
Cyclic GMP-Dependent Regulation of Vascular Tone and Blood Pressure Involves Cysteine-Rich LIM-Only Protein 4 (CRP4)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Expression Pattern of Vascular CRP4 and Different Components of the NO-GC/cGMP/cGKI Pathway
2.2. cGMP Via CRP4 Affects Norepinephrine-Induced (Ca2+)i Signals in VSMCs
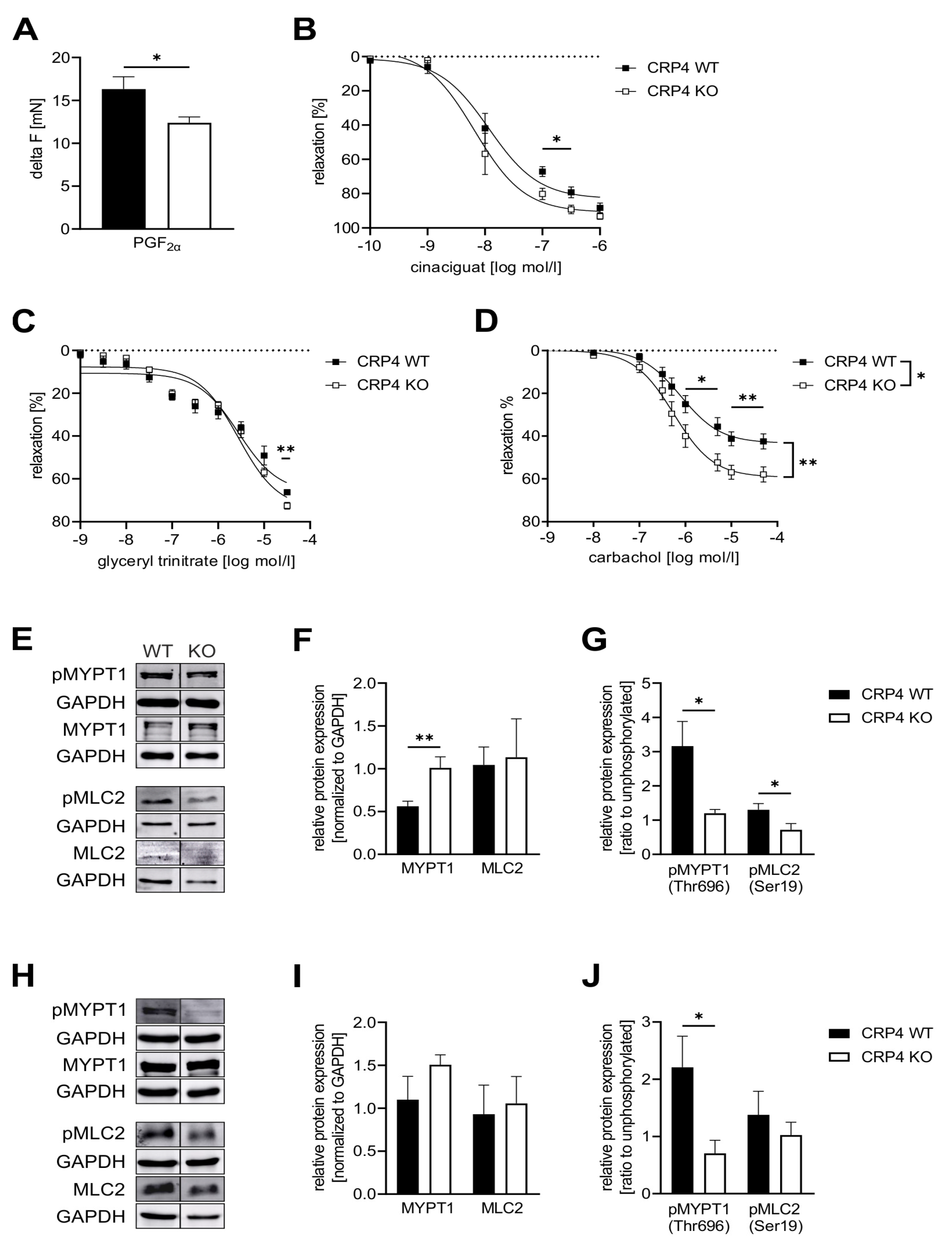
2.3. Lack of CRP4 Facilitates Vasorelaxant Actions of the NO-GC/cGMP Cascade
2.4. CRP4 Interferes with cGMP-Dependent Control of Blood Pressure
3. Discussion
4. Material and Methods
4.1. Animal Care
4.2. VSMC Culture
4.3. Immunohistochemistry
4.4. Immunoblot of VSMC Proteins
4.5. Organ Bath Experiments to Study Relaxation of Aortic Rings
4.6. In Vivo Phosphorylation of CRP4 in Murine Aorta
4.7. Telemetric Blood Pressure Measurement
4.8. Fluorescent-Based Ca2+ Measurements in VSMCs
4.9. Cyclic GMP ELISA Measurements
4.10. Statistical Analyses
5. Conclusions and Limitations of the Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Udan, R.S.; Culver, J.C.; Dickinson, M.E. Understanding Vascular Development. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 327–346. [Google Scholar] [CrossRef]
- Augustin, H.G.; Koh, G.Y. Organotypic Vasculature: From Descriptive Heterogeneity to Functional Pathophysiology. Science 2017, 357, eaal2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touyz, R.M.; Alves-Lopes, R.; Rios, F.; Camargo, L.D.L.; Anagnostopoulou, A.; Arner, A.; Montezano, A.C. Vascular Smooth Muscle Contraction in Hypertension. Cardiovasc. Res. 2018, 114, 529–539. [Google Scholar] [CrossRef] [Green Version]
- Sehgel, N.L.; Zhu, Y.; Sun, Z.; Trzeciakowski, J.P.; Hong, Z.; Hunter, W.C.; Vatner, D.E.; Meininger, G.A.; Vatner, S.F. Increased vascular smooth muscle cell stiffness: A novel mechanism for aortic stiffness in hypertension. Am. J. Physiol. Circ. Physiol. 2013, 305, H1281–H1287. [Google Scholar] [CrossRef]
- Michael, S.K.; Surks, H.K.; Wang, Y.; Zhu, Y.; Blanton, R.; Jamnongjit, M.; Aronovitz, M.; Baur, W.; Ohtani, K.; Wilkerson, M.K.; et al. High blood pressure arising from a defect in vascular function. Proc. Natl. Acad. Sci. USA 2008, 105, 6702–6707. [Google Scholar] [CrossRef] [Green Version]
- Brozovich, F.; Nicholson, C.; Degen, C.; Gao, Y.Z.; Aggarwal, M.; Morgan, K. Mechanisms of Vascular Smooth Muscle Contraction and the Basis for Pharmacologic Treatment of Smooth Muscle Disorders. Pharmacol. Rev. 2016, 68, 476–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sausbier, M.; Schubert, R.; Voigt, V.; Hirneiss, C.; Pfeifer, A.; Korth, M.; Kleppisch, T.; Ruth, P.; Hofmann, F. Mechanisms of NO/cGMP-dependent vasorelaxation. Circ. Res. 2000, 87, 825–830. [Google Scholar] [CrossRef] [Green Version]
