
The Renin–Angiotensin-Aldosterone System, Nitric Oxide, and Hydrogen Sulfide at the Crossroads of Hypertension and COVID-19: Racial Disparities and Outcomes



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
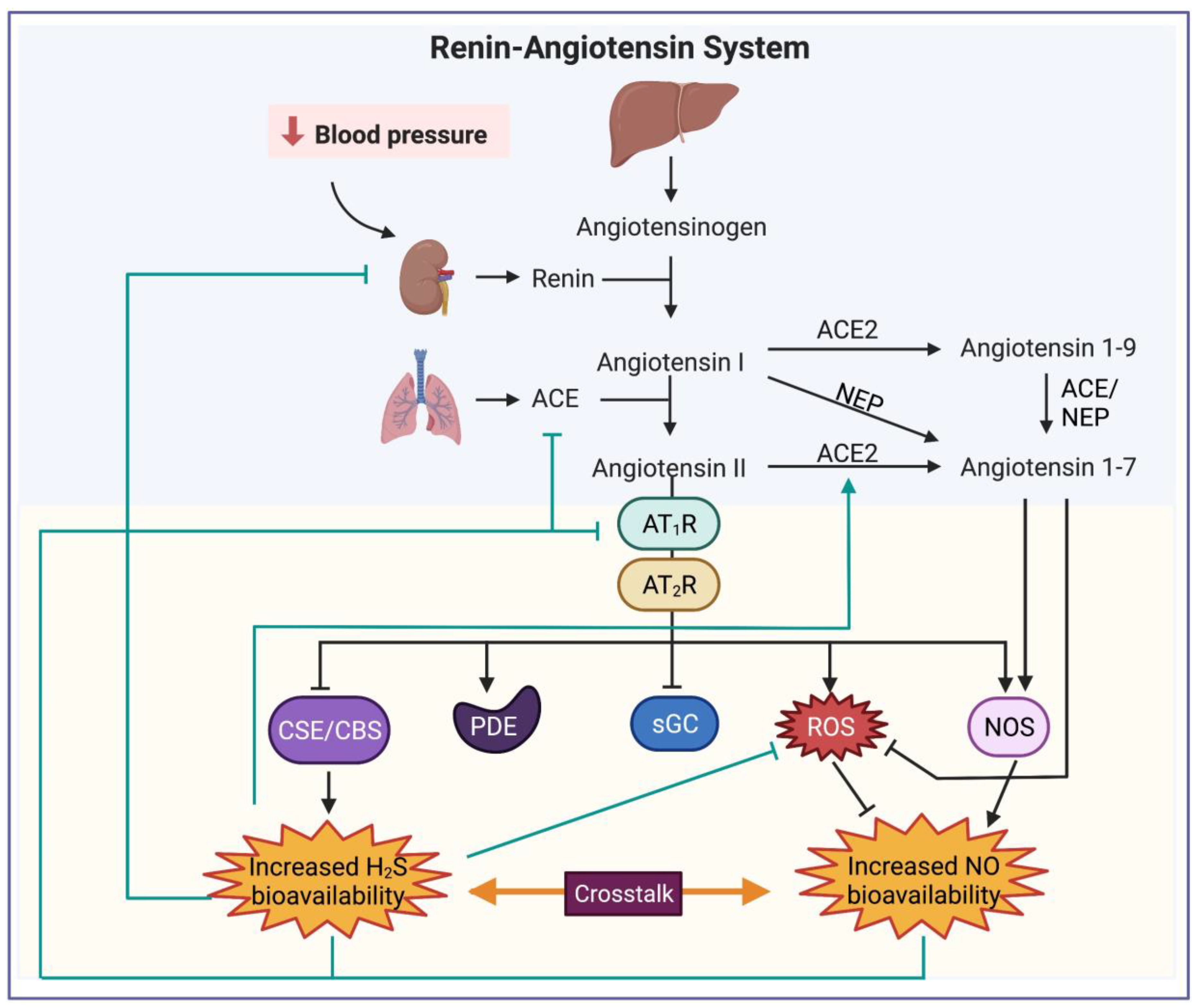
2. Renin–Angiotensin–Aldosterone System
Hypertension and RAAS Inhibitors
3. COVID-19
COVID-19 and RAAS
4. Nitric Oxide and Its Function in COVID-19
5. Hydrogen Sulfide and Its Function in COVID-19
6. Racial Disparities and COVID-19 Outcomes
7. Summary and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 3-MST | 3-mercaptopyruvate sulfurtransferase |
| ACE2 | angiotensin-converting enzyme 2 |
| ALI | acute lung injury |
| Ang | angiotensin |
| ARB | angiotensin receptor blockers |
| ARDS | acute respiratory distress syndrome |
| CAT | cysteine aminotransferase |
| CBS | cystathionine β-synthase |
| cGMP | cyclic guanosine monophosphate |
| COPD | chronic obstructive pulmonary disease |
| COVID-19 | coronavirus disease 2019 |
| CSE | cystathionine γ-lyase |
| eNOS | endothelial nitric oxide synthase |
| GSH | glutathione |
| H2S | hydrogen sulfide |
| iH2S | inhaled hydrogen sulfide |
| IL | interleukin |
| iNO | inhaled nitric oxide |
| iNOS | inducible nitric oxide synthase |
| MV | mechanical ventilation |
| NAC | N-acetylcysteine |
| nNOS | neuronal nitric oxide synthase |
| NO | nitric oxide |
| PDE5 | phosphodiesterase 5 |
| RAAS | renin–angiotensin–aldosterone system |
| ROS | reactive oxygen species |
| S protein | spike protein |
| SARS | severe acute respiratory syndrome |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| sGC | soluble guanylyl cyclase |
| SNP | sodium nitroprusside |
| SOD | superoxide dismutase |
| TMPRSS2 | transmembrane serine protease 2 |
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Godeau, D.; Petit, A.; Richard, I.; Roquelaure, Y.; Descatha, A. Return-to-work, disabilities and occupational health in the age of COVID-19. Scand. J. Work Environ. Health 2021, 47, 408–409. [Google Scholar] [CrossRef] [PubMed]
- CDC. About COVID-19: People with Certain Medical Conditions. Available online: https://www.cdc.gov/coronavirus/2019-ncov/need-extra-precautions/people-with-medical-conditions.html (accessed on 17 August 2022).
