
Molecular Dynamics Simulations of Phosphorylated Intrinsically Disordered Proteins: A Force Field Comparison


Abstract
:1. Introduction
2. Results and Discussion
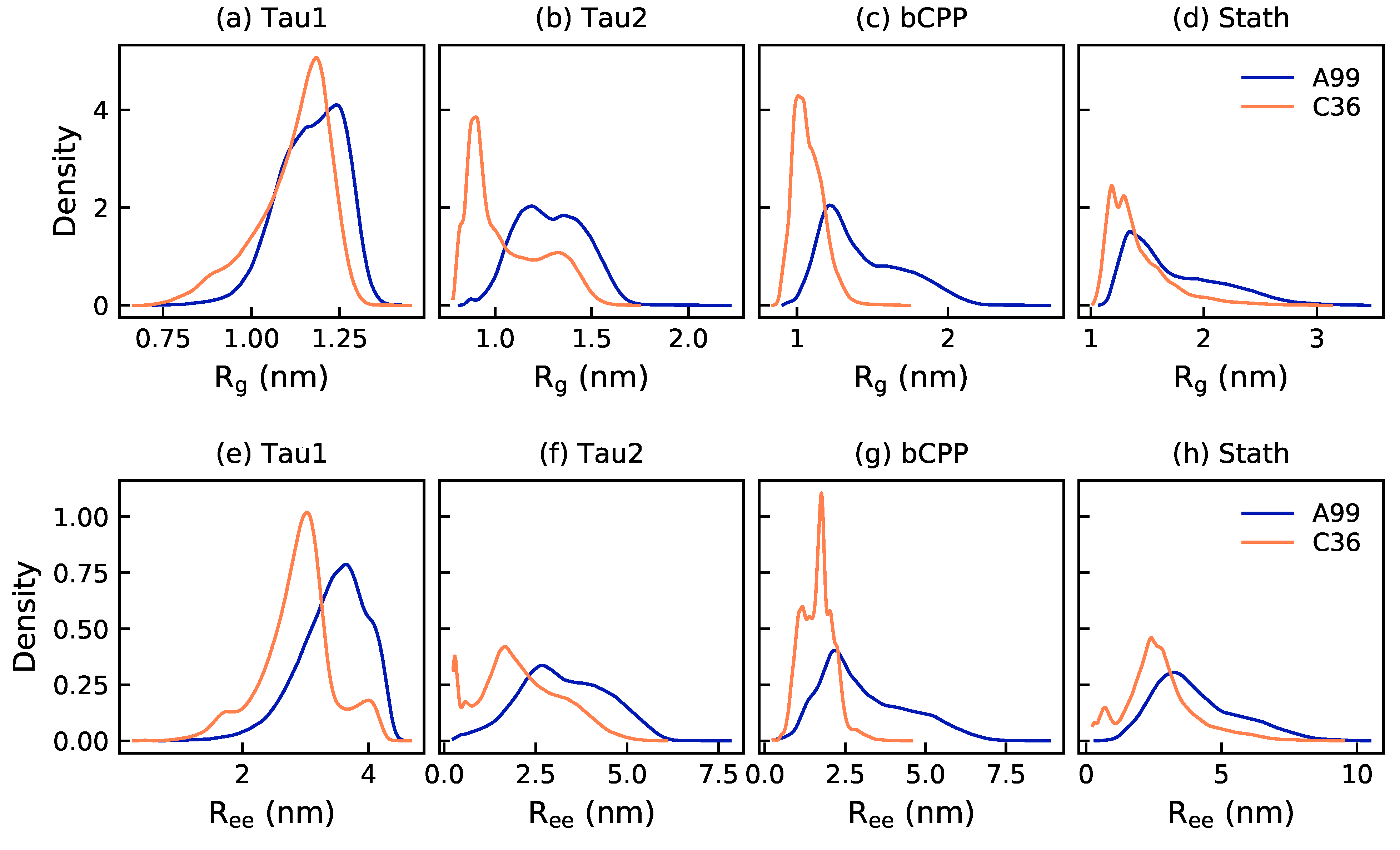
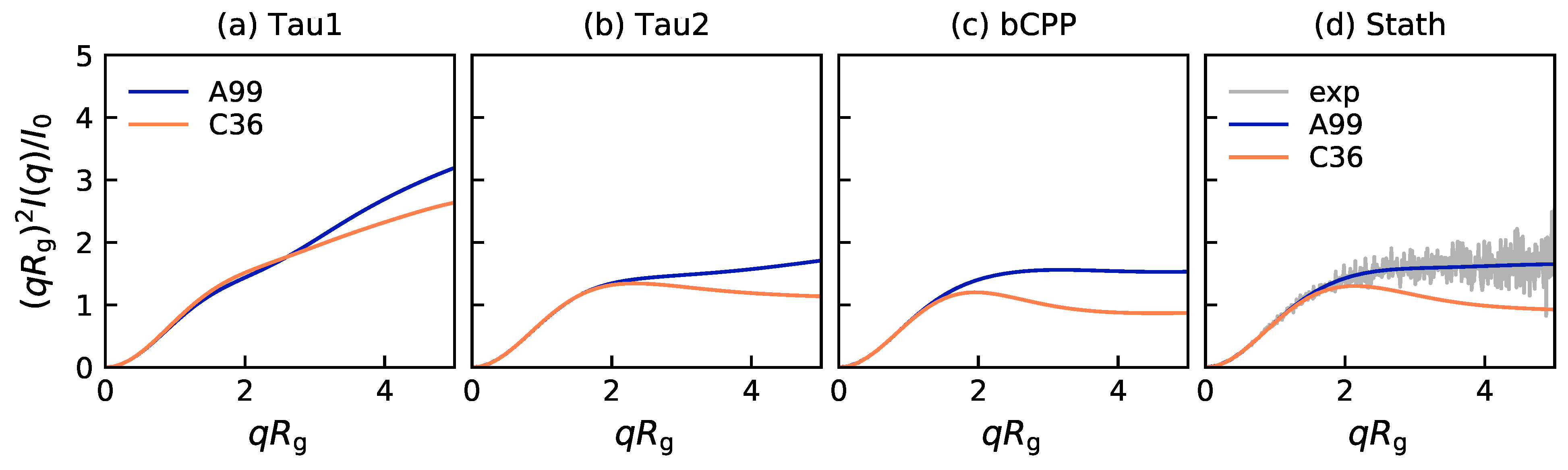
2.1. Size and Shape
2.2. Salt Bridges and Secondary Structure
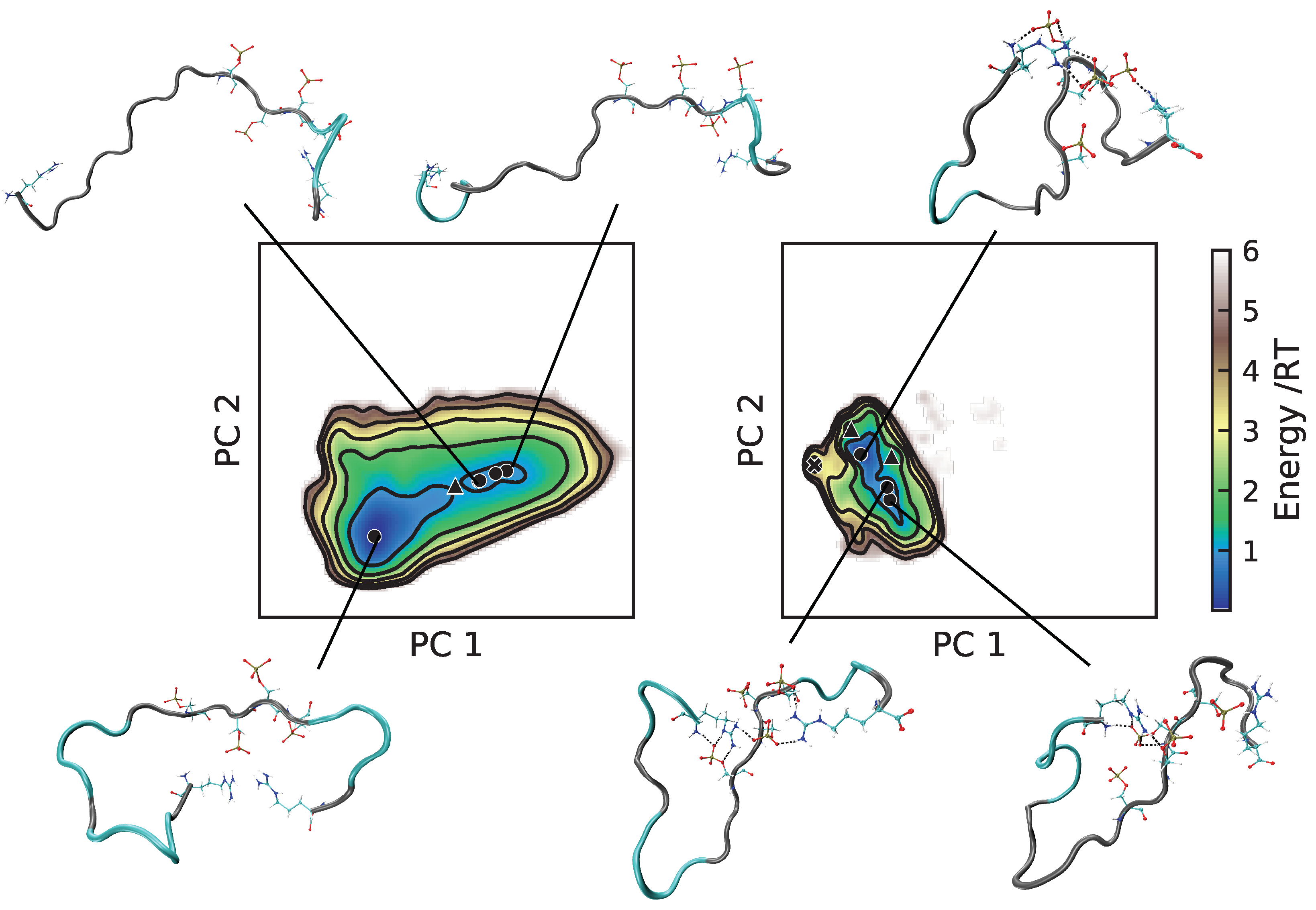
2.3. Energy Landscapes
2.4. Effect of Salt Concentration
3. Conclusions
4. Materials and Methods
Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| A99 | Amber ff99SB-ILDN with TIP4P-D water |
| C36 | CHARMM36m with CHARMM-modified TIP3P water |
| FRET | Fluorescence resonance energy transfer |
| IDP | Intrinsically disordered protein |
| NMR | Nuclear magnetic resonance |
| PPII | polyproline type II |
| Rg | Radius of gyration |
| Ree | End-to-end distance |
| SAXS | Small-angle X-ray scattering |
References
- Dunker, A.; Lawson, J.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef] [Green Version]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Fisher, C.K.; Stultz, C.M. Constructing ensembles for intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2011, 21, 426–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, L.N.; Lewis, R.J. Structural Basis for Control by Phosphorylation. Chem. Rev. 2001, 101, 2209–2242. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.X.; Iqbal, K. Hyperphosphorylation of microtubule-associated protein tau: A promising therapeutic target for Alzheimer disease. Curr. Med. Chem. 2008, 15, 2321–2328. [Google Scholar] [CrossRef]
- Raj, P.A.; Johnsson, M.; Levine, M.J.; Nancollas, G.H. Salivary statherin. Dependence on sequence, charge, hydrogen bonding potency, and helical conformation for adsorption to hydroxyapatite and inhibition of mineralization. J. Biol. Chem. 1992, 267, 5968–5976. [Google Scholar] [CrossRef]
- Makrodimitris, K.; Masica, D.L.; Kim, E.T.; Gray, J.J. Structure Prediction of Protein–Solid Surface Interactions Reveals a Molecular Recognition Motif of Statherin for Hydroxyapatite. J. Am. Chem. Soc. 2007, 129, 13713–13722. [Google Scholar] [CrossRef] [PubMed]
- De Kruif, C.G.; Holt, C. Casein Micelle Structure, Functions and Interactions. In Advanced Dairy Chemistry—1 Proteins: Part A/Part B; Fox, P.F., McSweeney, P.L.H., Eds.; Springer: Boston, MA, USA, 2003; pp. 233–276. [Google Scholar] [CrossRef]