- Ignarro, L.J.; Kadowitz, P.J. The pharmacological and physiological role of cyclic GMP in vascular smooth muscle relaxation. Annu. Rev. Pharmacol. Toxicol. 1985, 25, 171–191. [Google Scholar] [CrossRef] [PubMed]
- Hermann, M.; Flammer, A.; Luscher, T.F. Nitric oxide in hypertension. J. Clin. Hypertens. Greenwich 2006, 8 (Suppl. 4), 17–29. [Google Scholar] [CrossRef]
- Friebe, A.; Koesling, D. Regulation of Nitric Oxide-Sensitive Guanylyl Cyclase. Circ. Res. 2003, 93, 96–105. [Google Scholar] [CrossRef]
- Hofmann, F.; Wegener, J.W. cGMP-dependent protein kinases (cGK). Methods Mol. Biol. 2013, 1020, 17–50. [Google Scholar]
- Li, Y.; Madiraju, P.; Anand-Srivastava, M.B. Knockdown of natriuretic peptide receptor-A enhances receptor C expression and signalling in vascular smooth muscle cells. Cardiovasc. Res. 2012, 93, 350–359. [Google Scholar] [CrossRef]
- Moyes, A.J.; Hobbs, A.J. C-Type Natriuretic Peptide: A Multifaceted Paracrine Regulator in the Heart and Vasculature. Int. J. Mol. Sci. 2019, 20, 2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surks, H.K. cGMP-dependent protein kinase I and smooth muscle relaxation: A tale of two isoforms. Circ. Res. 2007, 101, 1078–1080. [Google Scholar] [CrossRef] [Green Version]
- Wall, M.E.; Francis, S.H.; Corbin, J.D.; Grimes, K.; Richie-Jannetta, R.; Kotera, J.; Macdonald, B.A.; Gibson, R.R.; Trewhella, J. Mechanisms associated with cGMP binding and activation of cGMP-dependent protein kinase. Proc. Natl. Acad. Sci. USA 2003, 100, 2380–2385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, S.; Bernhard, D.; Lukowski, R.; Weinmeister, P.; Worner, R.; Wegener, J.W.; Valtcheva, N.; Feil, S.; Schlossmann, J.; Hofmann, F.; et al. Rescue of cGMP Kinase I Knockout Mice by Smooth Muscle–Specific Expression of Either Isozyme. Circ. Res. 2007, 101, 1096–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orstavik, S.; Natarajan, V.; Tasken, K.; Jahnsen, T.; Sandberg, M. Characterization of the human gene encoding the type I alpha and type I beta cGMP-dependent protein kinase (PRKG1). Genomics 1997, 42, 311–318. [Google Scholar] [CrossRef]
- Geiselhoringer, A.; Gaisa, M.; Hofmann, F.; Schlossmann, J. Distribution of IRAG and cGKI-isoforms in murine tissues. FEBS Lett. 2004, 575, 19–22. [Google Scholar] [CrossRef] [Green Version]
- Schlossmann, J.; Desch, M. IRAG and novel PKG targeting in the cardiovascular system. Am. J. Physiol. Circ. Physiol. 2011, 301, H672–H682. [Google Scholar] [CrossRef] [Green Version]
