
A New Perspective on Cancer Therapy: Changing the Treaded Path?



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
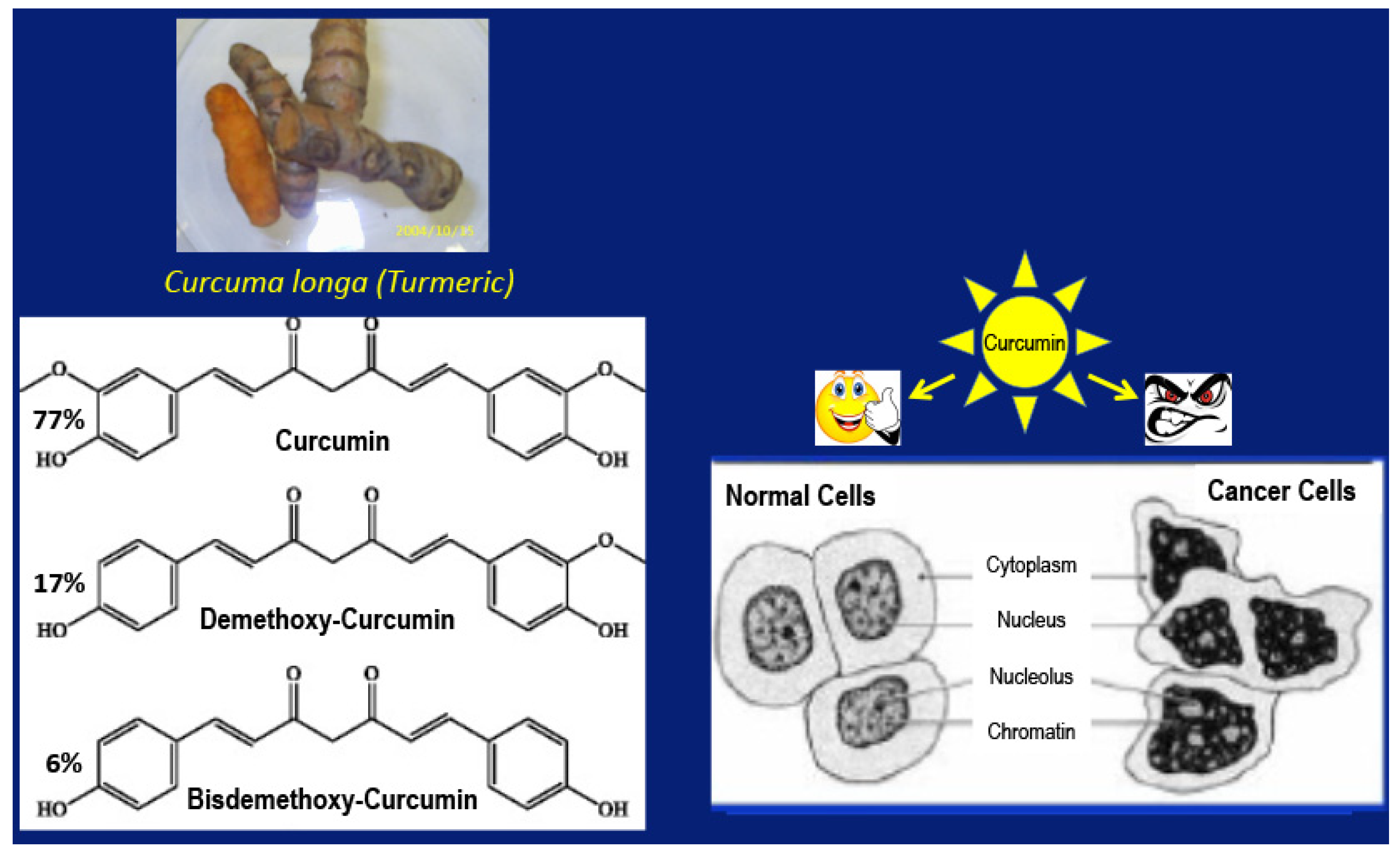
2. From the Anecdotal Use of a Remedy to the Clinic
3. Newly Appreciated Indirect Approaches in Cancer Therapy: Immunotherapy Using the Adaptive Immune System
4. Immunotherapy Using the Innate Immune System
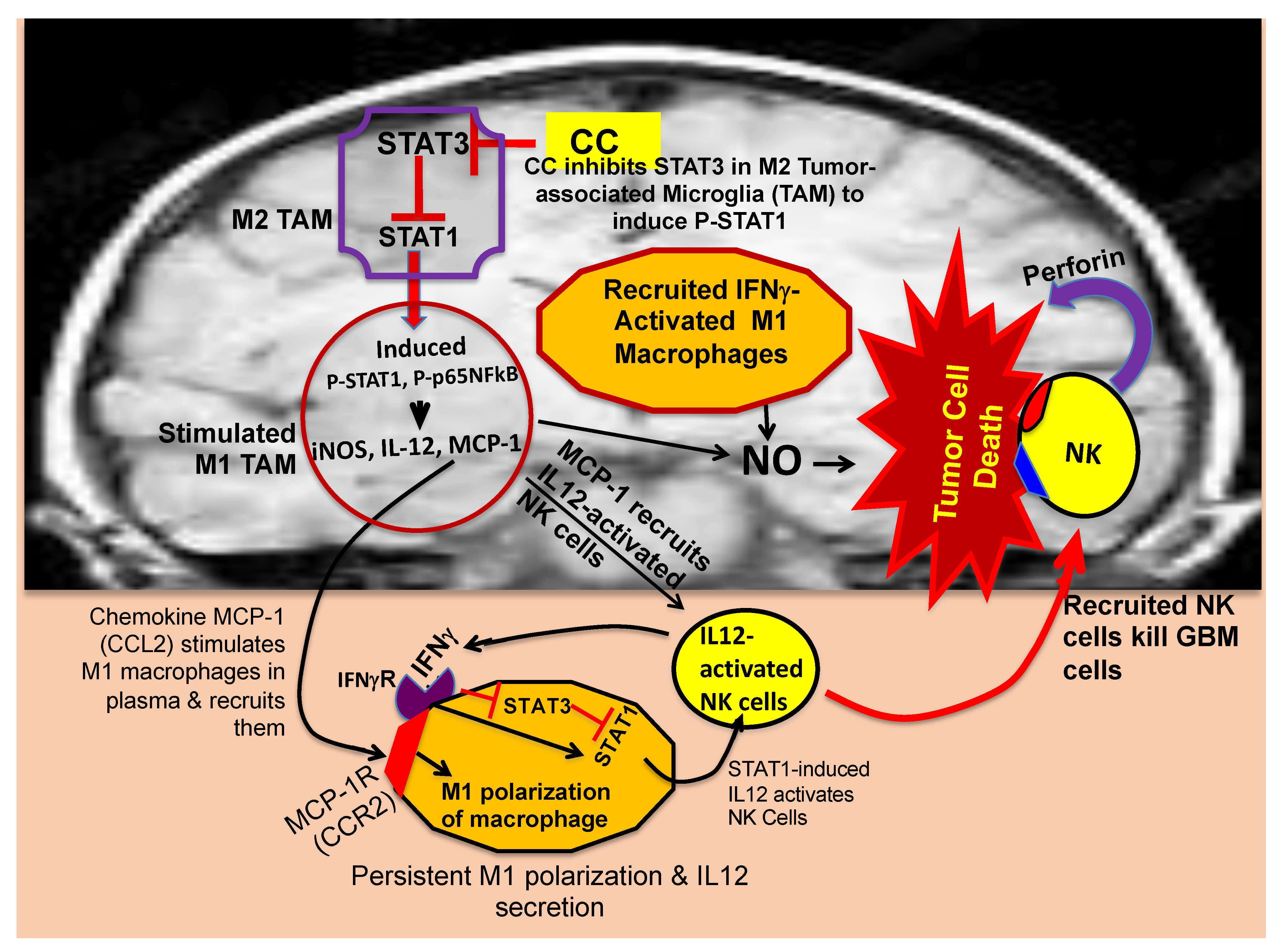
5. Proposed Mechanism of M2→M1 Repolarization of TAMs, Recruitment of Macrophages and NK Cells, and Tumor Elimination
6. Investigating Ways to Use the Innate Immune System in Cancer Therapy Requires the Use of Immune-Competent Animal Models
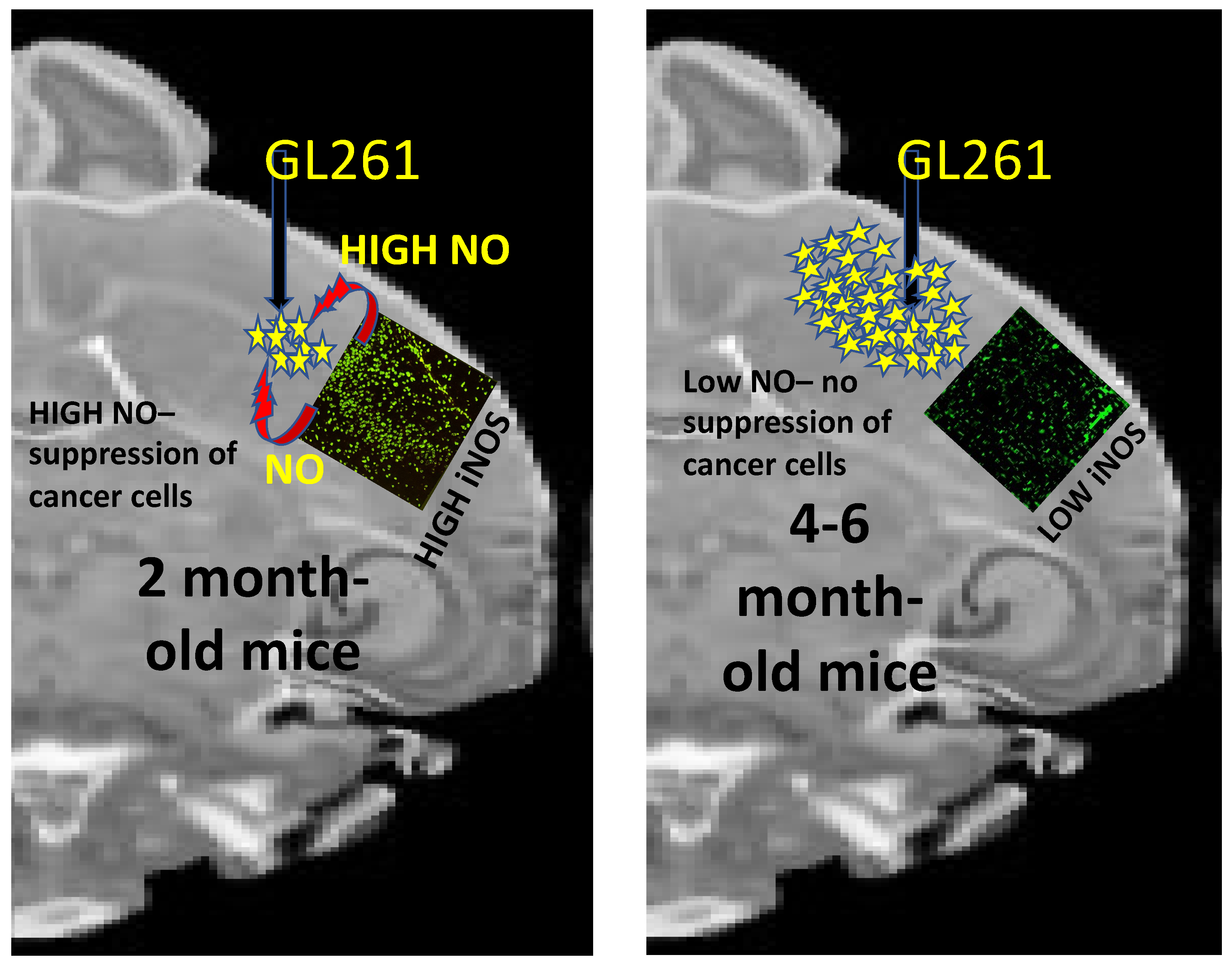
7. Nitric Oxide (NO) Production by Innate Immune Cells
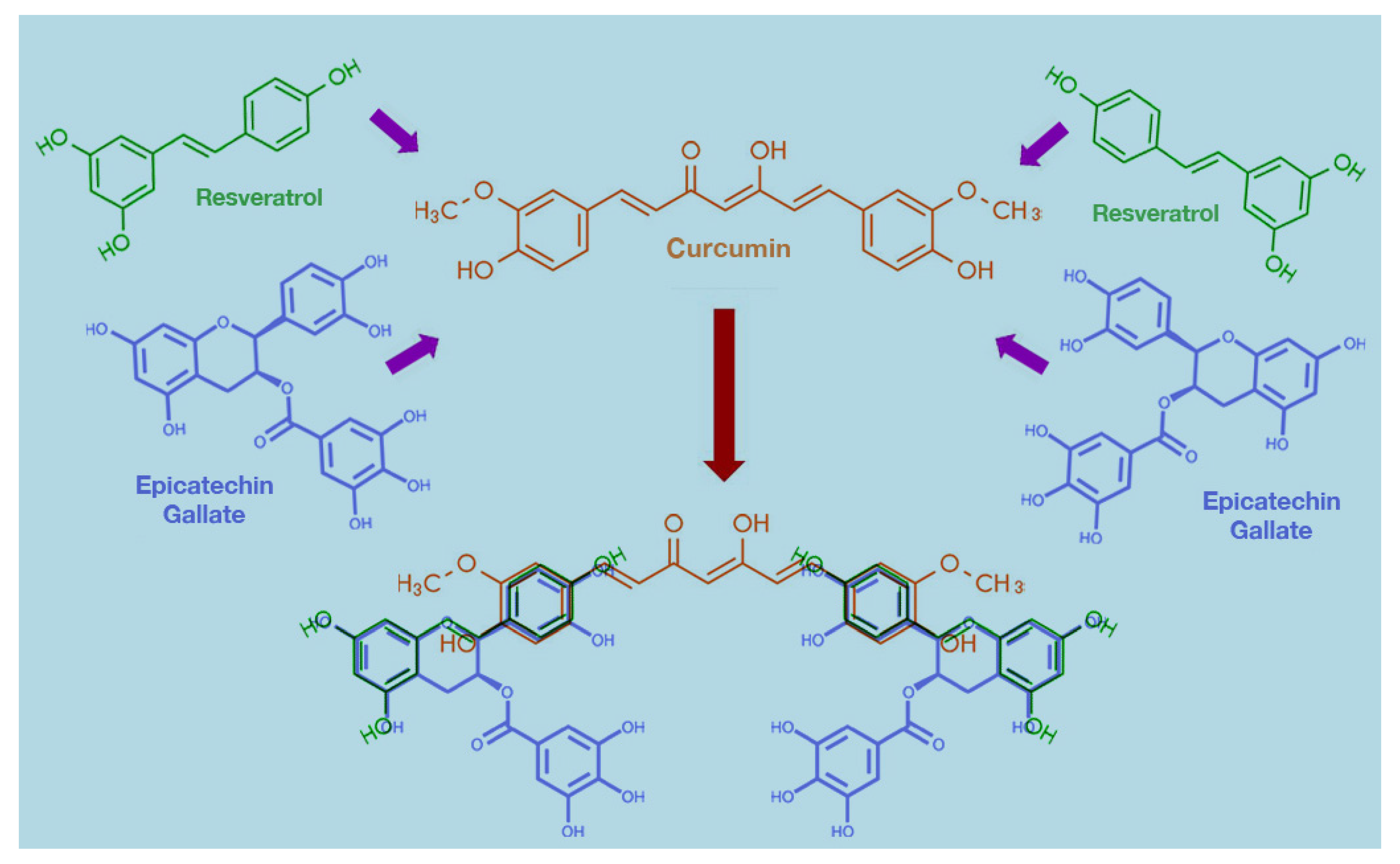
8. Possible Mechanisms of Synergism among Curcumin and Other Polyphenols
9. Chemoresistance Resulting from CA Treatment of Cancer Cells
10. The Role of Curcumin in Context-Dependent Regulation of iNOS and the Involvement of Resolvins in Inflammation and Tumor Clearance
11. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Förstermann, U.; Sessa, W. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2011, 33, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Förstermann, U.; Xloaa, E.J.; Pollock, J.S.; Nakane, M.; Schwarz, P.; Gath, I.; Kleinert, H. Nitric Oxide Synthase Isozymes: Characterization, Purification, Molecular Cloning, and Functions. Hypertension 1994, 21, 1121–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crane, B.R.; Arvai, A.S.; Ghosh, D.K.; Wu, C.; Getzoff, E.D.; Stuehr, D.J.; Tainer, J.A. Structure of Nitric Oxide Synthase Oxygenase Dimer with Pterin and Substrate. Science 1998, 279, 2121–2126. [Google Scholar] [CrossRef] [PubMed]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef] [PubMed]
- Li, L.M.; Kilbourn, R.G.; Adams, J.; Fidler, I.J. Role of nitric oxide in lysis of tumor cells by cytokine-activated endothelial cells. Cancer Res. 1991, 51, 2531–2535. [Google Scholar] [PubMed]
- Nathan, C.F.; Hibbs, J.B. Role of nitric oxide synthesis in macrophage antimicrobial activity. Curr. Opin. Immunol. 1991, 3, 65–70. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Mukherjee, S.; Baidoo, J.; Fried, A.; Atwi, D.; Dolai, S.; Boockvar, J.; Symons, M.; Ruggieri, R.; Raja, K.; Banerjee, P. Curcumin changes the polarity of tumor-associated microglia and eliminates glioblastoma. Int. J. Cancer 2016, 139, 2838–2849. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Baidoo, J.N.E.; Sampat, S.; Mancuso, A.; David, L.; Cohen, L.S.; Zhou, S.; Banerjee, P. Liposomal TriCurin, A Synergistic Combination of Curcumin, Epicatechin Gallate and Resveratrol, Repolarizes Tumor-Associated Microglia/Macrophages, and Eliminates Glioblastoma (GBM) and GBM Stem Cells. Molecules 2018, 23, 201. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Hussaini, R.; White, R.; Atwi, D.; Fried, A.; Sampat, S.; Piao, L.; Pan, Q.; Banerjee, P. TriCurin, a synergistic formulation of curcumin, resveratrol, and epicatechin gallate, repolarizes tumor-associated macrophages and triggers an immune response to cause suppression of HPV+ tumors. Cancer Immunol. Immunother. 2018, 67, 761–774. [Google Scholar] [CrossRef]
- Mukherjee, S.; Fried, A.; Hussaini, R.; White, R.; Baidoo, J.; Yalamanchi, S.; Banerjee, P. Phytosomal curcumin causes natural killer cell-dependent repolarization of glioblastoma (GBM) tumor-associated microglia/macrophages and elimination of GBM and GBM stem cells. J. Exp. Clin. Cancer Res. 2018, 37, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gantois, I.; Popic, J.; Khoutorsky, A.; Sonenberg, N. Metformin for Treatment of Fragile X Syndrome and Other Neurological Disorders. Annu. Rev. Med. 2019, 70, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, L.; Arvin, A. Chemothrapy-induced immunosuppression. Environ. Health Perspect. 1982, 43, 21–25. [Google Scholar] [CrossRef]
- Ding, Z.-C.; Lu, X.; Yu, M.; Lemos, H.; Huang, L.; Chandler, P.; Liu, K.; Walters, M.; Krasinski, A.; Mack, M.; et al. Immunosuppressive Myeloid Cells Induced by Chemotherapy Attenuate Antitumor CD4þ T-Cell Responses through the PD-1–PD-L1 Axis. Cancer Res. 2014, 74, 3441–3453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kersten, K.; Salvagno, C.; de Visser, K. Exploiting the immjunomodulatory properties of chemotherapeutic drugs to improve the success of cancer immunotherapy. Front. Immunol. 2015, 6, 516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Dong, J.; Haiech, J.; Kilhoffer, M.-C.; Zeniou, M. Cancer Stem Cell Quiescence and Plasticity as Major Challenges in Cancer Therapy. Stem Cells Int. 2016, 2016, 1740936. [Google Scholar] [CrossRef] [Green Version]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nat. Cell Biol. 2019, 575, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Sarkaria, J.N.; Kitange, G.J.; James, C.D.; Plummer, R.; Calvert, H.; Weller, M.; Wick, W. Mechanisms of Chemoresistance in Malignant Glioma. Clin. Cancer Res. 2008, 14, 2900–2908. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.-O.; Li, C.-W.; Xia, W.; Cha, J.-H.; Chan, L.-C.; Wu, Y.; Chang, S.-S.; Lin, W.