- Walubita, T.; Beccia, A.; Boama-Nyarko, E.; Goulding, M.; Herbert, C.; Kloppenburg, J.; Mabry, G.; Masters, G.; McCullers, A.; Forrester, S. Aging and COVID-19 in Minority Populations: A Perfect Storm. Curr. Epidemiol. Rep. 2021, 8, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Wilder, J.M. The Disproportionate Impact of COVID-19 on Racial and Ethnic Minorities in the United States. Clin. Infect. Dis. 2021, 72, 707–709. [Google Scholar] [CrossRef] [PubMed]
- Cagnoni, F.; Njwe, C.A.; Zaninelli, A.; Ricci, A.R.; Daffra, D.; D’Ospina, A.; Preti, P.; Destro, M. Blocking the RAAS at different levels: An update on the use of the direct renin inhibitors alone and in combination. Vasc. Health Risk Manag. 2010, 6, 549–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, G.; Aroor, A.R.; Hill, M.A.; Sowers, J.R. Role of Renin-Angiotensin-Aldosterone System Activation in Promoting Cardiovascular Fibrosis and Stiffness. Hypertension 2018, 72, 537–548. [Google Scholar] [CrossRef]
- Ferrario, C.M.; Strawn, W.B. Role of the renin-angiotensin-aldosterone system and proinflammatory mediators in cardiovascular disease. Am. J. Cardiol. 2006, 98, 121–128. [Google Scholar] [CrossRef]
- Patel, A.B.; Verma, A. COVID-19 and Angiotensin-Converting Enzyme Inhibitors and Angiotensin Receptor Blockers: What Is the Evidence? JAMA 2020, 323, 1769–1770. [Google Scholar] [CrossRef]
- Vaduganathan, M.; Vardeny, O.; Michel, T.; McMurray, J.J.V.; Pfeffer, M.A.; Solomon, S.D. Renin-Angiotensin-Aldosterone System Inhibitors in Patients with COVID-19. N. Engl. J. Med. 2020, 382, 1653–1659. [Google Scholar] [CrossRef]
- Touyz, R.M.; Li, H.; Delles, C. ACE2 the Janus-faced protein—From cardiovascular protection to severe acute respiratory syndrome-coronavirus and COVID-19. Clin. Sci. 2020, 134, 747–750. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, Y.; Liu, Y.; Li, L.; Chen, Y.; Liu, Y.; Feng, Y.; Yosypiv, I.V.; Song, R.; Peng, H. (Pro)renin receptor regulates lung development via the Wnt/beta-catenin signaling pathway. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L202–L211. [Google Scholar] [CrossRef] [PubMed]
- Fountain, J.H.; Lappin, S.L. Physiology, Renin Angiotensin System; StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Hall, J.E.; do Carmo, J.M.; da Silva, A.A.; Wang, Z.; Hall, M.E. Obesity, kidney dysfunction and hypertension: Mechanistic links. Nat. Rev. Nephrol. 2019, 15, 367–385. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Lu, X.; Danser, A.H.J. Revisiting the Brain Renin-Angiotensin System-Focus on Novel Therapies. Curr. Hypertens. Rep. 2019, 21, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, K.L.; Fralick, M.; Zipursky, J.S.; Stall, N.M. Renin-angiotensin-aldosterone system inhibitors and COVID-19. CMAJ 2020, 192, E553–E554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weir, M.R.; Dzau, V.J. The renin-angiotensin-aldosterone system: A specific target for hypertension management. Am. J. Hypertens. 1999, 12, 205S–213S. [Google Scholar] [CrossRef]
- Shimosawa, T. Salt, the renin-angiotensin-aldosterone system and resistant hypertension. Hypertens Res 2013, 36, 657–660. [Google Scholar] [CrossRef] [Green Version]
- Topouchian, J.; El Feghali, R.; Pannier, B.; Wang, S.; Zhao, F.; Smetana, K.; Teo, K.; Asmar, R. Arterial stiffness and pharmacological interventions--the TRanscend arterial stiffNess Substudy (TRANS study). Vasc. Health Risk Manag. 2007, 3, 381–387. [Google Scholar]
- Ma, T.K.; Kam, K.K.; Yan, B.P.; Lam, Y.Y. Renin-angiotensin-aldosterone system blockade for cardiovascular diseases: Current status. Br. J. Pharmacol. 2010, 160, 1273–1292. [Google Scholar] [CrossRef]
- Pradhan, A.; Vishwakarma, P.; Bhandari, M.; Sethi, R.; Narain, V.S. Differential Effects of Combination of Renin-Angiotensin-Aldosterone System Inhibitors on Central Aortic Blood Pressure: A Cross-Sectional Observational Study in Hypertensive Outpatients. Cardiovasc. Ther. 2020, 2020, 4349612. [Google Scholar] [CrossRef]
- Kearney, P.M.; Whelton, M.; Reynolds, K.; Muntner, P.; Whelton, P.K.; He, J. Global burden of hypertension: Analysis of worldwide data. Lancet 2005, 365, 217–223. [Google Scholar] [CrossRef]
- Paul, M.; Poyan Mehr, A.; Kreutz, R. Physiology of local renin-angiotensin systems. Physiol. Rev. 2006, 86, 747–803. [Google Scholar] [CrossRef]
- Macera, M.; De Angelis, G.; Sagnelli, C.; Coppola, N.; Vanvitelli, C.-G. Clinical Presentation of COVID-19: Case Series and Review of the Literature. Int. J. Environ. Res. Public Health 2020, 17, 5062. [Google Scholar] [CrossRef] [PubMed]
- Chathappady House, N.N.; Palissery, S.; Sebastian, H. Corona Viruses: A Review on SARS, MERS and COVID-19. Microbiol. Insights 2021, 14, 11786361211002481. [Google Scholar] [CrossRef] [PubMed]
- Chaplin, S. COVID-19: A brief history and treatments in development. Prescriber 2020, 31, 23–28. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Johns-Hopkins-University-Medicine. Corona Virus Resource Center. Available online: https://coronavirus.jhu.edu/map.html (accessed on 17 August 2022).
- Zou, L.; Ruan, F.; Huang, M.; Liang, L.; Huang, H.; Hong, Z.; Yu, J.; Kang, M.; Song, Y.; Xia, J.; et al. SARS-CoV-2 Viral Load in Upper Respiratory Specimens of Infected Patients. N. Engl. J. Med. 2020, 382, 1177–1179. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, J.; Rhee, C.; Klompas, M. Incidence, Characteristics, and Outcomes of Ventilator-associated Events during the COVID-19 Pandemic. Ann. Am. Thorac. Soc. 2021, 19, 82–89. [Google Scholar] [CrossRef]
- Batah, S.S.; Fabro, A.T. Pulmonary pathology of ARDS in COVID-19: A pathological review for clinicians. Respir. Med. 2021, 176, 106239. [Google Scholar] [CrossRef]
- Suarez-de-la-Rica, A.; Serrano, P.; De-la-Oliva, R.; Sánchez-Díaz, P.; Molinero, P.; Falces-Romero, I.; Ferrando, C.; Rello, J.; Maseda, E. Secondary infections in mechanically ventilated patients with COVID-19: An overlooked matter? Rev. Esp. Quimioter. 2021, 34, 330–336. [Google Scholar] [CrossRef]
- Yoshida, T.; Fujino, Y.; Amato, M.B.P.; Kavanagh, B.P. Fifty Years of Research in ARDS. Spontaneous Breathing during Mechanical Ventilation. Risks, Mechanisms, and Management. Am. J. Respir. Crit. Care Med. 2016, 195, 985–992. [Google Scholar] [CrossRef]