- Rybalkin, S.D.; Rybalkina, I.G.; Feil, R.; Hofmann, F.; Beavo, J.A. Regulation of cGMP-specific phosphodiesterase (PDE5) phosphorylation in smooth muscle cells. J. Biol. Chem. 2002, 277, 3310–3317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rybalkin, S.D.; Yan, C.; Bornfeldt, K.E.; Beavo, J.A. Cyclic GMP Phosphodiesterases and Regulation of Smooth Muscle Function. Circ. Res. 2003, 93, 280–291. [Google Scholar] [CrossRef]
- Zhang, L.; Bouadjel, K.; Manoury, B.; Vandecasteele, G.; Fischmeister, R.; Leblais, V. Cyclic nucleotide signalling compartmentation by PDEs in cultured vascular smooth muscle cells. Br. J. Pharmacol. 2019, 176, 1780–1792. [Google Scholar] [CrossRef]
- Krawutschke, C.; Koesling, D.; Russwurm, M. Cyclic GMP in Vascular Relaxation: Export Is of Similar Importance as Degradation. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2011–2019. [Google Scholar] [CrossRef] [Green Version]
- Ferrero, R.; Rodriguez-Pascual, F.; Miras-Portugal, M.T.; Torres, M. Nitric oxide-sensitive guanylyl cyclase activity inhibition through cyclic GMP-dependent dephosphorylation. J. Neurochem. 2000, 75, 2029–2039. [Google Scholar] [CrossRef]
- Schlossmann, J.; Schinner, E. cGMP becomes a drug target. Naunyn. Schmiedebergs Arch. Pharmacol. 2012, 385, 243–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adler, J.; Kuret, A.; Längst, N.; Lukowski, R. Targets of cGMP/cGKI in Cardiac Myocytes. J. Cardiovasc. Pharmacol. 2020, 75, 494–507. [Google Scholar] [CrossRef]
- Evora, P.R.; Evora, P.M.; Celotto, A.C.; Rodrigues, A.J.; Joviliano, E.E. Cardiovascular therapeutics targets on the NO-sGC-cGMP signaling pathway: A critical overview. Curr. Drug Targets 2012, 13, 1207–1214. [Google Scholar] [CrossRef]
- Herman, A.G.; Moncada, S. Therapeutic potential of nitric oxide donors in the prevention and treatment of atherosclerosis. Eur. Heart J. 2005, 26, 1945–1955. [Google Scholar] [CrossRef] [Green Version]
- Bruzziches, R.; Francomano, D.; Gareri, P.; Lenzi, A.; Aversa, A. An update on pharmacological treatment of erectile dysfunction with phosphodiesterase type 5 inhibitors. Expert Opin. Pharmacother. 2013, 14, 1333–1344. [Google Scholar] [CrossRef] [PubMed]
- Ghofrani, H.A.; Galie, N.; Grimminger, F.; Grunig, E.; Humbert, M.; Jing, Z.C.; Keogh, A.M.; Langleben, D.; Kilama, M.O.; Fritsch, A.; et al. Riociguat for the treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 330–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, P.W.; Pieske, B.; Anstrom, K.J.; Ezekowitz, J.; Hernandez, A.F.; Butler, J.; Lam, C.S.; Ponikowski, P.; Voors, A.A.; Jia, G.; et al. Vericiguat in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2020, 382, 1883–1893. [Google Scholar] [CrossRef]
- Markham, A.; Duggan, S. Vericiguat: First Approval. Drugs 2021, 81, 721–726. [Google Scholar] [CrossRef]