-C.; Hsu, J.-M.; Hsu, Y.-H.; et al. Deubiquitination and Stabilization of PD-L1 by CSN5. Cancer Cell 2016, 30, 925–939. [Google Scholar] [CrossRef] [Green Version]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Buchbinder, E.J.; Desai, A. CTLA-4 and PD-1 Pathways Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Merck Sharp & Dohme Corp. Keytruda. In Merck Oncology. 2019. Available online: https://www.keytruda.com/side-effects/?src=google&med=cpc&camp=Keytruda+Pan+Tumor_Brand_BRND_NA_ENGM_EXCT_TEXT_NA&adgrp=Side+Effects_General&kw=keytruda+side+effects&utm_kxconfid=sq7irm3mh&gclid=EAIaIQobChMIt4jO0K_i4gIViYCfCh3cNQ3KEAAYASAAEgJ4YPD_BwE&gclsrc=aw.ds#serious-side-effects (accessed on 8 September 2021).
- FDA Approves Opdivo as Single Agent for Melanoma. Oncol. Times 2016, 38, 41. [CrossRef]
- Purkayastha, S.; Berliner, A.; Fernando, S.S.; Ranasinghe, B.; Ray, I.; Tariq, H.; Banerjee, P. Curcumin blocks brain tumor formation. Brain Res. 2009, 1266, 130–138. [Google Scholar] [CrossRef]
- Langone, P.; Debata, P.R.; Inigo, J.D.R.; Dolai, S.; Mukherjee, S.; Halat, P.; Mastroianni, K.; Curcio, G.M.; Castellanos, M.R.; Raja, K.; et al. Coupling to a Glioblastoma-directed Antibody Potentiates Anti-tumor Activity of Curcumin. Int. J. Cancer 2014, 135, 710–719. [Google Scholar] [CrossRef] [PubMed]
- Marczylo, T.H.; Verschoyle, R.D.; Cooke, D.N.; Morazzoni, P.; Steward, W.P.; Gescher, A.J. Comparison of systemic availability of curcumin with that of curcumin formulated with phsphatidylcholine. Cancer Chemother Pharm. 2007, 60, 171–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, A.L.; Miska, J.; Wainwright, D.; Dey, M.; Rivetta, C.V.; Yu, D.; Kanojia, D.; Pituch, K.C.; Qiao, J.; Pytel, P.; et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer Res. 2016, 76, 5671–5682. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Tsirka, S.E. Monocyte chemoattractant protein-1 and the blood–brain barrier. Cell. Mol. Life Sci. 2013, 71, 683–697. [Google Scholar] [CrossRef] [Green Version]
- Gschwandtner, M.; Derler, R.; Midwood, K.S. More Than Just Attractive: How CCL2 Influences Myeloid Cell Behavior Beyond Chemotaxis. Front. Immunol. 2019, 10, 2759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, S.; Baidoo, J.N.; Fried, A.; Banerjee, P. Using curcumin to turn the innate immune system against cancer. Biochem. Pharmacol. 2020, 176, 113824. [Google Scholar] [CrossRef]
- Ito, S.; Ansari, P.; Sakatsume, M.; Dickensheets, H.; Vazquez, N.; Donnelly, R.P.; Larner, A.C.; Finbloom, D.S. Interleukin-10 inhibits expression of both interferon alpha- and interferon gamma- induced genes by suppressing tyrosine phosphorylation of STAT1. Blood 1999, 93, 1456–1463. [Google Scholar] [CrossRef] [PubMed]
