1. Introduction
Non-alcoholic fatty liver disease (NAFLD) is one of the most common chronic liver diseases in the world [
1]. NAFLD pathology is variable and can range from simple steatosis to steatohepatitis and cirrhosis [
2]. No effective treatments exist for NAFLD, making it a large unmet medical need. A better understanding of causes of NAFLD is important for developing novel therapeutic strategies for this disease. NAFLD is heritable and polygenic as evident by a growing list of NAFLD-related single nucleotide polymorphic risk variants identified from genome-wide association studies (GWAS) [
3]. One NAFLD associated variant, rs58542926, falls in the transmembrane 6 superfamily member 2 (
TM6SF2) gene and causes a nonsynonymous glutamic acid to lysine substitution at the amino acid residue 167 (E167K) [
4]. Presence of the E167K variant in humans is associated with higher hepatic lipid content and has been shown to be a strong genetic risk factor for NAFLD, fibrosis and cirrhosis [
4,
5,
6,
7,
8].
In vitro studies suggest that
TM6SF2 E167K variant results in significantly reduced expression of the TM6SF2 protein in liver [
4,
9]. Knocking out
Tm6sf2 in mice or inhibition of
TM6SF2 gene expression in hepatocytes recapitulates the human phenotype of E167K carriers, suggesting that the