- Hu, B.; Huang, S.; Yin, L. The cytokine storm and COVID-19. J. Med. Virol. 2021, 93, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Fara, A.; Mitrev, Z.; Rosalia, R.A.; Assas, B.M. Cytokine storm and COVID-19: A chronicle of pro-inflammatory cytokines. Open Biol. 2020, 10, 200160. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, T.; Cai, D.; Hu, Z.; Chen, J.a.; Liao, H.; Zhi, L.; Wei, H.; Zhang, Z.; Qiu, Y.; et al. Cytokine storm intervention in the early stages of COVID-19 pneumonia. Cytokine Growth Factor Rev. 2020, 53, 38–42. [Google Scholar] [CrossRef]
- Soy, M.; Keser, G.; Atagündüz, P.; Tabak, F.; Atagündüz, I.; Kayhan, S. Cytokine storm in COVID-19: Pathogenesis and overview of anti-inflammatory agents used in treatment. Clin. Rheumatol. 2020, 39, 2085–2094. [Google Scholar] [CrossRef] [PubMed]
- Klok, F.A.; Kruip, M.J.H.A.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.A.M.P.J.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020, 191, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Zhou, X.; Cheng, Z.; Luo, L.; Zhu, Y.; Lin, W.; Ming, Z.; Chen, W.; Hu, Y. Incidence and impact of disseminated intravascular coagulation in COVID-19 a systematic review and meta-analysis. Thromb. Res. 2021, 201, 23–29. [Google Scholar] [CrossRef]
- Ladikou, E.E.; Sivaloganathan, H.; Milne, K.M.; Arter, W.E.; Ramasamy, R.; Saad, R.; Stoneham, S.M.; Philips, B.; Eziefula, A.C.; Chevassut, T. Von Willebrand factor (vWF): Marker of endothelial damage and thrombotic risk in COVID-19? Clin. Med. 2020, 20, e178–e182. [Google Scholar] [CrossRef]
- Pearson, J.D. Endothelial cell function and thrombosis. Best Pract. Res. Clin. Haematol. 1999, 12, 329–341. [Google Scholar] [CrossRef]
- Melkumyants, A.; Buryachkovskaya, L.; Lomakin, N.; Antonova, O.; Serebruany, V. Mild COVID-19 and Impaired Blood Cell-Endothelial Crosstalk: Considering Long-Term Use of Antithrombotics? Thromb. Haemost. 2022, 122, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Sims, J.T.; Krishnan, V.; Chang, C.-Y.; Engle, S.M.; Casalini, G.; Rodgers, G.H.; Bivi, N.; Nickoloff, B.J.; Konrad, R.J.; de Bono, S.; et al. Characterization of the cytokine storm reflects hyperinflammatory endothelial dysfunction in COVID-19. J. Allergy Clin. Immunol. 2021, 147, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Krzemińska, J.; Wronka, M.; Młynarska, E.; Franczyk, B.; Rysz, J. Arterial Hypertension—Oxidative Stress and Inflammation. Antioxidants 2022, 11, 172. [Google Scholar] [CrossRef]
- Lontchi-Yimagou, E.; Sobngwi, E.; Matsha, T.E.; Kengne, A.P. Diabetes Mellitus and Inflammation. Curr. Diabetes Rep. 2013, 13, 435–444. [Google Scholar] [CrossRef]
- Abdi, A.; Jalilian, M.; Sarbarzeh, P.A.; Vlaisavljevic, Z. Diabetes and COVID-19: A systematic review on the current evidences. Diabetes Res. Clin. Pract. 2020, 166, 108347. [Google Scholar] [CrossRef]
- Savoia, C.; Volpe, M.; Kreutz, R. Hypertension, a Moving Target in COVID-19. Circ. Res. 2021, 128, 1062–1079. [Google Scholar] [CrossRef]
- de Abajo, F.J.; Rodriguez-Martin, S.; Lerma, V.; Mejia-Abril, G.; Aguilar, M.; Garcia-Luque, A.; Laredo, L.; Laosa, O.; Centeno-Soto, G.A.; Angeles Galvez, M.; et al. Use of renin-angiotensin-aldosterone system inhibitors and risk of COVID-19 requiring admission to hospital: A case-population study. Lancet 2020, 395, 1705–1714. [Google Scholar] [CrossRef]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: Molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020, 46, 586–590. [Google Scholar] [CrossRef] [Green Version]
- Turner, A.J.; Hiscox, J.A.; Hooper, N.M. ACE2: From vasopeptidase to SARS virus receptor. Trends Pharmacol. Sci. 2004, 25, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Bourgonje, A.R.; Abdulle, A.E.; Timens, W.; Hillebrands, J.L.; Navis, G.J.; Gordijn, S.J.; Bolling, M.C.; Dijkstra, G.; Voors, A.A.; Osterhaus, A.D.; et al. Angiotensin-converting enzyme 2 (ACE2), SARS-CoV-2 and the pathophysiology of coronavirus disease 2019 (COVID-19). J. Pathol. 2020, 251, 228–248. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Sanchis-Gomar, F.; Lavie, C.J.; Perez-Quilis, C.; Henry, B.M.; Lippi, G. Angiotensin-Converting Enzyme 2 and Antihypertensives (Angiotensin Receptor Blockers and Angiotensin-Converting Enzyme Inhibitors) in Coronavirus Disease 2019. Mayo Clin. Proc. 2020, 95, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, W.O.; Souza dos Santos, R.A.; Faria-Silva, R.; da Mata Machado, L.T.; Schiffrin, E.L.; Touyz, R.M. Angiotensin-(1-7) through receptor Mas mediates endothelial nitric oxide synthase activation via Akt-dependent pathways. Hypertension 2007, 49, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Lumpuy-Castillo, J.; Lorenzo-Almoros, A.; Pello-Lazaro, A.M.; Sanchez-Ferrer, C.; Egido, J.; Tunon, J.; Peiro, C.; Lorenzo, O. Cardiovascular Damage in COVID-19: Therapeutic Approaches Targeting the Renin-Angiotensin-Aldosterone System. Int. J. Mol. Sci. 2020, 21, 6471. [Google Scholar] [CrossRef]
- Hamming, I.; Cooper, M.E.; Haagmans, B.L.; Hooper, N.M.; Korstanje, R.; Osterhaus, A.D.; Timens, W.; Turner, A.J.; Navis, G.; van Goor, H. The emerging role of ACE2 in physiology and disease. J. Pathol. 2007, 212, 1–11. [Google Scholar] [CrossRef]
- Gupte, M.; Boustany-Kari, C.M.; Bharadwaj, K.; Police, S.; Thatcher, S.; Gong, M.C.; English, V.L.; Cassis, L.A. ACE2 is expressed in mouse adipocytes and regulated by a high-fat diet. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R781–R788. [Google Scholar] [CrossRef] [Green Version]
- Sinha, S.; Sehgal, A.; Sehgal, R. Association of ACE2 receptor and ACEIs/ARBs with disease severity in COVID-19. Drug Discov. Ther. 2020, 14, 161–170. [Google Scholar] [CrossRef]
- Bernardi, S.; Toffoli, B.; Zennaro, C.; Tikellis, C.; Monticone, S.; Losurdo, P.; Bellini, G.; Thomas, M.C.; Fallo, F.; Veglio, F.; et al. High-salt diet increases glomerular ACE/ACE2 ratio leading to oxidative stress and kidney damage. Nephrol. Dial. Transplant. 2012, 27, 1793–1800. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Tan, Z.; Zhou, L.; Yang, M.; Peng, L.; Liu, J.; Cai, J.; Yang, R.; Han, J.; Huang, Y.; et al. Effects of Angiotensin II Receptor Blockers and ACE (Angiotensin-Converting Enzyme) Inhibitors on Virus Infection, Inflammatory Status, and Clinical Outcomes in Patients With COVID-19 and Hypertension: A Single-Center Retrospective Study. Hypertension 2020, 76, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Rice, G.I.; Thomas, D.A.; Grant, P.J.; Turner, A.J.; Hooper, N.M. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem. J. 2004, 383, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, L.; Sakagami, H.; Miwa, N. ACE2: The key Molecule for Understanding the Pathophysiology of Severe and Critical Conditions of COVID-19: Demon or Angel? Viruses 2020, 12, 491. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, W.A.; Wyne, K. Renin-Angiotensin-aldosterone system in diabetes and hypertension. J. Clin. Hypertens. 2011, 13, 224–237. [Google Scholar] [CrossRef]