- Angermeier, E.; Domes, K.; Lukowski, R.; Schlossmann, J.; Rathkolb, B.; de Angelis, M.H.; Hofmann, F. Iron deficiency anemia in cyclic GMP kinase knockout mice. Haematologica 2016, 101, e48–e51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foller, M.; Feil, S.; Ghoreschi, K.; Koka, S.; Gerling, A.; Thunemann, M.; Hofmann, F.; Schuler, B.; Vogel, J.; Pichler, B.; et al. Anemia and splenomegaly in cGKI-deficient mice. Proc. Natl. Acad. Sci. USA 2008, 105, 6771–6776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Lukowski, R.; Gaertner, F.; Lorenz, M.; Legate, K.R.; Domes, K.; Angermeier, E.; Hofmann, F.; Massberg, S. Thrombocytosis as a response to high interleukin-6 levels in cGMP-dependent protein kinase I mutant mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1820–1828. [Google Scholar] [CrossRef] [Green Version]
- Kleppisch, T.; Wolfsgruber, W.; Feil, S.; Allmann, R.; Wotjak, C.T.; Goebbels, S.; Nave, K.A.; Hofmann, F.; Feil, R. Hippocampal cGMP-dependent protein kinase I supports an age- and protein synthesis-dependent component of long-term potentiation but is not essential for spatial reference and contextual memory. J. Neurosci. 2003, 23, 6005–6012. [Google Scholar] [CrossRef] [Green Version]
- Massberg, S.; Sausbier, M.; Klatt, P.; Bauer, M.; Pfeifer, A.; Siess, W.; Fassler, R.; Ruth, P.; Krombach, F.; Hofmann, F. Increased adhesion and aggregation of platelets lacking cyclic guanosine 3’,5’-monophosphate kinase I. J. Exp. Med. 1999, 189, 1255–1264. [Google Scholar] [CrossRef]
- Singh, A.K.; Spieβberger, B.; Zheng, W.; Xiao, F.; Lukowski, R.; Wegener, J.W.; Weinmeister, P.; Saur, D.; Klein, S.; Schemann, M.; et al. Neuronal cGMP kinase I is essential for stimulation of duodenal bicarbonate secretion by luminal acid. FASEB J. 2011, 26, 1745–1754. [Google Scholar] [CrossRef]
- Leiss, V.; Friebe, A.; Welling, A.; Hofmann, F.; Lukowski, R. Cyclic GMP Kinase I Modulates Glucagon Release From Pancreatic α-Cells. Diabetes 2010, 60, 148–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Methner, C.; Lukowski, R.; Grube, K.; Loga, F.; Smith, R.A.J.; Murphy, M.P.; Hofmann, F.; Krieg, T. Protection through postconditioning or a mitochondria-targeted S-nitrosothiol is unaffected by cardiomyocyte-selective ablation of protein kinase G. Basic Res. Cardiol. 2013, 108, 1–7. [Google Scholar] [CrossRef]
- Frantz, S.; Klaiber, M.; Baba, H.A.; Oberwinkler, H.; Völker, K.; Gaβner, B.; Bayer, B.; Abeβer, M.; Schuh, K.; Feil, R.; et al. Stress-dependent dilated cardiomyopathy in mice with cardiomyocyte-restricted inactivation of cyclic GMP-dependent protein kinase I. Eur. Heart J. 2011, 34, 1233–1244. [Google Scholar] [CrossRef] [Green Version]