TM6SF2 E167K variant acts by a loss-of-function mechanism [
4,
10]. The gene is predominantly expressed in the liver and small intestines and is mainly localized to the endoplasmic reticulum (ER) and the ER-Golgi intermediate compartment, where triglyceride (TG) rich lipoproteins (TRLs) are assembled [
4,
10]. TM6SF2 may have a role in the assembly of very-low density lipoproteins (VLDL) and studies have reported impaired lipidation of the VLDL particles and lower levels of plasma VLDL-TGs in human E167K carriers and in mice with
Tm6sf2 knockout [
11,
12,
13,
14,
15]. The mechanism by which E167K or the loss of TM6SF2 function causes hepatic steatosis, steatohepatitis, and fibrosis is less understood. While spontaneous increase in hepatic TGs under basal conditions is one of the main phenotypes associated with the
TM6SF2 variant, how the increased or decreased function of TM6SF2 protein affects lipid droplet properties, expression of lipid metabolizing genes or cellular lipid composition is not completely understood. Indeed, many argue that a neutral lipid like triglyceride is likely not the cause of NAFLD-related liver damage and such damage may be caused by the accumulation of more hepatotoxic lipids such as saturated fatty acids, diacylglycerols, ceramides, lysophophatidyl choline, phosphoinositol, and free cholesterols, which may also accumulate in NAFLD, albeit at lower amounts than TG [