- South, A.M.; Diz, D.I.; Chappell, M.C. COVID-19, ACE2, and the cardiovascular consequences. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H1084–H1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klimas, J.; Olvedy, M.; Ochodnicka-Mackovicova, K.; Kruzliak, P.; Cacanyiova, S.; Kristek, F.; Krenek, P.; Ochodnicky, P. Perinatally administered losartan augments renal ACE2 expression but not cardiac or renal Mas receptor in spontaneously hypertensive rats. J. Cell. Mol. Med. 2015, 19, 1965–1974. [Google Scholar] [CrossRef]
- Talreja, H.; Tan, J.; Dawes, M.; Supershad, S.; Rabindranath, K.; Fisher, J.; Valappil, S.; van der Merwe, V.; Wong, L.; van der Merwe, W.; et al. A consensus statement on the use of angiotensin receptor blockers and angiotensin converting enzyme inhibitors in relation to COVID-19 (corona virus disease 2019). N. Z. Med. J. 2020, 133, 85–87. [Google Scholar]
- Oza, P.P.; Kashfi, K. Utility of NO and H(2)S donating platforms in managing COVID-19: Rationale and promise. Nitric Oxide 2022, 128, 72–102. [Google Scholar] [CrossRef]
- Adusumilli, N.C.; Zhang, D.; Friedman, J.M.; Friedman, A.J. Harnessing nitric oxide for preventing, limiting and treating the severe pulmonary consequences of COVID-19. Nitric Oxide 2020, 103, 4–8. [Google Scholar] [CrossRef]
- Zamanian, R.T.; Pollack, C.V.; Gentile, M.A.; Rashid, M.; Fox, J.C.; Mahaffey, K.W.; de Jesus Perez, V. Outpatient Inhaled Nitric Oxide in a Patient with Vasoreactive Idiopathic Pulmonary Arterial Hypertension and COVID-19 Infection. Am. J. Respir. Crit. Care Med. 2020, 202, 130–132. [Google Scholar] [CrossRef]
- Martel, J.; Ko, Y.-F.; Young, J.D.; Ojcius, D.M. Could nasal nitric oxide help to mitigate the severity of COVID-19? Microbes Infect. 2020, 22, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Pieretti, J.C.; Rubilar, O.; Weller, R.B.; Tortella, G.R.; Seabra, A.B. Nitric oxide (NO) and nanoparticles—Potential small tools for the war against COVID-19 and other human coronavirus infections. Virus Res. 2021, 291, 198202. [Google Scholar] [CrossRef] [PubMed]
- Bruckdorfer, R. The basics about nitric oxide. Mol. Asp. Med. 2005, 26, 3–31. [Google Scholar] [CrossRef] [PubMed]
- Król, M.; Kepinska, M. Human Nitric Oxide Synthase—Its Functions, Polymorphisms, and Inhibitors in the Context of Inflammation, Diabetes and Cardiovascular Diseases. Int. J. Mol. Sci. 2021, 22, 56. [Google Scholar] [CrossRef]
- Guan, S.P.; Seet, R.C.S.; Kennedy, B.K. Does eNOS derived nitric oxide protect the young from severe COVID-19 complications? Ageing Res. Rev. 2020, 64, 101201. [Google Scholar] [CrossRef]
- Vassiliou, A.G.; Zacharis, A.; Keskinidou, C.; Jahaj, E.; Pratikaki, M.; Gallos, P.; Dimopoulou, I.; Kotanidou, A.; Orfanos, S.E. Soluble Angiotensin Converting Enzyme 2 (ACE2) Is Upregulated and Soluble Endothelial Nitric Oxide Synthase (eNOS) Is Downregulated in COVID-19-induced Acute Respiratory Distress Syndrome (ARDS). Pharmaceuticals 2021, 14, 695. [Google Scholar] [CrossRef]
- Guimarães, L.M.F.; Rossini, C.V.T.; Lameu, C. Implications of SARS-CoV-2 infection on eNOS and iNOS activity: Consequences for the respiratory and vascular systems. Nitric Oxide 2021, 111–112, 64–71. [Google Scholar] [CrossRef]
- Nikolaidis, A.; Kramer, R.; Ostojic, S. Nitric Oxide: The Missing Factor in COVID-19 Severity? Med. Sci. 2022, 10, 3. [Google Scholar] [CrossRef]
- Guzik, T.J.; Korbut, R.; Adamek-Guzik, T. Nitric oxide and superoxide in inflammation and immune regulation. J. Physiol. Pharmacol. 2003, 54, 469–487. [Google Scholar]
- Li, Q.; Youn, J.-Y.; Cai, H. Mechanisms and consequences of endothelial nitric oxide synthase dysfunction in hypertension. J. Hypertens. 2015, 33, 1128–1136. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.L. eNOS, metabolic syndrome and cardiovascular disease. Trends Endocrinol. Metab. 2009, 20, 295–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heitsch, H.; Brovkovych, S.; Malinski, T.; Wiemer, G. Angiotensin-(1-7)–Stimulated Nitric Oxide and Superoxide Release From Endothelial Cells. Hypertension 2001, 37, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Rees, C.A.; Rostad, C.A.; Mantus, G.; Anderson, E.J.; Chahroudi, A.; Jaggi, P.; Wrammert, J.; Ochoa, J.B.; Ochoa, A.; Basu, R.K.; et al. Altered amino acid profile in patients with SARS-CoV-2 infection. Proc. Natl. Acad. Sci. USA 2021, 118, e2101708118. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, A.; Thomas, T.; Akpan, I.J.; Reisz, J.A.; Cendali, F.I.; Gamboni, F.; Nemkov, T.; Thangaraju, K.; Katneni, U.; Tanaka, K.; et al. Biological and Clinical Factors Contributing to the Metabolic Heterogeneity of Hospitalized Patients with and without COVID-19. Cells 2021, 10, 2293. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, G.; Coppola, A.; Izzo, R.; Annunziata, A.; Bernardo, M.; Lombardi, A.; Trimarco, V.; Santulli, G.; Trimarco, B. Effects of adding L-arginine orally to standard therapy in patients with COVID-19: A randomized, double-blind, placebo-controlled, parallel-group trial. Results of the first interim analysis. EClinicalMedicine 2021, 40, 101125. [Google Scholar] [CrossRef] [PubMed]
- Saura, M.; Zaragoza, C.; McMillan, A.; Quick, R.A.; Hohenadl, C.; Lowenstein, J.M.; Lowenstein, C.J. An Antiviral Mechanism of Nitric Oxide: Inhibition of a Viral Protease. Immunity 1999, 10, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Regev-Shoshani, G.; Vimalanathan, S.; McMullin, B.; Road, J.; Av-Gay, Y.; Miller, C. Gaseous nitric oxide reduces influenza infectivity in vitro. Nitric Oxide 2013, 31, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Mokry, R.L.; Schumacher, M.L.; Hogg, N.; Terhune, S.S. Nitric Oxide Circumvents Virus-Mediated Metabolic Regulation during Human Cytomegalovirus Infection. mBio 2020, 11, e02630-20. [Google Scholar] [CrossRef]
- Xu, W.; Zheng, S.; Dweik, R.A.; Erzurum, S.C. Role of epithelial nitric oxide in airway viral infection. Free. Radic. Biol. Med. 2006, 41, 19–28. [Google Scholar] [CrossRef]
