, 13, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Byrne, M.H.; Stacey, A.; Goldring, M.B.; Birkhead, J.R.; Jaenisch, R.; Krane, S.M. Generation of collagenase-resistant collagen by site-directed mutagenesis of murine proα1(I) collagen gene. Proc. Natl. Acad. Sci. USA 1990, 87, 5888–5892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wu, H.; Byrne, M.; Jeffrey, J.; Krane, S.; Jaenisch, R. A targeted mutation at the known collagenase cleavage site in mouse type I collagen impairs tissue remodeling. J. Cell Biol. 1995, 130, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Wayner, E.A.; Carter, W.G. Identification of multiple cell adhesion receptors for collagen and fibronectin in human fibrosarcoma cells possessing unique α and common β subunits. J. Cell Biol. 1987, 105, 1873–1884. [Google Scholar] [CrossRef] [PubMed]
- Gullberg, D.; Gehlsen, K.R.; Turner, D.C.; Ahlen, K.; Zijenah, L.S.; Barnes, M.J.; Rubin, K. Analysis of α1β1, α2β1 and α3β1 integrins in cell-collagen interactions: Identification of conformation dependent α1β1 binding sites in collagen type I. EMBO J. 1992, 11, 3865–3873. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chow, L.H.; Pickering, J.G. Cell surface-bound collagenase-1 and focal substrate degradation stimulate the rear release of motile vascular smooth muscle cells. J. Biol. Chem. 2000, 275, 35384–35392. [Google Scholar] [CrossRef] [Green Version]
- Fera, E.; O’Neil, C.; Lee, W.; Li, S.; Pickering, J.G. Fibroblast growth factor-2 and remodeled type I collagen control membrane protrusion in human vascular smooth muscle cells: Biphasic activation of Rac1. J. Biol. Chem. 2004, 279, 35573–35582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Pickering, J.G. Cellular Senescence and Vascular Disease: Novel Routes to Better Understanding and Therapy. Can. J. Cardiol. 2016, 32, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Hornebeck, W.; Robert, L. Elastase-like enzymes in aortas and human breast carcinomas: Quantitative variations with age and pathology. Adv. Exp. Med. Biol. 1977, 79, 145–164. [Google Scholar] [CrossRef]
- Cohen, J.R.; Sarfati, I.; Danna, D.; Wise, L.; Hunter, G.; Mannick, J.A.; Tilson, M.D.; MacKenzie, J.W.; Darling, R.C. Smooth muscle cell elastase, atherosclerosis, and abdominal aortic aneurysms. Ann. Surg. 1992, 216, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Yoshida, T.; Tateno, K.; Miyauchi, H.; Zou, Y.; Toko, H.; Komuro, I. Ras Induces Vascular Smooth Muscle Cell Senescence and Inflammation in Human Atherosclerosis. Circulation 2003, 108, 2264–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Drozdov, I.; Shroff, R.; Beltran, L.E.; Shanahan, C.M. Prelamin A accelerates vascular calcification via activation of the DNA damage response and senescence-associated secretory phenotype in vascular smooth muscle cells. Circ. Res. 2013, 112, e99–e109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noureddine, H.; Gary-Bobo, G.; Alifano, M.; Marcos, E.; Saker, M.; Vienney, N.; Amsellem, V.; Maitre, B.; Chaouat, A.; Chouaid, C.; et al. Pulmonary Artery Smooth Muscle Cell Senescence Is a Pathogenic Mechanism for Pulmonary Hypertension in Chronic Lung Disease. Circ. Res. 2011, 109, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.G.A.; Giles, P.J.; Sheerin, A.N.P.; Smith, S.K.; Lawton, J.J.; Ostler, E.L.; Rhys-Williams, W.; Kipling, D.; Faragher, R.G.A. Microarray analysis of senescent vascular smooth muscle cells: A link to atherosclerosis and vascular calcification. Exp. Gerontol. 2009, 44, 659–665. [Google Scholar] [CrossRef] [Green Version]
- Spinetti, G.; Wang, M.; Monticone, R.; Zhang, J.; Zhao, D.; Lakatta, E.G. Rat aortic MCP-1 and its receptor CCR2 increase with age and alter vascular smooth muscle cell function. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1397–1402. [Google Scholar] [CrossRef] [Green Version]