- Bellora, F.; Castriconi, R.; Dondero, A.; Reggiardo, G.; Moretta, L.; Mantovani, A.; Bottino, C. The interaction of human natural killer cells with either unpolarized or polarized macrophages results in different functional outcomes. Proc. Natl. Acad. Sci. USA 2010, 107, 21659–21664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lünemann, A.; Lünemann, J.D.; Roberts, S.; Messmer, B.; Da Silva, R.B.; Raine, C.S.; Münz, C. Human NK cells kill resting but not activated microglia via NKG2D- and NKp46-mediated recognition. J. Immunol. 2008, 181, 6170–6177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castriconi, R.; Daga, A.; Dondero, A.; Zona, G.; Poliani, P.L.; Melotti, A.; Griffero, F.; Marubbi, D.; Spaziante, R.; Bellora, F.; et al. NK Cells Recognize and Kill Human Glioblastoma Cells with Stem Cell-Like Properties. J. Immunol. 2009, 182, 3530–3539. [Google Scholar] [CrossRef] [Green Version]
- Pegram, H.J.; Andrews, D.M.; Smyth, M.; Darcy, P.; Kershaw, M.H. Activating and inhibitory receptors of natural killer cells. Immunol. Cell Biol. 2010, 89, 216–224. [Google Scholar] [CrossRef]
- Prionisti, I.; Buhler, L.; Walker, P.R.; Jolivet, R.B. Harnessing Microglia and Macrophages for the Treatment of Glioblastoma. Front. Pharmacol. 2019, 10, 506. [Google Scholar] [CrossRef]
- van Dalen, F.J.; van Stevendaal, M.H.M.E.; Fennemann, F.L.; Verdoes, M.; Ilina, O. Molecular Repolarisation of Tumour-Associated Macrophages. Molecules 2019, 24, 9. [Google Scholar] [CrossRef] [Green Version]
- Parayath, N.; Parikh, A.; Amiji, M.M. Repolarization of Tumor-Associated Macrophages in a Genetically Engineered Nonsmall Cell Lung Cancer Model by Intraperitoneal Administration of Hyaluronic Acid-Based Nanoparticles Encapsulating MicroRNA-125b. Nano Lett. 2018, 18, 3571–3579. [Google Scholar] [CrossRef]
- Yang, Q.; Guo, N.; Zhou, Y.; Chen, J.; Wei, Q.; Han, M. The role of tumor-associated macrophages (TAMs) in tumor progression and relevant advance in targeted therapy. Acta Pharm. Sin. B 2020, 10, 2156–2170. [Google Scholar] [CrossRef]
- Kowal, J.; Kornete, M.; Joyce, J.A. Re-education of macrophages as a therapeutic strategy in cancer. Immunotherapy 2019, 11, 677–689. [Google Scholar] [CrossRef]
- Bolli, E.; Scherger, M.; Arnouk, S.M.; Antunes, A.R.P.; Straßburger, D.; Urschbach, M.; Stickdorn, J.; De Vlaminck, K.; Movahedi, K.; Räder, H.J.; et al. Targeted Repolarization of Tumor-Associated Macrophages via Imidazoquinoline-Linked Nanobodies. Adv. Sci. 2021, 8, 2004574. [Google Scholar] [CrossRef]