- Pfeifer, A.; Klatt, P.; Massberg, S.; Ny, L.; Sausbier, M.; Hirneiß, C.; Wang, G.; Korth, M.; Aszódi, A.; Andersson, K.; et al. Defective smooth muscle regulation in cGMP kinase I-deficient mice. EMBO J. 1998, 17, 3045–3051. [Google Scholar] [CrossRef]
- Groneberg, D.; König, P.; Wirth, A.; Offermanns, S.; Koesling, D.; Friebe, A. Smooth Muscle–Specific Deletion of Nitric Oxide–Sensitive Guanylyl Cyclase Is Sufficient to Induce Hypertension in Mice. Circulation 2010, 121, 401–409. [Google Scholar] [CrossRef] [Green Version]
- Wooldridge, A.A.; MacDonald, J.A.; Erdodi, F.; Ma, C.; Borman, M.A.; Hartshorne, D.J.; Haystead, T.A.J. Smooth Muscle Phosphatase Is Regulated in vivo by Exclusion of Phosphorylation of Threonine 696 of MYPT1 by Phosphorylation of Serine 695 in Response to Cyclic Nucleotides. J. Biol. Chem. 2004, 279, 34496–34504. [Google Scholar] [CrossRef] [Green Version]
- Surks, H.K.; Mochizuki, N.; Kasai, Y.; Georgescu, S.P.; Tang, K.M.; Ito, M.; Lincoln, T.M.; Mendelsohn, M.E. Regulation of Myosin Phosphatase by a Specific Interaction with cGMP- Dependent Protein Kinase, I. Science 1999, 286, 1583–1587. [Google Scholar] [CrossRef]
- Murthy, K.S.; Zhou, H. Selective phosphorylation of the IP3R-I in vivo by cGMP-dependent protein kinase in smooth muscle. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G221–G230. [Google Scholar] [CrossRef] [Green Version]
- Komalavilas, P.; Lincoln, T.M. Phosphorylation of the inositol 1,4,5-trisphosphate receptor. Cyclic GMP-dependent protein kinase mediates cAMP and cGMP dependent phosphorylation in the intact rat aorta. J. Biol. Chem. 1996, 271, 21933–21938. [Google Scholar] [CrossRef] [Green Version]
- Schlossmann, J.; Ammendola, A.; Ashman, K.; Zong, X.; Huber, A.; Neubauer, G.; Wang, G.-X.; Allescher, H.-D.; Korth, M.; Wilm, M.; et al. Regulation of intracellular calcium by a signalling complex of IRAG, IP3 receptor and cGMP kinase Iβ. Nature 2000, 404, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Geiselhöringer, A.; Werner, M.; Sigl, K.; Smital, P.; Wörner, R.; Acheo, L.; Stieber, J.; Weinmeister, P.; Feil, R.; Feil, S.; et al. IRAG is essential for relaxation of receptor-triggered smooth muscle contraction by cGMP kinase. EMBO J. 2004, 23, 4222–4231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, M.; Blanton, R.; Wang, G.-R.; Judson, T.J.; Abe, Y.; Myoishi, M.; Karas, R.H.; Mendelsohn, M.E. Direct Binding and Regulation of RhoA Protein by Cyclic GMP-dependent Protein Kinase Iα. J. Biol. Chem. 2012, 287, 41342–41351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauzeau, V.; Le Jeune, H.; Cario-Toumaniantz, C.; Smolenski, A.; Lohmann, S.M.; Bertoglio, J.; Chardin, P.; Pacaud, P.; Loirand, G. Cyclic GMP-dependent protein kinase signaling pathway inhibits RhoA-induced Ca2+ sensitization of contraction in vascular smooth muscle. J. Biol. Chem. 2000, 275, 21722–21729. [Google Scholar] [CrossRef] [Green Version]