- McCaffrey, T.A.; Nicholson, A.C.; Szabo, P.E.; Weksler, M.E.; Weksler, B.B. Aging and arteriosclerosis: The increased proliferation of arterial smooth muscle cells isolated from old rats is associated with increased platelet-derived growth factor-like activity. J. Exp. Med. 1988, 167, 163–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, L.; Zhang, K.; Chen, J.; Wang, J.; Huang, H. Roles of platelet-derived growth factor in vascular calcification. J. Cell. Physiol. 2018, 233, 2804–2814. [Google Scholar] [CrossRef] [PubMed]
- Stemerman, M.B.; Weinstein, R.; Rowe, J.W.; Maciag, T.; Fuhro, R.; Gardner, R. Vascular smooth muscle cell growth kinetics in vivo in aged rats. Proc. Natl. Acad. Sci. USA 1982, 79, 3863–3866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, S.M.; Majesky, M.W.; Murry, C.E. The intima: Development and monoclonal responses to injury. Atherosclerosis 1995, 118, S125–S140. [Google Scholar] [CrossRef]
- Fu, Z.; Wang, M.; Gucek, M.; Zhang, J.; Wu, J.; Jiang, L.; Monticone, R.E.; Khazan, B.; Telljohann, R.; Mattison, J.; et al. Milk Fat Globule Protein Epidermal Growth Factor-8. Circ. Res. 2009, 104, 1337–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Fu, Z.; Wu, J.; Zhang, J.; Jiang, L.; Khazan, B.; Telljohann, R.; Zhao, M.; Krug, A.W.; Pikilidou, M.; et al. MFG-E8 activates proliferation of vascular smooth muscle cells via integrin signaling. Aging Cell 2012, 11, 500–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, H.Y.; Chu, P.H.; Lee, T.H. MFG-E8 mediates arterial aging by promoting the proinflammatory phenotype of vascular smooth muscle cells. J. Biomed. Sci. 2019, 26, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Cheng, M.; Li, B.Y.; Li, X.L.; Wang, Q.; Zhang, J.H.; Jing, X.J.; Gao, H.Q. Correlation between serum lactadherin and pulse wave velocity and cardiovascular risk factors in elderly patients with type 2 diabetes mellitus. Diabetes Res. Clin. Pract. 2012, 95, 125–131. [Google Scholar] [CrossRef]
- Sarkar, D.; Fisher, P.B. Molecular mechanisms of aging-associated inflammation. Cancer Lett. 2006, 236, 13–23. [Google Scholar] [CrossRef]
- Peppin, G.J.; Weiss, S.J. Activation of the endogenous metalloproteinase, gelatinase, by triggered human neutrophils. Proc. Natl. Acad. Sci. USA 1986, 83, 4322–4326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular Matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef]
- Ala-aho, R.; Kähäri, V.-M. Collagenases in cancer. Biochimie 2005, 87, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Nagase, H.; Woessner, J.F. Matrix Metalloproteinases. J. Biol. Chem. 1999, 274, 21491–21494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stout, L.C.; Whorton, E.B.; Vaghela, M. Pathogenesis of diffuse intimal thickening (DIT) in non-human primate thoracic aortas. Atherosclerosis 1983, 47, 1–6. [Google Scholar] [CrossRef]
- McNulty, M.; Spiers, P.; McGovern, E.; Feely, J. Aging is associated with increased matrix metalloproteinase-2 activity in the human aorta. Am. J. Hypertens. 2005, 18, 504–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauly, R.R.; Passaniti, A.; Bilato, C.; Monticone, R.; Cheng, L.; Papadopoulos, N.; Gluzband, Y.A.; Smith, L.; Weinstein, C.; Lakatta, E.G.; et al. Migration of cultured vascular smooth muscle cells through a basement membrane barrier requires type IV collagenase activity and is inhibited by cellular differentiation. Circ. Res. 1994, 75, 41–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senior, R.M.; Griffin, G.L.; Fliszar, C.J.; Shapiro, S.D.; Goldberg, G.I.; Welgus, H.G. Human 92- and 72-kilodalton type IV collagenases are elastases. J. Biol. Chem. 1991, 266, 7870–7875. [Google Scholar] [CrossRef]
- Berton, A.; Rigot, V.; Huet, E.; Decarme, M.; Eeckhout, Y.; Patthy, L.; Godeau, G.; Hornebeck, W.; Bellon, G.; Emonard, H. Involvement of Fibronectin Type II Repeats in the Efficient Inhibition of Gelatinases A and B by Long-chain Unsaturated Fatty Acids. J. Biol. Chem. 2001, 276, 20458–20465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zieman, S.J.; Melenovsky, V.; Kass, D.A. Mechanisms, Pathophysiology, and Therapy of Arterial Stiffness. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 932–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Zhang, J.; Telljohann, R.; Jiang, L.; Wu, J.; Monticone, R.E.; Kapoor, K.; Talan, M.; Lakatta, E.G. Chronic matrix metalloproteinase inhibition retards age-Associated arterial proinflammation and increase in blood pressure. Hypertension 2012, 60, 459–466. [Google Scholar] [CrossRef] [Green Version]