- Martin, E.W.; Holehouse, A.S.; Grace, C.R.; Hughes, A.; Pappu, R.V.; Mittag, T. Sequence Determinants of the Conformational Properties of an Intrinsically Disordered Protein Prior to and upon Multisite Phosphorylation. J. Am. Chem. Soc. 2016, 138, 15323–15335. [Google Scholar] [CrossRef] [Green Version]
- Mittag, T.; Marsh, J.; Grishaev, A.; Orlicky, S.; Lin, H.; Sicheri, F.; Tyers, M.; Forman-Kay, J.D. Structure/Function Implications in a Dynamic Complex of the Intrinsically Disordered Sic1 with the Cdc4 Subunit of an SCF Ubiquitin Ligase. Structure 2010, 18, 494–506. [Google Scholar] [CrossRef] [Green Version]
- Chin, A.; Toptygin, D.; Elam, W.; Schrank, T.; Hilser, V. Phosphorylation Increases Persistence Length and End-to-End Distance of a Segment of Tau Protein. Biophys. J. 2016, 110, 362–371. [Google Scholar] [CrossRef] [Green Version]
- Schwalbe, M.; Kadavath, H.; Biernat, J.; Ozenne, V.; Blackledge, M.; Mandelkow, E.; Zweckstetter, M. Structural Impact of Tau Phosphorylation at Threonine 231. Structure 2015, 23, 1448–1458. [Google Scholar] [CrossRef] [Green Version]
- RFarrell, H.; Qi, P.; Wickham, E.; Unruh, J. Secondary Structural Studies of Bovine Caseins: Structure and Temperature Dependence of β-Casein Phosphopeptide (1–25) as Analyzed by Circular Dichroism, FTIR Spectroscopy, and Analytical Ultracentrifugation. J. Protein Chem. 2002, 21, 307–321. [Google Scholar] [CrossRef]
- Brister, M.A.; Pandey, A.K.; Bielska, A.A.; Zondlo, N.J. OGlcNAcylation and Phosphorylation Have Opposing Structural Effects in tau: Phosphothreonine Induces Particular Conformational Order. J. Am. Chem. Soc. 2014, 136, 3803–3816. [Google Scholar] [CrossRef] [PubMed]
- Cragnell, C.; Rieloff, E.; Skepö, M. Utilizing Coarse-Grained Modeling and Monte Carlo Simulations to Evaluate the Conformational Ensemble of Intrinsically Disordered Proteins and Regions. J. Mol. Biol. 2018, 430, 2478–2492. [Google Scholar] [CrossRef]
- Sieradzan, A.K.; Bogunia, M.; Mech, P.; Ganzynkowicz, R.; Giełdoń, A.; Liwo, A.; Makowski, M. Introduction of Phosphorylated Residues into the UNRES Coarse-Grained Model: Toward Modeling of Signaling Processes. J. Phys. Chem. B 2019, 123, 5721–5729. [Google Scholar] [CrossRef] [PubMed]