- Sausbier, M.; Arntz, C.; Bucurenciu, I.; Zhao, H.; Zhou, X.-B.; Sausbier, U.; Feil, S.; Kamm, S.; Essin, K.; Sailer, C.A.; et al. Elevated Blood Pressure Linked to Primary Hyperaldosteronism and Impaired Vasodilation in BK Channel–Deficient Mice. Circulation 2005, 112, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.M.; Wang, G.R.; Lu, P.; Karas, R.H.; Aronovitz, M.; Heximer, S.P.; Kaltenbronn, K.M.; Blumer, K.J.; Siderovski, D.P.; Zhu, Y.; et al. Regulator of G-protein signaling-2 mediates vascular smooth muscle relaxation and blood pressure. Nat. Med. 2003, 9, 1506–1512. [Google Scholar] [CrossRef]
- Sun, X.; Kaltenbronn, K.M.; Steinberg, T.H.; Blumer, K.J. RGS2 Is a Mediator of Nitric Oxide Action on Blood Pressure and Vasoconstrictor Signaling. Mol. Pharmacol. 2004, 67, 631–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halbrugge, M.; Walter, U. Purification of a vasodilator-regulated phosphoprotein from human platelets. JBIC J. Biol. Inorg. Chem. 1989, 185, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Aszodi, A.; Pfeifer, A.; Ahmad, M.; Glauner, M.; Zhou, X.H.; Ny, L.; Andersson, K.E.; Kehrel, B.; Offermanns, S.; Fassler, R. The vasodilator-stimulated phosphoprotein (VASP) is involved in cGMP- and cAMP-mediated inhibition of agonist-induced platelet aggregation, but is dispensable for smooth muscle function. EMBO J. 1999, 18, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Straubinger, J.; Boldt, K.; Kuret, A.; Deng, L.; Krattenmacher, D.; Bork, N.; Desch, M.; Feil, R.; Feil, S.; Nemer, M.; et al. Amplified pathogenic actions of angiotensin II in cysteine-rich LIM-only protein 4-negative mouse hearts. FASEB J. 2017, 31, 1620–1638. [Google Scholar] [CrossRef] [Green Version]
- Huber, A.; Neuhuber, W.L.; Klugbauer, N.; Ruth, P.; Allescher, H.-D. Cysteine-rich Protein 2, a Novel Substrate for cGMP Kinase I in Enteric Neurons and Intestinal Smooth Muscle. J. Biol. Chem. 2000, 275, 5504–5511. [Google Scholar] [CrossRef] [Green Version]
- Schmidtko, A.; Gao, W.; Sausbier, M.; Rauhmeier, I.; Sausbier, U.; Niederberger, E.; Scholich, K.; Huber, A.; Neuhuber, W.; Allescher, H.D.; et al. Cysteine-rich protein 2, a novel downstream effector of cGMP/cGMP-dependent protein kinase I-mediated persistent inflammatory pain. J. Neurosci. 2008, 28, 1320–1330. [Google Scholar] [CrossRef]