- Thomas, D.D.; Liu, X.; Kantrow, S.P.; Lancaster, J.R., Jr. The biological lifetime of nitric oxide: Implications for the perivascular dynamics of NO and O2. Proc. Natl. Acad Sci. USA 2001, 98, 355–360. [Google Scholar] [CrossRef]
- Vahora, H.; Khan, M.A.; Alalami, U.; Hussain, A. The Potential Role of Nitric Oxide in Halting Cancer Progression through Chemoprevention. J. Cancer Prev. 2016, 21, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Debata, P.R.; Hussaini, R.; Chatterjee, K.; Baidoo, J.N.; Sampat, S.; Szerszen, A.; Navarra, J.P.; Fata, J.; Severinova, E.; et al. Unique synergistic formulation of curcumin, epicatechin gallate and resveratrol, tricurin, suppresses HPV E6, eliminates HPV+ cancer cells, and inhibits tumor progression. Oncotarget 2017, 8, 60904–60916. [Google Scholar] [CrossRef] [PubMed]
- Osuka, S.; Van Meir, E.G. Overcoming therapeutic resistance in glioblastoma: The way forward. J. Clin. Investig. 2017, 127, 415–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, M.; Cloughesy, T.; Perry, J.R.; Wick, W. Standards of care for treatment of recurrent glioblastoma—Are we there yet? Neuro Oncol. 2013, 15, 4–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qazi, M.A.; Vora, P.; Venugopal, C.; McFarlane, N.; Subapanditha, M.K.; Murty, N.K.; Hassell, J.A.; Hallett, R.M.; Singh, S.K. A novel stem cell culture model of recurrent glioblastoma. J. Neuro Oncol. 2015, 126, 57–67. [Google Scholar] [CrossRef]
- Hasan, S.; Dinh, K.; Lombardo, F.; Kark, J. Doxorubicin cardiotoxicity in African Americans. J. Natl. Med. Assoc. 2004, 96, 196–199. [Google Scholar]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Howells, L.M.; Sale, S.; Sriramareddy, S.N.; Irving, G.R.; Jones, D.J.; Ottley, C.J.; Pearson, D.G.; Mann, C.D.; Manson, M.M.; Berry, D.P.; et al. Curcumin ameliorates oxaliplatin-induced chemoresistance in HCT116 colorectal cancer cells in vitro and in vivo. Int. J. Cancer 2010, 129, 476–486. [Google Scholar] [CrossRef]
- Saha, S.; Adhikary, A.; Bhattacharyya, P.; Das, T.; Sa, G. Death by design: Where curcumin sensitizes drug-resistant tumours. Anticancer Res. 2012, 32, 2567–2584. [Google Scholar]