16,
17].
Here, our objective is to explore how changes in TM6SF2 expression affect lipid metabolism in hepatic cells under lipid exposure. For this, we overexpressed wild-type or E167K variant forms of TM6SF2 or knocked down TM6SF2 in oleic acid-treated Huh-7 cells. We used biochemical assays, untargeted lipidomic analysis, RNAseq transcriptome analysis and high-throughput fluorescent microscopic image analysis to determine the cellular changes associated with altering TM6SF2 protein.
3. Discussion
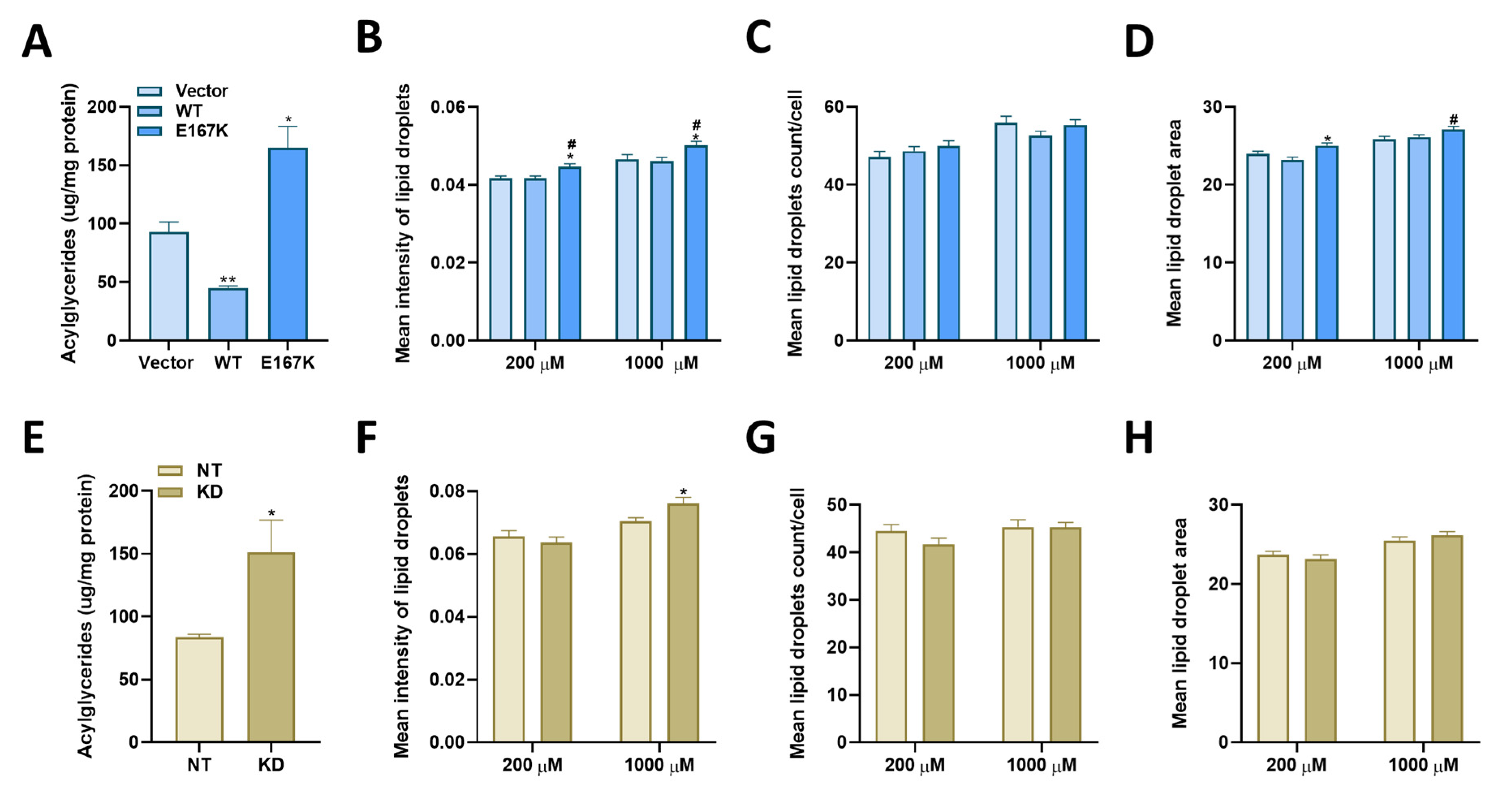
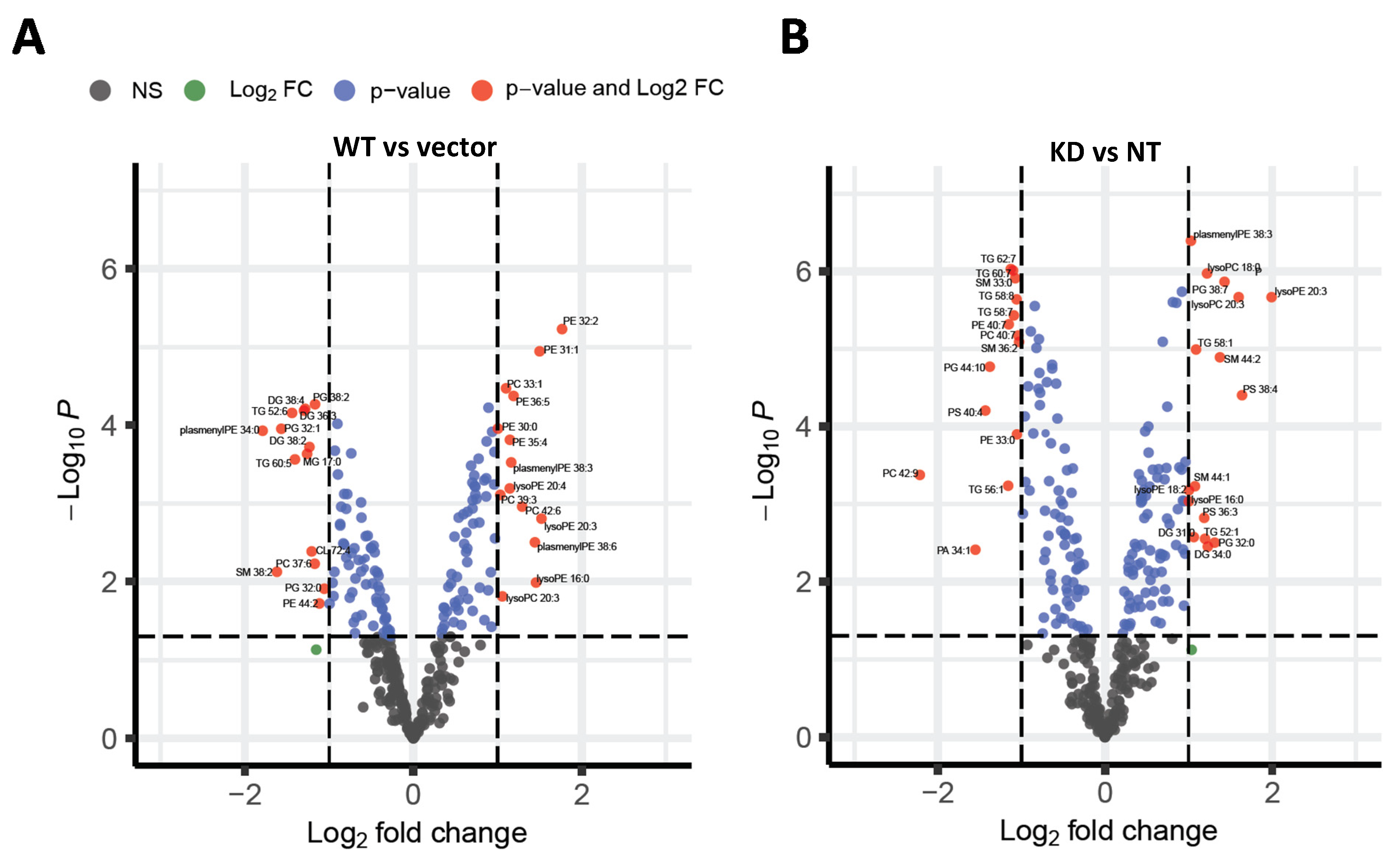
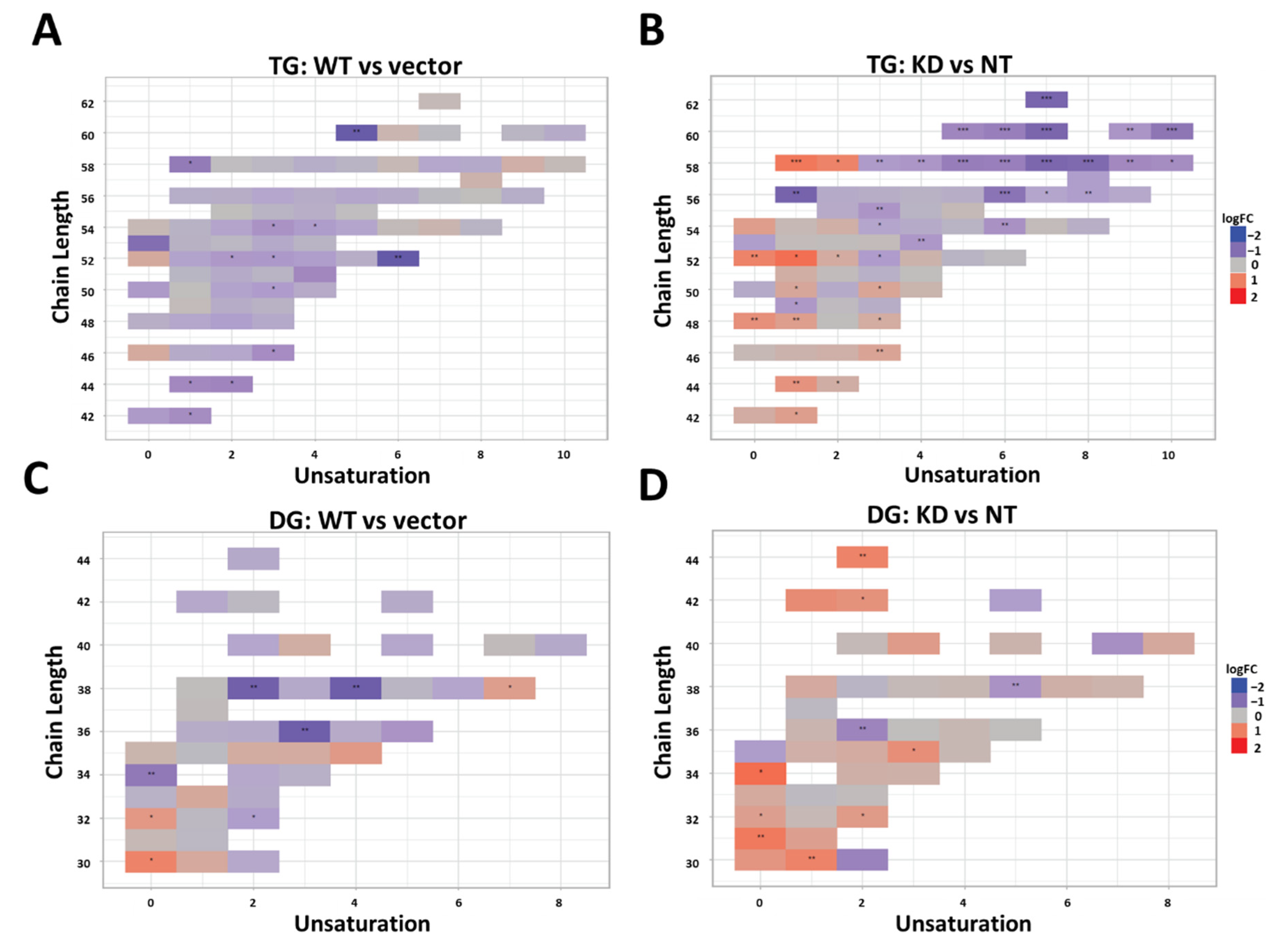
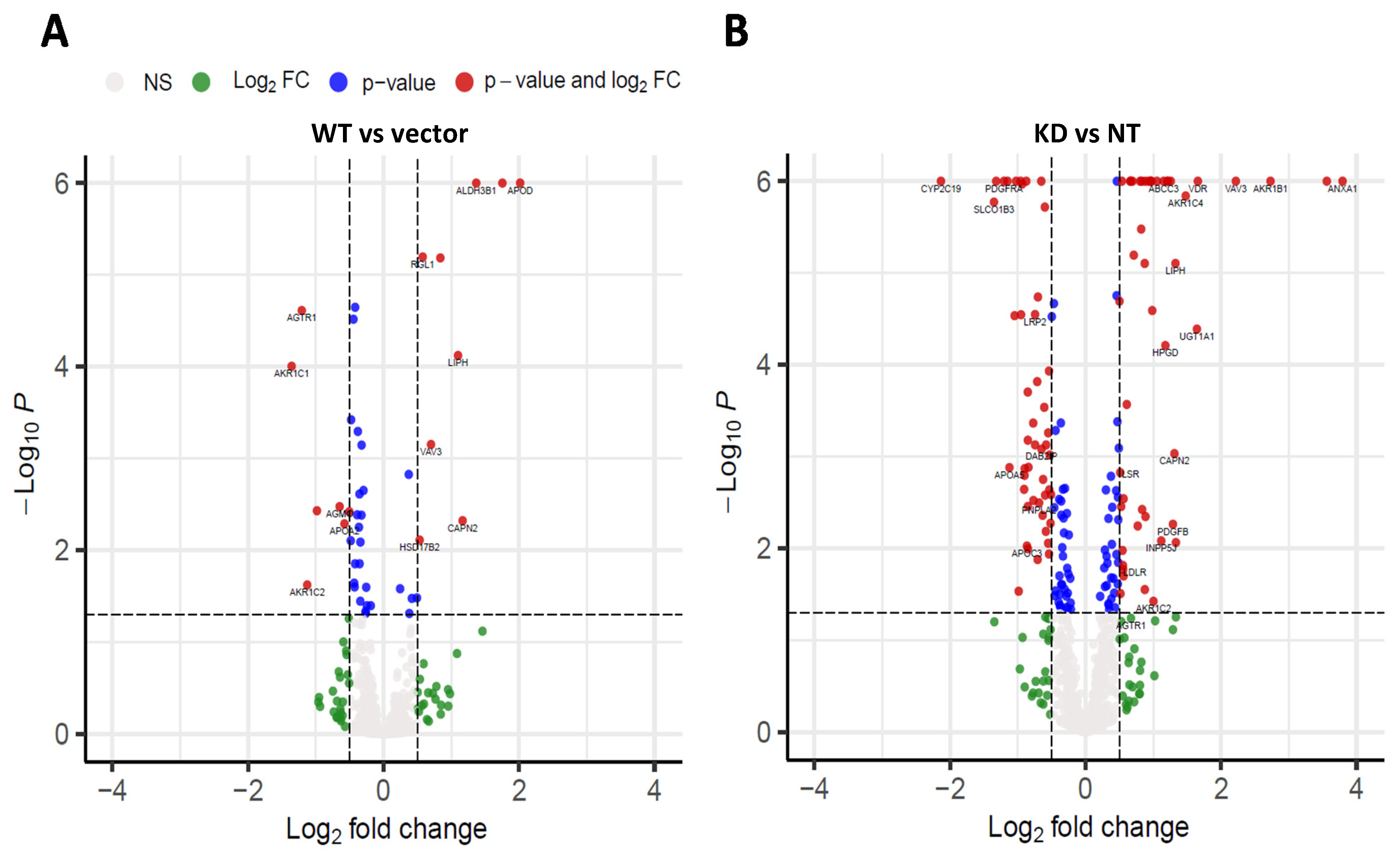
The primary objective of this study was to determine the differential effects of TM6SF2 overexpression and loss on hepatic lipid metabolism, composition and lipid droplet dynamics under steatogenic conditions. We utilized four parallel experimental approaches to determine changes in total levels of acylglycerides, gene expression, individual lipid species, and lipid droplet phenotype in oleic acid-treated Huh-7 cells. Our data shows that knockdown of TM6SF2 and overexpression of the E167K mutant variant increases whereas overexpression of the wild-type lowers acylglycerol accumulation in lipid-loaded hepatocytes. While we did not see changes across lipid classes, we observed lipid chain remodeling for TGs and DGs by TM6SF2 knockdown, leading to a relative increase in shorter and more saturated side chains on glycerol lipids. Importantly, TM6SF2 knockdown and overexpression lead to significant changes in the abundance of several lipid species, including phosphoplipids, lysophospholipids, and acylglycerides. RNA sequencing revealed differential expression of lipid metabolizing genes belonging to the AKR1 family and lipases with TM6SF2 knockdown causing most of the significant changes. Characterization of lipid droplets profile through high-content image analysis determined that overexpression of the E167K variant and TM6SF2 knockdown significantly increase the staining intensity and size of lipid droplets. Taken together, our data shows that overexpression of TM6SF2 gene or its loss of function significantly and differentially affects intrahepatic lipid metabolism to change lipid species composition, expression of lipid metabolizing genes and lipid droplet profile in oleic acid- treated Huh-7 cells.