- Gamba, G.; Cavalieri, H.; Courreges, M.C.; Massouh, E.J.; Benencia, F. Early inhibition of nitric oxide production increases HSV-1 intranasal infection. J. Med. Virol. 2004, 73, 313–322. [Google Scholar] [CrossRef]
- Sanders, S.P.; Siekierski, E.S.; Porter, J.D.; Richards, S.M.; Proud, D. Nitric oxide inhibits rhinovirus-induced cytokine production and viral replication in a human respiratory epithelial cell line. J. Virol. 1998, 72, 934–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klingström, J.; Åkerström, S.; Hardestam, J.; Stoltz, M.; Simon, M.; Falk, K.I.; Mirazimi, A.; Rottenberg, M.; Lundkvist, Å. Nitric oxide and peroxynitrite have different antiviral effects against hantavirus replication and free mature virions. Eur. J. Immunol. 2006, 36, 2649–2657. [Google Scholar] [CrossRef] [PubMed]
- Zaragoza, C.; Ocampo, C.J.; Saura, M.; McMillan, A.; Lowenstein, C.J. Nitric oxide inhibition of coxsackievirus replication in vitro. J. Clin. Investig. 1997, 100, 1760–1767. [Google Scholar] [CrossRef] [PubMed]
- Flodström, M.; Horwitz, M.S.; Maday, A.; Balakrishna, D.; Rodriguez, E.; Sarvetnick, N. A Critical Role for Inducible Nitric Oxide Synthase in Host Survival Following Coxsackievirus B4 Infection. Virology 2001, 281, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, N.; Buller, R.M.; Karupiah, G. Gamma interferon-induced, nitric oxide-mediated inhibition of vaccinia virus replication. J. Virol. 1995, 69, 910–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akerström, S.; Mousavi-Jazi, M.; Klingström, J.; Leijon, M.; Lundkvist, A.; Mirazimi, A. Nitric oxide inhibits the replication cycle of severe acute respiratory syndrome coronavirus. J. Virol. 2005, 79, 1966–1969. [Google Scholar] [CrossRef] [Green Version]
- Akerström, S.; Gunalan, V.; Keng, C.T.; Tan, Y.-J.; Mirazimi, A. Dual effect of nitric oxide on SARS-CoV replication: Viral RNA production and palmitoylation of the S protein are affected. Virology 2009, 395, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Keyaerts, E.; Vijgen, L.; Chen, L.; Maes, P.; Hedenstierna, G.; Van Ranst, M. Inhibition of SARS-coronavirus infection in vitro by S-nitroso-N-acetylpenicillamine, a nitric oxide donor compound. Int. J. Infect. Dis. 2004, 8, 223–226. [Google Scholar] [CrossRef] [Green Version]
- Akaberi, D.; Krambrich, J.; Ling, J.; Luni, C.; Hedenstierna, G.; Järhult, J.D.; Lennerstrand, J.; Lundkvist, Å. Mitigation of the replication of SARS-CoV-2 by nitric oxide in vitro. Redox Biol. 2020, 37, 101734. [Google Scholar] [CrossRef]
- Winchester, S.; John, S.; Jabbar, K.; John, I. Clinical efficacy of nitric oxide nasal spray (NONS) for the treatment of mild COVID-19 infection. J. Infect. 2021, 83, 237–279. [Google Scholar] [CrossRef]
- Tsai, M.-L.; Tsou, C.-C.; Liaw, W.-F. Dinitrosyl Iron Complexes (DNICs): From Biomimetic Synthesis and Spectroscopic Characterization toward Unveiling the Biological and Catalytic Roles of DNICs. Acc. Chem. Res. 2015, 48, 1184–1193. [Google Scholar] [CrossRef] [PubMed]
- Pectol, D.C.; Delaney, C.R.; Zhu, J.; Mellott, D.M.; Katzfuss, A.; Taylor, Z.W.; Meek, T.D.; Darensbourg, M.Y. Dinitrosyl iron complexes (DNICs) as inhibitors of the SARS-CoV-2 main protease. Chem. Commun. 2021, 57, 8352–8355. [Google Scholar] [CrossRef] [PubMed]
- Vanin, A.F.; Pekshev, A.V.; Vagapov, A.B.; Sharapov, N.A.; Lakomkin, V.L.; Abramov, A.A.; Timoshin, A.A.; Kapelko, V.I. Gaseous Nitric Oxide and Dinitrosyl Iron Complexes with Thiol-Containing Ligands as Potential Medicines that Can Relieve COVID-19. Biophysics 2021, 66, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Parikh, R.; Wilson, C.; Weinberg, J.; Gavin, D.; Murphy, J.; Reardon, C.C. Inhaled nitric oxide treatment in spontaneously breathing COVID-19 patients. Ther. Adv. Respir. Dis. 2020, 14, 1753466620933510. [Google Scholar] [CrossRef]
- Garfield, B.; McFadyen, C.; Briar, C.; Bleakley, C.; Vlachou, A.; Baldwin, M.; Lees, N.; Price, S.; Ledot, S.; McCabe, C.; et al. Potential for personalised application of inhaled nitric oxide in COVID-19 pneumonia. Br. J. Anaesth. 2021, 126, e72–e75. [Google Scholar] [CrossRef]
- Wiegand, S.B.; Safaee Fakhr, B.; Carroll, R.W.; Zapol, W.M.; Kacmarek, R.M.; Berra, L. Rescue Treatment With High-Dose Gaseous Nitric Oxide in Spontaneously Breathing Patients With Severe Coronavirus Disease 2019. Crit. Care Explor. 2020, 2, e0277. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.; Kirschenbaum, L.A.; LaRow, C.; Berna, G.; Griffin, K.; Astiz, M.E. Augmentation of Platelet and Endothelial Cell Enos Activity Decreases Sepsis-Related Neutrophil-Endothelial Cell Interactions. Shock 2010, 33, 242–246. [Google Scholar] [CrossRef]
- Kosutova, P.; Kolomaznik, M.; Calkovska, A.; Mokra, D.; Mikolka, P. Nitric-Oxide-Releasing Dexamethasone Derivative NCX-1005 Improves Lung Function and Attenuates Inflammation in Experimental Lavage-Induced ARDS. Pharmaceutics 2021, 13, 2092. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, P.; Tripathi, P.; Kashyap, L.; Singh, V. The role of nitric oxide in inflammatory reactions. FEMS Immunol. Med. Microbiol. 2007, 51, 443–452. [Google Scholar] [CrossRef] [Green Version]
- Connelly, L.; Palacios-Callender, M.; Ameixa, C.; Moncada, S.; Hobbs, A.J. Biphasic Regulation of NF-κB Activity Underlies the Pro- and Anti-Inflammatory Actions of Nitric Oxide. J. Immunol. 2001, 166, 3873–3881. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Peleli, M.; Zollbrecht, C.; Giulietti, A.; Terrando, N.; Lundberg, J.O.; Weitzberg, E.; Carlström, M. Inorganic nitrite attenuates NADPH oxidase-derived superoxide generation in activated macrophages via a nitric oxide-dependent mechanism. Free. Radic. Biol. Med. 2015, 83, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lu, J.; Shi, J.; Lin, X.; Dong, J.; Zhang, S.; Liu, Y.; Tong, Q. Central administration of angiotensin-(1–7) stimulates nitric oxide release and upregulates the endothelial nitric oxide synthase expression following focal cerebral ischemia/reperfusion in rats. Neuropeptides 2008, 42, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Cerrato, B.D.; Frasch, A.P.; Nakagawa, P.; Longo-Carbajosa, N.; Peña, C.; Höcht, C.; Gironacci, M.M. Angiotensin-(1–7) upregulates central nitric oxide synthase in spontaneously hypertensive rats. Brain Res. 2012, 1453, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.-J.; Shih, C.-C.; Chang, K.-Y.; Liao, M.-H.; Liaw, W.-J.; Wu, C.-C.; Tsao, C.-M. Angiotensin-(1–7) treatment blocks lipopolysaccharide-induced organ damage, platelet dysfunction, and IL-6 and nitric oxide production in rats. Sci. Rep. 2021, 11, 610. [Google Scholar] [CrossRef] [PubMed]