- Okano, I.; Yamamoto, T.; Kaji, A.; Kimura, T.; Mizuno, K.; Nakamura, T. Cloning of CRP2, a novel member of the cysteine-rich protein family with two repeats of an unusual LIM/double zinc-finger motif. FEBS Lett. 1993, 333, 51–55. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Zhuang, S.; Casteel, D.E.; Looney, D.J.; Boss, G.R.; Pilz, R.B. A cysteine-rich LIM-only protein mediates regulation of smooth muscle-specific gene expression by cGMP-dependent protein kinase. J. Biol. Chem. 2007, 282, 33367–33380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yet, S.F.; Folta, S.C.; Jain, M.K.; Hsieh, C.M.; Maemura, K.; Layne, M.D.; Zhang, D.; Marria, P.B.; Yoshizumi, M.; Chin, M.T.; et al. Molecular cloning, characterization, and promoter analysis of the mouse Crp2/SmLim gene. Preferential expression of its promoter in the vascular smooth muscle cells of transgenic mice. J. Biol. Chem. 1998, 273, 10530–10537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, D.F.; Belaguli, N.S.; Iyer, D.; Roberts, W.B.; Wu, S.-P.; Dong, X.-R.; Marx, J.G.; Moore, M.S.; Beckerle, M.C.; Majesky, M.W.; et al. Cysteine-Rich LIM-Only Proteins CRP1 and CRP2 Are Potent Smooth Muscle Differentiation Cofactors. Dev. Cell 2003, 4, 107–118. [Google Scholar] [CrossRef] [Green Version]
- Weiskirchen, R.; Günther, K. The CRP/MLP/TLP family of LIM domain proteins: Acting by connecting. BioEssays 2003, 25, 152–162. [Google Scholar] [CrossRef]
- Lilly, B.; Clark, K.A.; Yoshigi, M.; Pronovost, S.; Wu, M.-L.; Periasamy, M.; Chi, M.; Paul, R.J.; Yet, S.-F.; Beckerle, M.C. Loss of the Serum Response Factor Cofactor, Cysteine-Rich Protein 1, Attenuates Neointima Formation in the Mouse. Arter. Thromb. Vasc. Biol. 2010, 30, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Knöll, R.; Hoshijima, M.; Hoffman, H.M.; Person, V.; Lorenzen-Schmidt, I.; Bang, M.-L.; Hayashi, T.; Shiga, N.; Yasukawa, H.; Schaper, W.; et al. The Cardiac Mechanical Stretch Sensor Machinery Involves a Z Disc Complex that Is Defective in a Subset of Human Dilated Cardiomyopathy. Cell 2002, 111, 943–955. [Google Scholar] [CrossRef] [Green Version]
- Weinmeister, P.; Lukowski, R.; Linder, S.; Traidl-Hoffmann, C.; Hengst, L.; Hofmann, F.; Feil, R. Cyclic Guanosine Monophosphate-dependent Protein Kinase I Promotes Adhesion of Primary Vascular Smooth Muscle Cells. Mol. Biol. Cell 2008, 19, 4434–4441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolenski, A.; Bachmann, C.; Reinhard, K.; Hönig-Liedl, P.; Jarchau, T.; Hoschuetzky, H.; Walter, U. Analysis and Regulation of Vasodilator-stimulated Phosphoprotein Serine 239 Phosphorylation in Vitro and in Intact Cells Using a Phosphospecific Monoclonal Antibody. J. Biol. Chem. 1998, 273, 20029–20035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feil, R.; Gappa, N.; Rutz, M.; Schlossmann, J.; Rose, C.R.; Konnerth, A.; Brummer, S.; Kühbandner, S.; Hofmann, F. Functional Reconstitution of Vascular Smooth Muscle Cells With cGMP-Dependent Protein Kinase I Isoforms. Circ. Res. 2002, 90, 1080–1086. [Google Scholar] [CrossRef] [Green Version]