- Li, M.; Zhang, Z.; Hill, D.L.; Wang, H.; Zhang, R. Curcumin, a Dietary Component, Has Anticancer, Chemosensitization, and Radiosensitization Effects by Down-regulating the MDM2 Oncogene through the PI3K/mTOR/ETS2 Pathway. Cancer Res. 2007, 67, 1988–1996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, S.; Zaher, H.; Hartz, A.; Bauer, B.; Ware, J.A.; Ambudkar, S.V. Curcumin Inhibits the Activity of ABCG2/BCRP1, a Multidrug Resistance-Linked ABC Drug Transporter in Mice. Pharm. Res. 2008, 26, 480–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, S.; Mandal, D.P.; Saha, B.; Sen, G.S.; Das, T.; Sa, G. Curcumin Prevents Tumor-induced T Cell Apoptosis through Stat-5a-mediated Bcl-2 Induction. J. Biol. Chem. 2007, 282, 15954–15964. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.-S.; Weng, S.-H.; Kuo, Y.-H.; Chiu, Y.-F.; Lin, Y.-W. Synergistic Effect of Curcumin and Cisplatin via Down-Regulation of Thymidine Phosphorylase and Excision Repair Cross-Complementary 1 (ERCC1). Mol. Pharmacol. 2011, 80, 136–146. [Google Scholar] [CrossRef] [Green Version]
- Ting, C.-Y.; Wang, H.-E.; Yu, C.-C.; Liu, H.-C.; Liu, Y.-C.; Chiang, I.-T. Curcumin Triggers DNA Damage and Inhibits Expression of DNA Repair Proteins in Human Lung Cancer Cells. Anticancer Res. 2015, 35, 3867–3873. [Google Scholar] [PubMed]
- Chearwae, W.; Shukla, S.; Limtrakul, P.; Ambudkar, S.V. Modulation of the function of the multidrug resistance–linked ATP-binding cassette transporter ABCG2 by the cancer chemopreventive agent curcumin. Mol. Cancer Ther. 2006, 5, 1995–2006. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Chen, Q.; Wang, Y.; Peng, W.; Cai, H. Effects of curcumin on ion channels and transporters. Front. Physiol. 2014, 5, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tivnan, A.; Zakaria, Z.; O’Leary, C.; Kogel, D.; Pokomy, J.L.; Sarkaria, J.N.; Prehn, J.H.M. Inhibition of multidrug resistance protein1 (MRP1) improves chemotherapy drug response in primary and recurrent glioblastoma multiforme. Front. Neurosci. 2015, 9, 218. [Google Scholar] [CrossRef] [Green Version]
- Jin, W.; Liao, X.; Lv, Y.; Pang, Z.; Wang, Y.; Li, Q.; Liao, Y.; Ye, Q.; Chen, G.; Zhao, K.; et al. MUC1 induces acquired chemoresistance by upregulating ABCB1 in EGFR-dependent manner. Cell Death Dis. 2017, 8, e2980. [Google Scholar] [CrossRef] [Green Version]
- Chan, M.M.-Y.; Huang, H.-I.; Fenton, M.R.; Fong, D. In Vivo Inhibition of Nitric Oxide Synthase Gene Expression by Curcumin, a Cancer Preventive Natural Product with Anti-Inflammatory Properties. Biochem. Pharmacol. 1998, 55, 1955–1962. [Google Scholar] [CrossRef]
- Yu, Y.; Shen, Q.; Lai, Y.; Park, S.Y.; Ou, X.; Lin, D.; Jin, M.; Zhang, W. Anti-inflammatory Effects of Curcumin in Microglial Cells. Front. Pharmacol. 2018, 9, 386. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.Y.; Park, E.J.; Joe, E.-H.; Jou, I. Curcumin Suppresses Janus Kinase-STAT Inflammatory Signaling through Activation of Src Homology 2 Domain-Containing Tyrosine Phosphatase 2 in Brain Microglia. J. Immunol. 2003, 171, 6072–6079. [Google Scholar] [CrossRef] [Green Version]