- Zavaczki, E.; Jeney, V.; Agarwal, A.; Zarjou, A.; Oros, M.; Katkó, M.; Varga, Z.; Balla, G.; Balla, J. Hydrogen sulfide inhibits the calcification and osteoblastic differentiation of vascular smooth muscle cells. Kidney Int. 2011, 80, 731–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagenhäuser, M.U.; Schellinger, I.N.; Yoshino, T.; Toyama, K.; Kayama, Y.; Deng, A.; Guenther, S.P.; Petzold, A.; Mulorz, J.; Mulorz, P.; et al. Chronic nicotine exposure induces murine aortic remodeling and stiffness segmentation-implications for abdominal aortic aneurysm susceptibility. Front. Physiol. 2018, 9, 1459. [Google Scholar] [CrossRef] [PubMed]
- Hickson, L.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, A.S.; Gubbi, S.; Barzilai, N. Benefits of Metformin in Attenuating the Hallmarks of Aging. Cell Metab. 2020, 32, 15–30. [Google Scholar] [CrossRef]
- Alfaras, I.; Di Germanio, C.; Bernier, M.; Csiszar, A.; Ungvari, Z.; Lakatta, E.G.; de Cabo, R. Pharmacological Strategies to Retard Cardiovascular Aging. Circ. Res. 2016, 118, 1626–1642. [Google Scholar] [CrossRef] [PubMed]
- Safar, M.E. Arterial aging—hemodynamic changes and therapeutic options. Nat. Rev. Cardiol. 2010, 7, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Xian, J.Z.; Lu, M.; Fong, F.; Qiao, R.; Patel, N.R.; Abeydeera, D.; Iriana, S.; Demer, L.L.; Tintut, Y. Statin Effects on Vascular Calcification: Microarchitectural Changes in Aortic Calcium Deposits in Aged Hyperlipidemic Mice. Arterioscler. Thromb. Vasc. Biol. 2021, 41, E185–E192. [Google Scholar] [CrossRef] [PubMed]
- Sparavigna, A. Role of the extracellular matrix in skin aging and dedicated treatment—State of the art. Plast. Aesthetic Res. 2020, 2020, 14. [Google Scholar] [CrossRef]
- Koyama, H.; Raines, E.W.; Bornfeldt, K.E.; Roberts, J.M.; Ross, R. Fibrillar Collagen Inhibits Arterial Smooth Muscle Proliferation through Regulation of Cdk2 Inhibitors. Cell 1996, 87, 1069–1078. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, C.J.; Seta, F.; Lee, S.; Morgan, K.G. MicroRNA-203 mimics age-related aortic smooth muscle dysfunction of cytoskeletal pathways. J. Cell. Mol. Med. 2017, 21, 81–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engler, A.J.; Sen, S.; Sweeney, H.L.; Discher, D.E. Matrix Elasticity Directs Stem Cell Lineage Specification. Cell 2006, 126, 677–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnsdorf, E.J.; Tummala, P.; Kwon, R.Y.; Jacobs, C.R. Mechanically induced osteogenic differentiation—The role of RhoA, ROCKII and cytoskeletal dynamics. J. Cell Sci. 2009, 122, 546–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerthoffer, W.T. Mechanisms of Vascular Smooth Muscle Cell Migration. Circ. Res. 2007, 100, 607–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribeiro-Silva, J.C.; Nolasco, P.; Krieger, J.E.; Miyakawa, A.A. Dynamic Crosstalk between Vascular Smooth Muscle Cells and the Aged Extracellular Matrix. Int. J. Mol. Sci. 2021, 22, 10175. https://doi.org/10.3390/ijms221810175
Ribeiro-Silva JC, Nolasco P, Krieger JE, Miyakawa AA. Dynamic Crosstalk between Vascular Smooth Muscle Cells and the Aged Extracellular Matrix. International Journal of Molecular Sciences. 2021; 22(18):10175. https://doi.org/10.3390/ijms221810175
Chicago/Turabian StyleRibeiro-Silva, Joao Carlos, Patricia Nolasco, Jose Eduardo Krieger, and Ayumi Aurea Miyakawa. 2021. "Dynamic Crosstalk between Vascular Smooth Muscle Cells and the Aged Extracellular Matrix" International Journal of Molecular Sciences 22, no. 18: 10175. https://doi.org/10.3390/ijms221810175
APA StyleRibeiro-Silva, J. C., Nolasco, P., Krieger, J. E., & Miyakawa, A. A. (2021). Dynamic Crosstalk between Vascular Smooth Muscle Cells and the Aged Extracellular Matrix. International Journal of Molecular Sciences, 22(18), 10175. https://doi.org/10.3390/ijms221810175