While data from multiple human studies have established the strong association of
TM6SF2 E167K mutant variant with NAFLD, the exact mechanism how the loss of TM6SF2 function causes hepatic steatosis is not entirely clear. Study in NAFLD patients and in animal models have largely focused on understanding how the E167K variant or the
TM6SF2 knockdown affects very-low density lipoprotein (VLDL) and secretion of VLDL TG, and on TGs levels in plasma or in the liver [
12,
13,
14,
15,
18,
19,
20]. Similarly, most of the in vitro studies on
TM6SF2 knockdown or overexpression explored the effects of gene expression modification in hepatic lipid accumulation under basal conditions and in the absence of lipid exposure [
10,
21]. Here, we evaluated the effects of
TM6SF2 overexpression or its loss of function on intrahepatic lipid metabolism under a steatogenic environment and show that increased accumulation of intracellular lipids is clearly evident in oleic acid-treated Huh-7 cells with
TM6SF2 knockdown or E167K variant overexpression. Additionally, in the present study, we utilized high-throughput imaging with high-content image analysis to determine changes in hepatic lipid accumulation at individual lipid droplet level. Our results show significant increases in intensity and area of individual lipid droplets with
TM6SF2 knockdown or E167K variant overexpression, suggesting the presence of larger, more TG rich lipid droplets in the cells. One possible mechanism behind this observation could be the previously demonstrated role of TM6SF2 on lipidation of very low density lipoproteins and data showing that its loss of function results in intrahepatic retention of TGs [
13]. Previous studies that investigated the effects of E167K variant overexpression or
TM6SF2 knockdown did so separately in animal or cell-based models and suggest a loss of function effects for the variant [
4,
10]. Recently, Li et al. showed that overexpression of E167K in Huh-7 cells reduces TM6SF2 protein expression compared to wild-type overexpression and act similarly to
TM6SF2 knockdown to destabilize APOB48 expression [
20]. In this study, we show that E167K variant overexpression recapitulates the effects of
TM6SF2 knockdown on hepatic lipid accumulation and lipid droplets’ profile suggesting E167K protein may possibly have a dominant negative mechanism of action on hepatic lipid metabolism when overexpressed.
TM6SF2 E167K variant has also been associated with progression of NAFLD to NASH [
7,
8]. Although accumulation of TGs is the major phenotype in E167K carriers, neutral lipids like TGs may not lead to NAFLD-associated liver damage, but rather the accumulation of other more toxic lipids may determine severity and progression of NAFLD. In the current study, overexpression of the wild-type
TM6SF2 did not cause any major changes in lipid species that have been previously shown to cause liver damage in NAFLD. However,
TM6SF2 knockdown significantly increased in lysoPC and lysoPE, both of which were recently identified as early biomarkers of NAFLD and are increased in NALFD patients with hepatitis [
22]. LysoPC has also been previously shown to be increased in liver of NASH patients [
23,
24,
25]. However, lysoPE, but not lysoPCs, were also increased by wild-type overexpression; therefore, further study is needed to determine the significance of these changes.