- Sieradzan, A.K.; Korneev, A.; Begun, A.; Kachlishvili, K.; Scheraga, H.A.; Molochkov, A.; Senet, P.; Niemi, A.J.; Maisuradze, G.G. Investigation of Phosphorylation-Induced Folding of an Intrinsically Disordered Protein by Coarse-Grained Molecular Dynamics. J. Chem. Theory Comput. 2021, 17, 3203–3220. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.H.; Chatterjee, P.; Ham, S. Computer Simulations of Intrinsically Disordered Proteins. Annu. Rev. Phys. Chem. 2017, 68, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; MacKerell, A.D. Force field development and simulations of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2018, 48, 40–48. [Google Scholar] [CrossRef]
- Rieloff, E.; Skepö, M. Phosphorylation of a Disordered Peptide–Structural Effects and Force Field Inconsistencies. J. Chem. Theory Comput. 2020, 16, 1924–1935. [Google Scholar] [CrossRef]
- Jin, F.; Gräter, F. How multisite phosphorylation impacts the conformations of intrinsically disordered proteins. PLoS Comput. Biol. 2021, 17, e1008939. [Google Scholar] [CrossRef]
- Ahmed, M.C.; Papaleo, E.; Lindorff-Larsen, K. How well do force fields capture the strength of salt bridges in proteins? PeerJ 2018, 6, e4967. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piana, S.; Donchev, A.G.; Robustelli, P.; Shaw, D.E. Water Dispersion Interactions Strongly Influence Simulated Structural Properties of Disordered Protein States. J. Phys. Chem. B 2015, 119, 5113–5123. [Google Scholar] [CrossRef]
- Homeyer, N.; Horn, A.H.C.; Lanig, H.; Sticht, H. AMBER force-field parameters for phosphorylated amino acids in different protonation states: phosphoserine, phosphothreonine, phosphotyrosine, and phosphohistidine. J. Mol. Model. 2006, 12, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Steinbrecher, T.; Latzer, J.; Case, D.A. Revised AMBER Parameters for Bioorganic Phosphates. J. Chem. Theory Comput. 2012, 8, 4405–4412. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2016, 14, 71–73. [Google Scholar]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Henriques, J.; Arleth, L.; Lindorff-Larsen, K.; Skepö, M. On the Calculation of SAXS Profiles of Folded and Intrinsically Disordered Proteins from Computer Simulations. J. Mol. Biol. 2018, 430, 2521–2539. [Google Scholar] [CrossRef] [PubMed]