- Karlstetter, M.; Lippe, E.; Walczak, Y.; Moehle, C.; Aslanidis, A.; Mirza, M.; Langmann, T. Curcumin is a potent modulator of microglial gene expression and migration. J. Neuroinflamm. 2011, 8, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuelsson, B. Leukotrienes: Mediators of immediate hypersensitivity reactions and inflammation. Science 1983, 220, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Flower, R.J. Prostaglandins, bioassay and inflammation. Br. J. Pharmacol. 2006, 147, S182–S192. [Google Scholar] [CrossRef]
- Nolan, E.; O’Meara, Y.M.; Godson, C. Lipid mediators of inflammation in obesity-related glomerulopathy. Nephrol. Dial. Transplant. 2013, 28, iv22–iv29. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Brain, S.D.; Buckley, C.D.; Gilroy, D.W.; Haslett, C.; O’Neill, L.A.J.; Perretti, M.; Rossi, A.G.; Wallace, J.L. Resolution of inflammation: State of the art, definitions and terms. FASEB J. 2006, 672271. [Google Scholar] [CrossRef]
- Schwab, J.; Chiang, N.; Arita, M.; Serhan, C.N. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 2007, 447, 869–874. [Google Scholar] [CrossRef] [Green Version]
- Gronert, K.; Maheshwari, N.; Khan, N.; Hassan, I.R.; Dunn, M.; Schwartzman, M.L. A role for the mouse 12/15-lipoxygenase pathway in promoting epithelial wound healing and host defense. J. Biol. Chem. 2005, 280, 15267–15278. [Google Scholar] [CrossRef] [Green Version]
- Titos, E.; Rius, B.; González-Périz, A.; López-Vicario, C.; Morán-Salvador, E.; Martínez-Clemente, M.; Arroyo, V.; Claria, J. Resolvin D1 and Its Precursor Docosahexaenoic Acid Promote Resolution of Adipose Tissue Inflammation by Eliciting Macrophage Polarization toward an M2-Like Phenotype. J. Immunol. 2011, 187, 5408–5418. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Clish, C.; Brannon, J.; Colgan, S.P.; Chiang, N.; Gronert, K. Novel Functional Sets of Lipid-Derived Mediators with Antiinflammatory Actions Generated from Omega-3 Fatty Acids via Cyclooxygenase 2–Nonsteroidal Antiinflammatory Drugs and Transcellular Processing. J. Exp. Med. 2000, 192, 1197–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nat. Cell Biol. 2014, 510, 92–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, Y.; Matsumiya, M.; Matsuura, T.; Oishi, M.; Kaibori, M.; Okumura, T.; Nishizawa, M.; Takada, H.; Kwon, A.