Biochemical assessment showed significant increases in the total acylglyceride levels in our cells with
TM6SF2 knockdown compared to non-target controls. Similarly, the levels of DGs were generally increased by lipidomic analysis in
TM6SF2 knockdown cells compared to non-target controls. We observed mixed (some increased and some decreased) patterns of changes for particular TG species in
TM6SF2 knockdown compared to non-target controls. Previous studies show that hepatic TGs or DGs extracted from the VLDLs in liver of E167K mutant carriers have shorter and more saturated fatty acids [
15,
26]. In accord with these findings, we observed relative increase in the abundance of more saturated acylglycerides with shorter side chain lengths in cells with
TM6SF2 knockdown. In addition to TGs, previous studies also suggest that
TM6SF2 knockdown may lead to a global shift in lipids in terms of saturation, including PCs, which are major membrane phospholipids [
27]. At the same time, studies show variable results on
TM6SF2 knockdown-mediated changes in the total abundance of hepatic PCs with data showing both decrease or increase in Huh-7 cells, no change in mice and decrease in human E167K carriers [
13,
26,
27]. Luukkonen et al. also reported that PCs synthesis from polyunsaturated fatty acids is impaired in E167K variant carriers thereby lowering total PCs in liver. Although we did not see total class level changes for PCs in our study, abundance of two of the PC species were significantly decreased by
TM6SF2 knockdown, while opposite effects were observed for wild-type
TM6SF2 overexpression where abundance of three out of four significantly changed PCs were increased. Similar effects were also observed for significantly changed PE species, all of which were decreased by
TM6SF2 knockdown and increased by wild-type overexpression. Changes at absolute levels of acyglycerides biochemically may not be reflective of changes in total acylglycerides measured in lipidomic analyses as the lipidomic analyses do not measure all possible acylglyceride species. Members of aldo-keto reductase 1 (AKR1) family, enzymes that regulate steroid metabolism, have been previously known to play a major role in hepatocarcinoma [
28]. Increasing evidence suggest that several members of the AKR1 family such as AKR1B7 and AKR1B10 might play important role in the development of NAFLD or NASH [
29,
30,
31]. Here, expression of several AKR1 family of enzymes were either increased or decreased in all of our cell lines compared to controls, with most changes observed for cells with
TM6SF2 knockdown. We also found differential gene expression for lipases
PNPLA2 and
LIPH in our study along with changes in the expression of lipoprotein receptor genes (
LDLR, LSR, LRP2), which could be cellular response to increased TG levels in the cells. Further investigation is needed to reveal any mechanistic association of these genes in
TM6SF2-mediated changes in hepatic lipid levels.
In this study, we combined systems biology, biochemical assays and high-throughput high-content image analysis to get a more complete picture of hepatic lipid metabolism-associated changes resulting from alterations in TM6SF2 gene expression than currently available. Our data shows some novel changes in lipid composition and expression of lipid metabolizing genes; however, the mechanism of how these changes manifest and their role, if any, on hepatic steatosis and other NAFLD-related phenotypes needs further investigation. In summary, the current study shows that overexpression of wild-type TM6SF2 or loss of TM6SF2 function can effectively and differentially modulate hepatic lipid metabolism to produce significant changes in intracellular lipid levels and lipid species composition. Importantly, loss of TM6SF2 function may produce more profound and overall changes in lipid metabolic pathways under a steatogenic environment. Finally, we have shown that the in vitro model used in this study, particularly the TM6SF2 knockdown studies, recapitulates many of the lipid-metabolism associated phenotypes that were observed in human E167K carriers. Thus, use of this in vitro model along with high-throughput experimental study design such as high-content image analysis used in this study could be a valuable tool to perform further functional analysis on TM6SF2 or other GWAS identified genetic modifiers of hepatic steatosis.
4. Methods
4.1. Cell Line Generation
4.1.1. Cloning Human TM6SF2 cDNA into pCMV6-Entry Vector
All experiments using lentivirus were performed at the University of Michigan under Biosafety Level 2 (BSL2) protocols in compliance with containment procedures in laboratories approved for use by the University of Michigan Institutional Biosafety Committee (IBC) and Environment, Health and Safety (EHS). Huh-7 human hepatocarcinoma cell line was used for functional analyses [
32].
Human wild-type TM6SF2 was amplified by PCR from a human cDNAs library using primers containing Mlu I and EcoR I restriction sites and was sub cloned into the pCMV6-Entry vector (OriGene, Rockville, MD, USA) using the Gibson cloning kit with Q5 High-Fidelity DNA Polymerase (New England Biolabs, Ipswich, MA, USA). The sequences of TM6SF2 insert was confirmed by Sanger sequencing with 3X coverage of sequencing (University of Michigan Sequencing Core, Ann Arbor, MI, USA).
4.1.2. Preparation of Human pCMV6-TM6SF2 (E167K) Mutant cDNA Plasmid
The coding sequence for the human TM6SF2 mutant variant (E167K) cDNA was amplified from wild- type TM6SF2 cDNA plasmid (pCMV6-TM6SF2 plasmid DNA as PCR template) by PCR site-directed mutagenesis using QuikChangeTM Site-Directed Mutagenesis Kit (Stratagene, San Diego, CA, USA). The sequences of TM6SF2 mutant variant (E167K) construct was confirmed by Sanger sequencing with 3X coverage of sequencing (University of Michigan Sequencing Core).