- Regenhardt, R.W.; Desland, F.; Mecca, A.P.; Pioquinto, D.J.; Afzal, A.; Mocco, J.; Sumners, C. Anti-inflammatory effects of angiotensin-(1-7) in ischemic stroke. Neuropharmacology 2013, 71, 154–163. [Google Scholar] [CrossRef] [Green Version]
- Rajendran, S.; Chirkov, Y.Y.; Horowitz, J.D. Potentiation of platelet responsiveness to nitric oxide by angiotensin-(1–7) is associated with suppression of superoxide release. Platelets 2007, 18, 158–164. [Google Scholar] [CrossRef]
- Tesanovic, S.; Vinh, A.; Gaspari, T.A.; Casley, D.; Widdop, R.E. Vasoprotective and Atheroprotective Effects of Angiotensin (1–7) in Apolipoprotein E–Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1606–1613. [Google Scholar] [CrossRef] [Green Version]
- Crassous, P.-A.; Couloubaly, S.; Huang, C.; Zhou, Z.; Baskaran, P.; Kim, D.D.; Papapetropoulos, A.; Fioramonti, X.; Durán, W.N.; Beuve, A. Soluble guanylyl cyclase is a target of angiotensin II-induced nitrosative stress in a hypertensive rat model. Am. J. Physiol.-Heart Circ. Physiol. 2012, 303, H597–H604. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Rybalkin, S.D.; Pi, X.; Wang, Y.; Zhang, C.; Munzel, T.; Beavo, J.A.; Berk, B.C.; Yan, C. Upregulation of Phosphodiesterase 1A1 Expression Is Associated With the Development of Nitrate Tolerance. Circulation 2001, 104, 2338–2343. [Google Scholar] [CrossRef] [Green Version]
- Galougahi, K.K.; Liu, C.C.; Gentile, C.; Kok, C.; Nunez, A.; Garcia, A.; Fry, N.A.S.; Davies, M.J.; Hawkins, C.L.; Rasmussen, H.H.; et al. Glutathionylation Mediates Angiotensin II–Induced eNOS Uncoupling, Amplifying NADPH Oxidase–Dependent Endothelial Dysfunction. J. Am. Heart Assoc. 2014, 3, e000731. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, E.; Tamamaki, N.; Nakamura, T.; Kataoka, K.; Tokutomi, Y.; Dong, Y.-F.; Fukuda, M.; Matsuba, S.; Ogawa, H.; Kim-Mitsuyama, S. Excess Salt Causes Cerebral Neuronal Apoptosis and Inflammation in Stroke-Prone Hypertensive Rats Through Angiotensin II-Induced NADPH Oxidase Activation. Stroke 2008, 39, 3049–3056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.-Y.; Wauquier, F.; Eid, A.A.; Roman, L.J.; Ghosh-Choudhury, G.; Khazim, K.; Block, K.; Gorin, Y. Nox4 NADPH Oxidase Mediates Peroxynitrite-dependent Uncoupling of Endothelial Nitric-oxide Synthase and Fibronectin Expression in Response to Angiotensin II: ROLE OF MITOCHONDRIAL REACTIVE OXYGEN SPECIES *. J. Biol. Chem. 2013, 288, 28668–28686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashi, Y.; Oshima, T.; Ono, N.; Hiraga, H.; Yoshimura, M.; Watanabe, M.; Matsuura, H.; Kambe, M.; Kajiyama, G. Intravenous administration of L-arginine inhibits angiotensin-converting enzyme in humans. J. Clin. Endocrinol. Metab. 1995, 80, 2198–2202. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, A.; Fernández-Alfonso, M.S.; Sánchez de Rojas, R.; Ortega, T.; Paul, M.; González, C. Modulation of angiotensin-converting enzyme by nitric oxide. Br. J. Pharmacol. 1998, 124, 291–298. [Google Scholar] [CrossRef] [Green Version]
- Kumar, K.V.; Das, U.N. Effect of cis-Unsaturated Fatty Acids, Prostaglandins, and Free Radicals on Angiotensin-Converting Enzyme Activity in Vitro. Proc. Soc. Exp. Biol. Med. 1997, 214, 374–379. [Google Scholar] [CrossRef]
- Ichiki, T.; Usui, M.; Kato, M.; Funakoshi, Y.; Ito, K.; Egashira, K.; Takeshita, A. Downregulation of Angiotensin II Type 1 Receptor Gene Transcription by Nitric Oxide. Hypertension 1998, 31, 342–346. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.M.; Zheng, H.; Li, Y.-F.; Patel, K.P. Nitric oxide inhibits the expression of AT1 receptors in neurons. Am. J. Physiol.-Cell Physiol. 2012, 302, C1162–C1173. [Google Scholar] [CrossRef] [Green Version]
- Nägele, M.P.; Haubner, B.; Tanner, F.C.; Ruschitzka, F.; Flammer, A.J. Endothelial dysfunction in COVID-19: Current findings and therapeutic implications. Atherosclerosis 2020, 314, 58–62. [Google Scholar] [CrossRef]
- Wallace, J.L.; McKnight, W.; Del Soldato, P.; Baydoun, A.R.; Cirino, G. Anti-thrombotic effects of a nitric oxide-releasing, gastric-sparing aspirin derivative. J. Clin. Investig. 1995, 96, 2711–2718. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, D.R.; Beigi, F.; Treuer, A.V.; Hare, J.M. Deficient ryanodine receptor S-nitrosylation increases sarcoplasmic reticulum calcium leak and arrhythmogenesis in cardiomyocytes. Proc. Natl. Acad. Sci. USA 2007, 104, 20612–20617. [Google Scholar] [CrossRef] [Green Version]
- Iravanian, S.; Dudley, S.C. The renin-angiotensin-aldosterone system (RAAS) and cardiac arrhythmias. Heart Rhythm 2008, 5, S12–S17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutler, M.J.; Plummer, B.N.; Wan, X.; Sun, Q.A.; Hess, D.; Liu, H.; Deschenes, I.; Rosenbaum, D.S.; Stamler, J.S.; Laurita, K.R. Aberrant S-nitrosylation mediates calcium-triggered ventricular arrhythmia in the intact heart. Proc. Natl. Acad. Sci. USA 2012, 109, 18186–18191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iciek, M.; Bilska-Wilkosz, A.; Kozdrowicki, M.; Górny, M. Reactive Sulfur Compounds in the Fight against COVID-19. Antioxidants 2022, 11, 1053. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.D.; Snyder, S.H. Gasotransmitter hydrogen sulfide signaling in neuronal health and disease. Biochem. Pharmacol. 2018, 149, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Bazhanov, N.; Escaffre, O.; Freiberg, A.N.; Garofalo, R.P.; Casola, A. Broad-Range Antiviral Activity of Hydrogen Sulfide Against Highly Pathogenic RNA Viruses. Sci. Rep. 2017, 7, 41029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Ma, Y.; Escaffre, O.; Ivanciuc, T.; Komaravelli, N.; Kelley, J.P.; Coletta, C.; Szabo, C.; Rockx, B.; Garofalo, R.P.; et al. Role of hydrogen sulfide in paramyxovirus infections. J. Virol. 2015, 89, 5557–5568. [Google Scholar] [CrossRef] [Green Version]
- Pozzi, G.; Masselli, E.; Gobbi, G.; Mirandola, P.; Taborda-Barata, L.; Ampollini, L.; Carbognani, P.; Micheloni, C.; Corazza, F.; Galli, D.; et al. Hydrogen Sulfide Inhibits TMPRSS2 in Human Airway Epithelial Cells: Implications for SARS-CoV-2 Infection. Biomedicines 2021, 9, 1273. [Google Scholar] [CrossRef]
- Kimura, Y.; Goto, Y.-I.; Kimura, H. Hydrogen Sulfide Increases Glutathione Production and Suppresses Oxidative Stress in Mitochondria. Antioxid. Redox Signal. 2009, 12, 1–13. [Google Scholar] [CrossRef]