- Naganagowda, G.A.; Gururaja, T.L.; Levine, M.J. Delineation of Conformational Preferences in Human Salivary Statherin by 1H, 31P NMR and CD Studies: Sequential Assignment and Structure-Function Correlations. J. Biomol. Struct. Dyn. 1998, 16, 91–107. [Google Scholar] [CrossRef]
- Robustelli, P.; Piana, S.; Shaw, D.E. Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. USA 2018, 115, E4758–E4766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan-Yao-Chong, M.; Deville, C.; Pinet, L.; van Heijenoort, C.; Durand, D.; Ha-Duong, T. Structural Characterization of N-WASP Domain V Using MD Simulations with NMR and SAXS Data. Biophys. J. 2019, 116, 1216–1227. [Google Scholar] [CrossRef] [Green Version]
- Das, R.K.; Pappu, R.V. Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc. Natl. Acad. Sci. USA 2013, 110, 13392–13397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piana, S.; Lindorff-Larsen, K.; Shaw, D.E. How Robust Are Protein Folding Simulations with Respect to Force Field Parameterization? Biophys. J. 2011, 100, L47–L49. [Google Scholar] [CrossRef] [Green Version]
- Debiec, K.T.; Gronenborn, A.M.; Chong, L.T. Evaluating the Strength of Salt Bridges: A Comparison of Current Biomolecular Force Fields. J. Phys. Chem. B 2014, 118, 6561–6569. [Google Scholar] [CrossRef]
- Best, R.B. Atomistic Force Fields for Proteins. In Biomolecular Simulations: Methods and Protocols; Bonomi, M., Camilloni, C., Eds.; Springer: New York, NY, USA, 2019; pp. 3–19. [Google Scholar] [CrossRef]
- Bienkiewicz, E.A.; Lumb, K.J. Random-coil chemical shifts of phosphorylated amino acids. J. Biomol. NMR 1999, 15, 203–206. [Google Scholar] [CrossRef]
- Kawade, R.; Kuroda, D.; Tsumoto, K. How the protonation state of a phosphorylated amino acid governs molecular recognition: Insights from classical molecular dynamics simulations. FEBS Lett. 1999, 594, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Holehouse, A.S.; Das, R.K.; Ahad, J.N.; Richardson, M.O.G.; Pappu, R.V. CIDER: Resources to Analyze Sequence-Ensemble Relationships of Intrinsically Disordered Proteins. Biophys. J. 2017, 112, 16–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berendsen, H.