-H. Peroxidation of n-3 Polyunsaturated Fatty Acids Inhibits the Induction of iNOS Gene Expression in Proinflammatory Cytokine-Stimulated Hepatocytes. J. Nutr. Metab. 2011, 2011, 374542. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Noble, E.E.; Tyagi, E.; Ying, Z.; Zhuang, Y.; Gomez-Pinilla, F. Curcumin boosts DHA in the brain: Implications for the prevention of anxiety disorders. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1852, 951–961. [Google Scholar] [CrossRef] [Green Version]
- Sulciner, M.L.; Serhan, C.N.; Gilligan, M.M.; Mudge, D.K.; Chang, J.; Gartung, A.; Lehner, K.A.; Bielenberg, D.R.; Schmidt, B.; Dalli, J.; et al. Resolvins suppress tumor growth and enhance cancer therapy. J. Exp. Med. 2017, 215, 115–140. [Google Scholar] [CrossRef]
- Fiala, M. Curcumin and Omega-3 Fatty Acids Enhance NK Cell-Induced Apoptosis of Pancreatic Cancer Cells but Curcumin Inhibits Interferon-γ Production: Benefits of Omega-3 with Curcumin against Cancer. Molecules 2015, 20, 3020–3026. [Google Scholar] [CrossRef] [Green Version]
- Hallisey, V.M.; Kipper, F.C.; Moore, J.; Gartung, A.; Bielenberg, D.R.; Petrik, J.; Lawler, J.; Panigrahy, D.; Serhan, C.N. Pro-Resolving Lipid Mediators and Anti-Angiogenic Therapy Exhibit Synergistic Anti-Tumor Activity via Resolvin Receptor Activation. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Mattoscio, D.; Isopi, E.; Lamolinara, A.; Patruno, S.; Medda, A.; De Cecco, F.; Chiocca, S.; Iezzi, M.; Romano, M.; Recchiuti, A. Resolvin D1 reduces cancer growth stimulating a protective neutrophil-dependent recruitment of anti-tumor monocytes. J. Exp. Clin. Cancer Res. 2021, 40, 129. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baidoo, J.N.E.; Mukherjee, S.; Kashfi, K.; Banerjee, P. A New Perspective on Cancer Therapy: Changing the Treaded Path? Int. J. Mol. Sci. 2021, 22, 9836. https://doi.org/10.3390/ijms22189836
Baidoo JNE, Mukherjee S, Kashfi K, Banerjee P. A New Perspective on Cancer Therapy: Changing the Treaded Path? International Journal of Molecular Sciences. 2021; 22(18):9836. https://doi.org/10.3390/ijms22189836
Chicago/Turabian StyleBaidoo, Juliet N. E., Sumit Mukherjee, Khosrow Kashfi, and Probal Banerjee. 2021. "A New Perspective on Cancer Therapy: Changing the Treaded Path?" International Journal of Molecular Sciences 22, no. 18: 9836. https://doi.org/10.3390/ijms22189836
APA StyleBaidoo, J. N. E., Mukherjee, S., Kashfi, K., & Banerjee, P. (2021). A New Perspective on Cancer Therapy: Changing the Treaded Path? International Journal of Molecular Sciences, 22(18), 9836. https://doi.org/10.3390/ijms22189836