4.1.3. Overexpression of TM6SF2 (Wild-Type and E167K Mutant) Genes in Cultured Human Hepatoma Cells
Huh-7 cells were grown to 70% confluence in DMEM medium with 10% FBS plus 100 IU/mL penicillin and 100 µg/mL streptomycin. For TM6SF2 overexpression, Huh-7 cells were transfected with the pCMV6-Entry vector expressing TM6SF2 (wild-type or E167K mutant) genes or a control empty pCMV6-Entry vector using FuGENE transfection reagent (Thermo Fisher Scientific, Waltham, MA, USA). Post transfection (48 h), cells stably expressing these genes were selected with G-418 (10 µg/mL) for 72 h. Culture DMEM medium was freshly changed and fresh G-418 was added every 2 days. After 3 days, cells stably overexpressing the TM6SF2 (wild-type or E167K mutant) gene were collected, and the total cellular RNA was extracted using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA). Overexpression was measured and confirmed by RT-qPCR.
4.1.4. Knockdown of Endogenous TM6SF2 Gene Expression in Cultured Human Hepatoma Cells
Huh-7 cells were grown to 70% confluence in DMEM medium with 10% FBS plus 100 IU/mL penicillin and 100 µg/mL streptomycin and infected with human TM6SF2 shRNAs (clone 382425, clone 382426) lentivirus (Sigma-Aldrich Company, St. Louis, MO, USA). At 72 h post infection, Huh-7 cells stably expressing TM6SF2 shRNAs lentivirus vector were selected in new C-DMEM medium containing 10 µg/mL puromycin (Sigma-Aldrich Company) for 7 days. The culture medium was changed, and fresh puromycin (10 µg/mL) was added every 2 days. After 7 days, Huh-7 cells with stable TM6SF2 knock down were collected, and total cellular RNA was extracted using TRIzol reagent. Knockdown of TM6SF2 mRNA was confirmed by RT-PCR.
4.2. Experimental Design
For our studies, Huh-7 cells (controls or genetically modified cells) were cultured in high glucose (25 mM) DMEM with 10% FBS and 1% PenStrep along in the presence of antibiotics. After 24 h, the growth medium was replaced with DMEM containing 10% delipidated FBS. Twenty-four hours after lipid starvation, 200 µM of BSA-conjugated oleic acid was added to DMEM culture medium. Cells were harvested after 24 h and samples were collected for RNA, protein, and intracellular triglyceride measurements and lipids were extracted for LC-MS/MS untargeted lipidomics analysis. Similarly treated cells were also fixed and stained for image analysis using high-throughput microscopy.
4.3. Gene Expression Analysis
TM6SF2 gene expression was measured by real time quantitative RT-qPCR method, and total cellular RNA was prepared from stable overexpression/knockdown Huh-7 cells using TRIzol reagent (Thermo Fisher Scientific) and purified by ZYMO Clean Kit. Superscript VILO (Life Technologies, Carlsbad, CA, USA) reverse transcriptase kit was used to synthesize the first strand cDNA, and reverse transcribed cDNA served as template in polymerase chain reactions. The real-time relative quantitative PCR was performed using TaqMan® Gene Expression Assay (Life Technologies) FAM probes for TM6SF2 gene, respectively, with TagMan® Gene Expression Master Mix (Life Technologies) following the manufacturer’s protocol. The TaqMan® ELF1 FAM Gene Expression Assay probes (Life Technologies) were used as endogenous gene control.
4.4. Acylglyceride Assay
After harvesting, total cellular triglycerides were extracted from cells using the Folch extraction method and quantified by using a Serum Triglyceride Determination Kit (Sigma-Aldrich Company). The absorbance was read at 540 nm using an Eppendorf Biospectrometer and 2.5 mg/mL glycerol was used as the standard (Sigma-Aldrich Company). The total cellular triglyceride content was normalized to total cellular protein.
4.5. Sample Preparation for Untargeted Lipidomics
Twenty-four hours after oleic acid exposure, cell culture medium was removed and cells were washed twice with 1X PBS buffer. Cells were then collected by 0.05% Trypsin-EDTA lysis, counted using LUNATM automated cell counter (Annandale, VA, USA) and stored at −80 °C until analyzed. The lipids were extracted using a modified Bligh–Dyer method. The extraction was carried using 2:2:2 (v/v/v) water/methanol/dichloromethane at room temperature after spiking internal standards. The organic layer was collected and dried completely under the stream of nitrogen. Dried extract was re-suspended in 100 µL of 10 mM ammonium acetate.