- Dugbartey, G.J.; Alornyo, K.K.; Ohene, B.O.; Boima, V.; Antwi, S.; Sener, A. Renal consequences of the novel coronavirus disease 2019 (COVID-19) and hydrogen sulfide as a potential therapy. Nitric Oxide 2022, 120, 16–25. [Google Scholar] [CrossRef]
- Rushworth, G.F.; Megson, I.L. Existing and potential therapeutic uses for N-acetylcysteine: The need for conversion to intracellular glutathione for antioxidant benefits. Pharmacol. Ther. 2014, 141, 150–159. [Google Scholar] [CrossRef]
- Hsu, C.-N.; Hou, C.-Y.; Chang-Chien, G.-P.; Lin, S.; Tain, Y.-L. Maternal N-Acetylcysteine Therapy Prevents Hypertension in Spontaneously Hypertensive Rat Offspring: Implications of Hydrogen Sulfide-Generating Pathway and Gut Microbiota. Antioxidants 2020, 9, 856. [Google Scholar] [CrossRef] [PubMed]
- De Flora, S.; Balansky, R.; La Maestra, S. Rationale for the use of N-acetylcysteine in both prevention and adjuvant therapy of COVID-19. FASEB J. 2020, 34, 13185–13193. [Google Scholar] [CrossRef] [PubMed]
- Manček-Keber, M.; Hafner-Bratkovič, I.; Lainšček, D.; Benčina, M.; Govednik, T.; Orehek, S.; Plaper, T.; Jazbec, V.; Bergant, V.; Grass, V.; et al. Disruption of disulfides within RBD of SARS-CoV-2 spike protein prevents fusion and represents a target for viral entry inhibition by registered drugs. FASEB J. 2021, 35, e21651. [Google Scholar] [CrossRef]
- Li, T.; Zhao, B.; Wang, C.; Wang, H.; Liu, Z.; Li, W.; Jin, H.; Tang, C.; Du, J. Regulatory Effects of Hydrogen Sulfide on IL-6, IL-8 and IL-10 Levels in the Plasma and Pulmonary Tissue of Rats with Acute Lung Injury. Exp. Biol. Med. 2008, 233, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Faller, S.; Seiler, R.; Donus, R.; Engelstaedter, H.; Hoetzel, A.; Spassov, S.G. Pre- and posttreatment with hydrogen sulfide prevents ventilator-induced lung injury by limiting inflammation and oxidation. PLoS ONE 2017, 12, e0176649. [Google Scholar] [CrossRef] [Green Version]
- Gaynitdinova, V.; Avdeev, S.; Merzhoeva, Z.; Berikkhanov, Z.; Medvedeva, I.; Gorbacheva, T. N-acetylcysteine as a part of complex treatment of moderate COVID-associated pneumonia. Pulmonologiya 2021, 31, 21–29. [Google Scholar] [CrossRef]
- Ibrahim, H.; Perl, A.; Smith, D.; Lewis, T.; Kon, Z.; Goldenberg, R.; Yarta, K.; Staniloae, C.; Williams, M. Therapeutic blockade of inflammation in severe COVID-19 infection with intravenous N-acetylcysteine. Clin. Immunol. 2020, 219, 108544. [Google Scholar] [CrossRef]
- Pagliaro, P.; Penna, C. ACE/ACE2 Ratio: A Key Also in 2019 Coronavirus Disease (COVID-19)? Front. Med. 2020, 7, 335. [Google Scholar] [CrossRef]
- Banu, N.; Panikar, S.S.; Leal, L.R.; Leal, A.R. Protective role of ACE2 and its downregulation in SARS-CoV-2 infection leading to Macrophage Activation Syndrome: Therapeutic implications. Life Sci. 2020, 256, 117905. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C.; et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020, 63, 364–374. [Google Scholar] [CrossRef] [Green Version]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Qi, Y.; Chen, S.; Wang, J.; Tang, C.; Du, J.; Jin, H.; Huang, Y. Angiotensin II downregulates vascular endothelial cell hydrogen sulfide production by enhancing cystathionine γ-lyase degradation through ROS-activated ubiquitination pathway. Biochem. Biophys. Res. Commun. 2019, 514, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Zeng, H.; Gao, L.; Gu, T.; Wang, C.; Zhang, H. Hydrogen Sulfide Attenuates Atherosclerosis in a Partially Ligated Carotid Artery Mouse model via Regulating Angiotensin Converting Enzyme 2 Expression. Front. Physiol. 2017, 8, 782. [Google Scholar] [CrossRef]
- Li, Z.; Organ Chelsea, L.; Kang, J.; Polhemus David, J.; Trivedi Rishi, K.; Sharp Thomas, E.; Jenkins Jack, S.; Tao, Y.-x.; Xian, M.; Lefer David, J. Hydrogen Sulfide Attenuates Renin Angiotensin and Aldosterone Pathological Signaling to Preserve Kidney Function and Improve Exercise Tolerance in Heart Failure. JACC Basic Transl. Sci. 2018, 3, 796–809. [Google Scholar] [CrossRef]
- Laggner, H.; Hermann, M.; Esterbauer, H.; Muellner, M.K.; Exner, M.; Gmeiner, B.M.K.; Kapiotis, S. The novel gaseous vasorelaxant hydrogen sulfide inhibits angiotensin-converting enzyme activity of endothelial cells. J. Hypertens. 2007, 25, 2100–2104. [Google Scholar] [CrossRef]
- Liu, S.-Y.; Duan, X.-C.; Jin, S.; Teng, X.; Xiao, L.; Xue, H.-M.; Wu, Y.-M. Hydrogen Sulfide Improves Myocardial Remodeling via Downregulated Angiotensin Ⅱ/AT1R Pathway in Renovascular Hypertensive Rats. Am. J. Hypertens. 2017, 30, 67–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Q.; Feng, X.; Xue, H.; Teng, X.; Jin, S.; Duan, X.; Xiao, L.; Wu, Y. Maternal Renovascular Hypertensive Rats Treatment With Hydrogen Sulfide Increased the Methylation of AT1b Gene in Offspring. Am. J. Hypertens. 2017, 30, 1220–1227. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Liu, Y.-H.; Goh, H.S.; Wang, J.J.X.; Yong, Q.-C.; Wang, R.; Bian, J.-S. Hydrogen Sulfide Inhibits Plasma Renin Activity. J. Am. Soc. Nephrol. 2010, 21, 993. [Google Scholar] [CrossRef] [Green Version]
- Altaany, Z.; Ju, Y.; Yang, G.; Wang, R. The coordination of S-sulfhydration, S-nitrosylation, and phosphorylation of endothelial nitric oxide synthase by hydrogen sulfide. Sci. Signal. 2014, 7, ra87. [Google Scholar] [CrossRef]
- Qin, Y.-R.; You, S.-J.; Zhang, Y.; Li, Q.; Wang, X.-H.; Wang, F.; Hu, L.-F.; Liu, C.-F. Hydrogen sulfide attenuates ferric chloride-induced arterial thrombosis in rats. Free. Radic. Res. 2016, 50, 654–665. [Google Scholar] [CrossRef]
- Grambow, E.; Leppin, C.; Leppin, K.; Kundt, G.; Klar, E.; Frank, M.; Vollmar, B. The effects of hydrogen sulfide on platelet-leukocyte aggregation and microvascular thrombolysis. Platelets 2017, 28, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Grambow, E.; Mueller-Graf, F.; Delyagina, E.; Frank, M.; Kuhla, A.; Vollmar, B. Effect of the hydrogen sulfide donor GYY4137 on platelet activation and microvascular thrombus formation in mice. Platelets 2014, 25, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.-L.; Liu, X.-H.; Gong, Q.-H.; Wu, D.; Zhu, Y.-Z. Hydrogen Sulfide Attenuated Tumor Necrosis Factor-α-Induced Inflammatory Signaling and Dysfunction in Vascular Endothelial Cells. PLoS ONE 2011, 6, e19766. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, F.; Davinelli, S.; Krishnan, S.; Gallo, R.C.; Scapagnini, G.; Zella, D.; Curreli, S. Sulfur compounds block MCP-1 production by Mycoplasma fermentans-infected macrophages through NF-κB inhibition. J. Transl. Med. 2014, 145. [Google Scholar] [CrossRef] [Green Version]