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Páll, S.; Abraham, M.J.; Kutzner, C.; Hess, B.; Lindahl, E. Tackling Exascale Software Challenges in Molecular Dynamics Simulations with GROMACS. In Solving Software Challenges for Exascale; Markidis, S., Laure, E., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 3–27. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hockney, R.W.; Eastwood, J.W. Computer Simulation Using Particles; McGraw-Hill: New York, NY, USA, 1981. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Svergun, D.; Barberato, C.; Koch, M.H.J. CRYSOL—A Program to Evaluate X-ray Solution Scattering of Biological Macromolecules from Atomic Coordinates. J. Appl. Crystallogr. 1995, 28, 768–773. [Google Scholar] [CrossRef]
- Nelder, J.A.; Mead, R. A Simplex Method for Function Minimization. Comput. J. 1965, 7, 308–313. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Franke, D.; Petoukhov, M.V.; Konarev, P.V.; Panjkovich, A.; Tuukkanen, A.; Mertens, H.D.T.; Kikhney, A.G.; Hajizadeh, N.R.; Franklin, J.M.; Jeffries, C.M.; et al. ATSAS 2.8: A comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 2017, 50, 1212–1225. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Mansiaux, Y.; Joseph, A.P.; Gelly, J.C.; de Brevern, A.G. Assignment of PolyProline II Conformation and Analysis of Sequence—Structure Relationship. PLoS ONE 2011, 6, e18401. [Google Scholar] [CrossRef]
- Chebrek, R.; Leonard, S.; de Brevern, A.G.; Gelly, J.C. PolyprOnline: Polyproline helix II and secondary structure assignment database. Database 2014, 2014, bau102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernández, C.X.; Schwantes, C.R.; Wang, L.P.; Lane, T.J.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef] [Green Version]
- Wernet, P.; Nordlund, D.; Bergmann, U.; Cavalleri, M.; Odelius, M.; Ogasawara, H.; Näslund, L.Å.; Hirsch, T.K.; Ojamäe, L.; Glatzel, P.; et al. The Structure of the First Coordination Shell in Liquid Water. Science 2004, 304, 995–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campos, S.R.R.; Baptista, A.M. Conformational Analysis in a Multidimensional Energy Landscape: Study of an Arginylglutamate Repeat. J. Phys. Chem. B 2009, 113, 15989–16001. [Google Scholar] [CrossRef]