4.6. LC-MS/MS
Lipid extract was injected onto a 1.8 μm particle 50 × 2.1 mm id Waters Acquity HSS T3 column (Waters, Milford, MA, USA) heated to 55 °C. The column was eluted with acetonitrile/water (40:60, v/v) with 10 mM ammonium acetate as solvent A and acetonitrile/water/isopropanol (10:5:85 v/v) with 10 mM ammonium acetate as solvent B. Gradient is 0 min 40% B, 10 min 100% B, 12 min 100% B, 12.1 min 40% B, and 15 min 40% B. Data were acquired in positive and negative mode using data-dependent MSMS with dynamic mass exclusion at the University of Michigan Metabolomics core. Pooled human plasma sample and pooled experimental sample (prepared by combining small aliquots of all client’s samples) were used to control the quality of sample preparation and analysis. Randomization scheme was used to distribute pooled samples within the set. Mixture of pure authentic standards is used to monitor the instrument performance on a regular basis.
4.7. LC-MS/MS Data Analysis
Lipids are identified using LIPIDBLAST package (
http://fiehnlab.ucdavis.edu/projects/LipidBlast; accessed on 16 June 2017), computer-generated tandem MS library of 212,516 spectra covering 119,200 compounds representing 26 lipid classes, including phospholipids, glycerolipids, bacterial lipoglycans and plant glycolipids. Quantification of lipids is done by Multiquant software (AB-SCIEX). Data was normalized using internal standards first and cell number. Data from positive and negative ion mode runs were combined with repeats removed and filtered by RSD (<30%).
4.8. Lipidomics Data Analysis
Combined normalized data for each run was imported into the R package lipidr (
https://github.com/ahmohamed/lipidr; accessed 19 January 2021). Data was imported as a lipidomics object, summarized, annotated, and normalized using the PQN method. Lipidr was used for analysis and to generate graphs.
4.9. High-Throughput Lipid Droplet Imaging and Analysis
Huh-7 cells were plated into 96-well optical-bottom plates (Thermo Scientific, 1256670) at the density of 5000 cells/well in conditions similar to other studies. Then, 24 h after oleic acid treatment, cells were fixed in 4% paraformaldehyde for 10 min in RT, washed three times with 1× PBS, and permeabilized with 0.1% Triton-X for 10 min. Intracellular lipid droplets were stained with the LipidTOX Green neutral lipid stain (1:1000; Thermo Scientific, H34475), the nucleus was stained with DAPI (1:1000; Thermo Fisher Scientific, D1306) for 30 min, and cells were then washed twice in PBS. Cells were imaged using the Cellomics Array Scan VT1 (Thermo Fisher Scientific) at a magnification of 20×, and 8 non-overlapping images were captured per well. The images were then imported into the CellProfiler software and processed through automated image analysis pipeline using different analysis modules. Within each image, the pipeline first identifies the nuclei. For each identified nuclei, the pipeline identifies the cell boundary using the propagation algorithm and determines the cell perimeter. Within each identified cell, the program then quantitates lipid droplets’ related variables such as area, diameter, radius, intensity, number of total droplets in cells, etc. For each image, measurements for mean intensity and area of lipid droplets as well as mean lipid droplet number per cell were quantitated. Mean lipid droplet area and mean intensity of droplets represent the average area or intensity for each individual lipid droplet across all image fields for each sample and are independent to the number of cells per field. Combined values across three independent experiments are graphically presented. The pipeline used for CellProfiler imaging is added as
Supplementary Method S1.
4.10. RNA Sequencing and Transcriptome Analysis
Total cellular RNA was prepared from Huh-7 cells using TRIzol reagent (Thermo Fisher Scientific) and purified by ZYMO Clean Kit (Life Technologies) and submitted to the University of Michigan DNA Sequencing Core for quality control, library preparation, and sequencing. RNA sequencing was performed using the Illumina Hi-Seq 4000 platform. The raw sequencing data were 125 base, paired-end reads. Transcriptome analysis was carried out by the University of Michigan bioinformatics core. Briefly, quality of the raw reads data for each sample was checked using FastQC, and the Tuxedo Suite software package was used for alignment and post-analysis diagnostics. FastQC was then used for a second round of quality control (post-alignment), to ensure that only high quality data would be input to expression quantitation and differential expression analysis. Differential gene expression analysis was performed using DESeq2, using a negative binomial generalized linear model (Benjamini–Hochberg FDR (Padj) < 0.05). Genes were annotated with NCBI Entrez Gene IDs and differential expression analysis for lipid metabolizing genes was then performed by using the ipathway Guide (Advaita).


{kind=link}
{kind=link}
{kind=link}
{kind=link}