- Oh, G.-S.; Pae, H.-O.; Lee, B.-S.; Kim, B.-N.; Kim, J.-M.; Kim, H.-R.; Jeon, S.B.; Jeon, W.K.; Chae, H.-J.; Chung, H.-T. Hydrogen sulfide inhibits nitric oxide production and nuclear factor-κB via heme oxygenase-1 expression in RAW264.7 macrophages stimulated with lipopolysaccharide. Free. Radic. Biol. Med. 2006, 41, 106–119. [Google Scholar] [CrossRef]
- Liu, Z.; Han, Y.; Li, L.; Lu, H.; Meng, G.; Li, X.; Shirhan, M.; Peh, M.T.; Xie, L.; Zhou, S.; et al. The hydrogen sulfide donor, GYY4137, exhibits anti-atherosclerotic activity in high fat fed apolipoprotein E(-/-) mice. Br. J. Pharmacol. 2013, 169, 1795–1809. [Google Scholar] [CrossRef] [Green Version]
- Lin, F.; Yang, Y.; Wei, S.; Huang, X.; Peng, Z.; Ke, X.; Zeng, Z.; Song, Y. Hydrogen Sulfide Protects Against High Glucose-Induced Human Umbilical Vein Endothelial Cell Injury Through Activating PI3K/Akt/eNOS Pathway. Drug Des. Dev. Ther. 2020, 14, 621–633. [Google Scholar] [CrossRef] [Green Version]
- Al-Magableh, M.R.; Kemp-Harper, B.K.; Hart, J.L. Hydrogen sulfide treatment reduces blood pressure and oxidative stress in angiotensin II-induced hypertensive mice. Hypertens. Res. 2015, 38, 13–20. [Google Scholar] [CrossRef]
- Merai, R.; Siegel, C.; Rakotz, M.; Basch, P.; Wright, J.; Wong, B.; Dhsc; Thorpe, P. CDC Grand Rounds: A Public Health Approach to Detect and Control Hypertension. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 1261–1264. [Google Scholar] [CrossRef] [Green Version]
- Bosworth, H.B.; Powers, B.; Grubber, J.M.; Thorpe, C.T.; Olsen, M.K.; Orr, M.; Oddone, E.Z. Racial differences in blood pressure control: Potential explanatory factors. J. Gen. Intern. Med. 2008, 23, 692–698. [Google Scholar] [CrossRef]
- Helmer, A.; Slater, N.; Smithgall, S. A Review of ACE Inhibitors and ARBs in Black Patients With Hypertension. Ann. Pharmacother. 2018, 52, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Flaten, H.K.; Monte, A.A. The Pharmacogenomic and Metabolomic Predictors of ACE Inhibitor and Angiotensin II Receptor Blocker Effectiveness and Safety. Cardiovasc. Drugs Ther. 2017, 31, 471–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtz, T.W.; DiCarlo, S.E.; Pravenec, M.; Morris, R.C., Jr. An Appraisal of Methods Recently Recommended for Testing Salt Sensitivity of Blood Pressure. J. Am. Heart Assoc. 2017, 6, e005653. [Google Scholar] [CrossRef] [PubMed]
- Ogedegbe, G.; Shah, N.R.; Phillips, C.; Goldfeld, K.; Roy, J.; Guo, Y.; Gyamfi, J.; Torgersen, C.; Capponi, L.; Bangalore, S. Comparative Effectiveness of Angiotensin-Converting Enzyme Inhibitor-Based Treatment on Cardiovascular Outcomes in Hypertensive Blacks Versus Whites. J. Am. Coll. Cardiol. 2015, 66, 1224–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bangalore, S.; Ogedegbe, G.; Gyamfi, J.; Guo, Y.; Roy, J.; Goldfeld, K.; Torgersen, C.; Capponi, L.; Phillips, C.; Shah, N.R. Outcomes with Angiotensin-converting Enzyme Inhibitors vs Other Antihypertensive Agents in Hypertensive Blacks. Am. J. Med. 2015, 128, 1195–1203. [Google Scholar] [CrossRef] [Green Version]
- Barreras, A.; Gurk-Turner, C. Angiotensin II receptor blockers. Bayl. Univ. Med. Cent. Proc. 2003, 16, 123–126. [Google Scholar] [CrossRef] [Green Version]
- Kostis, J.B.; Kim, H.J.; Rusnak, J.; Casale, T.; Kaplan, A.; Corren, J.; Levy, E. Incidence and characteristics of angioedema associated with enalapril. Arch. Intern. Med. 2005, 165, 1637–1642. [Google Scholar] [CrossRef] [Green Version]
- Kirby, T. Evidence mounts on the disproportionate effect of COVID-19 on ethnic minorities. Lancet Respir. Med. 2020, 8, 547–548. [Google Scholar] [CrossRef]
- Dorn, A.V.; Cooney, R.E.; Sabin, M.L. COVID-19 exacerbating inequalities in the US. Lancet 2020, 395, 1243–1244. [Google Scholar] [CrossRef]
- Liverpool, L. Why are ethnic minorities worse affected? New Sci. 2020, 246, 11. [Google Scholar] [CrossRef]
- Fuschillo, S.; Palomba, L.; Capparelli, R.; Motta, A.; Maniscalco, M. Nitric Oxide and Hydrogen Sulfide: A Nice Pair in the Respiratory System. Curr. Med. Chem. 2020, 27, 7136–7148. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assimakopoulos, S.F.; Marangos, M. N-acetyl-cysteine may prevent COVID-19-associated cytokine storm and acute respiratory distress syndrome. Med. Hypotheses 2020, 140, 109778. [Google Scholar] [CrossRef] [PubMed]
- Smyth, L.J.; Canadas-Garre, M.; Cappa, R.C.; Maxwell, A.P.; McKnight, A.J. Genetic associations between genes in the renin-angiotensin-aldosterone system and renal disease: A systematic review and meta-analysis. BMJ Open 2019, 9, e026777. [Google Scholar] [CrossRef] [Green Version]
- Rossaint, R.; Falke, K.J.; Lopez, F.; Slama, K.; Pison, U.; Zapol, W.M. Inhaled nitric oxide for the adult respiratory distress syndrome. N. Engl. J. Med. 1993, 328, 399–405. [Google Scholar] [CrossRef]
- Gonzalez-Reyes, S.; Guzman-Beltran, S.; Medina-Campos, O.N.; Pedraza-Chaverri, J. Curcumin pretreatment induces Nrf2 and an antioxidant response and prevents hemin-induced toxicity in primary cultures of cerebellar granule neurons of rats. Oxid. Med. Cell. Longev. 2013, 2013, 801418. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranjbar, T.; Oza, P.P.; Kashfi, K. The Renin–Angiotensin-Aldosterone System, Nitric Oxide, and Hydrogen Sulfide at the Crossroads of Hypertension and COVID-19: Racial Disparities and Outcomes. Int. J. Mol. Sci. 2022, 23, 13895. https://doi.org/10.3390/ijms232213895
Ranjbar T, Oza PP, Kashfi K. The Renin–Angiotensin-Aldosterone System, Nitric Oxide, and Hydrogen Sulfide at the Crossroads of Hypertension and COVID-19: Racial Disparities and Outcomes. International Journal of Molecular Sciences. 2022; 23(22):13895. https://doi.org/10.3390/ijms232213895
Chicago/Turabian StyleRanjbar, Tara, Palak P. Oza, and Khosrow Kashfi. 2022. "The Renin–Angiotensin-Aldosterone System, Nitric Oxide, and Hydrogen Sulfide at the Crossroads of Hypertension and COVID-19: Racial Disparities and Outcomes" International Journal of Molecular Sciences 23, no. 22: 13895. https://doi.org/10.3390/ijms232213895
APA StyleRanjbar, T., Oza, P. P., & Kashfi, K. (2022). The Renin–Angiotensin-Aldosterone System, Nitric Oxide, and Hydrogen Sulfide at the Crossroads of Hypertension and COVID-19: Racial Disparities and Outcomes. International Journal of Molecular Sciences, 23(22), 13895. https://doi.org/10.3390/ijms232213895