- Henriques, J.; Cragnell, C.; Skepö, M. Molecular Dynamics Simulations of Intrinsically Disordered Proteins: Force Field Evaluation and Comparison with Experiment. J. Chem. Theory Comput. 2015, 11, 3420–3431. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Stone, J. An Efficient Library for Parallel Ray Tracing and Animation. Master’s Thesis, Computer Science Department, University of Missouri-Rolla, Rolla, MO, USA, 1998. [Google Scholar]
- Frishman, D.; Argos, P. Knowledge-based secondary structure assignment. Proteins 1995, 23, 566–579. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Protein | Sequence |
|---|---|---|
| Tau1 | Tau173-183 | CAKTPPAPKTPPAW |
| Tau2 | Tau225-246 | KVAVVRTPPKSPSSAKSRLQTA |
| bCPP | β-casein1-25 | RELEELNVPGEIVESLSSSEESITR |
| Stath | Statherin | DSSEEKFLRRIGRFGYGYGPYQPVPEQPLYPQPYQPQYQQYTF |
| Peptide | Radius of Gyration (nm) | End-to-End Distance (nm) | ||||
|---|---|---|---|---|---|---|
| A99 | C36 | Difference (%) | A99 | C36 | Difference (%) | |
| Tau1 | 4 | 16 | ||||
| Tau2 | 18 | 36 | ||||
| bCPP | 24 | 47 | ||||
| Stath | 18 | 32 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rieloff, E.; Skepö, M. Molecular Dynamics Simulations of Phosphorylated Intrinsically Disordered Proteins: A Force Field Comparison. Int. J. Mol. Sci. 2021, 22, 10174. https://doi.org/10.3390/ijms221810174
Rieloff E, Skepö M. Molecular Dynamics Simulations of Phosphorylated Intrinsically Disordered Proteins: A Force Field Comparison. International Journal of Molecular Sciences. 2021; 22(18):10174. https://doi.org/10.3390/ijms221810174
Chicago/Turabian StyleRieloff, Ellen, and Marie Skepö. 2021. "Molecular Dynamics Simulations of Phosphorylated Intrinsically Disordered Proteins: A Force Field Comparison" International Journal of Molecular Sciences 22, no. 18: 10174. https://doi.org/10.3390/ijms221810174
APA StyleRieloff, E., & Skepö, M. (2021). Molecular Dynamics Simulations of Phosphorylated Intrinsically Disordered Proteins: A Force Field Comparison. International Journal of Molecular Sciences, 22(18), 10174. https://doi.org/10.3390/ijms221810174