- Kai, H.; Kanaide, H.; Matsumoto, T.; Nakamura, M. 8-Bromoguanosine 3′:5′-cyclic monophosphate decreases intracellular free calcium concentrations in cultured vascular smooth muscle cells from rat aorta. FEBS Lett. 1987, 221, 284–288. [Google Scholar] [CrossRef] [Green Version]
- Chacko, S.; Conti, M.A.; Adelstein, R. Effect of phosphorylation of smooth muscle myosin on actin activation and Ca2+ regulation. Proc. Natl. Acad. Sci. USA 1977, 74, 129–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SOMLYO, A.P.; SOMLYO, A.V. Ca2+ Sensitivity of Smooth Muscle and Nonmuscle Myosin II: Modulated by G Proteins, Kinases, and Myosin Phosphatase. Physiol. Rev. 2003, 83, 1325–1358. [Google Scholar] [CrossRef] [Green Version]
- Khatri, J.J.; Joyce, K.M.; Brozovich, F.V.; Fisher, S.A. Role of Myosin Phosphatase Isoforms in cGMP-mediated Smooth Muscle Relaxation. J. Biol. Chem. 2001, 276, 37250–37257. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Koga, Y.; Sakai, H.; Homma, K.; Ikebe, M. cGMP-Dependent Relaxation of Smooth Muscle Is Coupled With the Change in the Phosphorylation of Myosin Phosphatase. Circ. Res. 2007, 101, 712–722. [Google Scholar] [CrossRef] [Green Version]
- Lukowski, R.; Rybalkin, S.D.; Loga, F.; Leiss, V.; Beavo, J.A.; Hofmann, F. Cardiac hypertrophy is not amplified by deletion of cGMP-dependent protein kinase I in cardiomyocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 5646–5651. [Google Scholar] [CrossRef] [Green Version]
- Sheng, H.; Ishii, K.; Murad, F. Generation of an endothelium-derived relaxing factor-like substance in bovine tracheal smooth muscle. Am. J. Physiol. Cell. Mol. Physiol. 1991, 260, L489–L493. [Google Scholar] [CrossRef]
- Lopez, M.J.; Wong, S.K.-F.; Kishimoto, I.; Dubois, S.; Mach, V.; Friesen, J.; Garbers, D.L.; Beuve, A. Salt-resistant hypertension in mice lacking the guanylyl cyclase-A receptor for atrial natriuretic peptide. Nature 1995, 378, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, N.; Alfie, M.E.; Sigmon, D.H.; Rhaleb, N.-E.; Shesely, E.G.; Carretero, O.A. Role of nNOS in Blood Pressure Regulation in eNOS Null Mutant Mice. Hypertension 1998, 32, 856–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfertstetter, S.; Huettner, J.P.; Schlossmann, J. cGMP-Dependent Protein Kinase Inhibitors in Health and Disease. Pharmaceuticals 2013, 6, 269–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kung, C.F.; Moreau, P.; Takase, H.; Luscher, T.F. L-NAME Hypertension Alters Endothelial and Smooth Muscle Function in Rat Aorta. Prevention by trandolapril and verapamil. Hypertension 1995, 26, 744–751. [Google Scholar] [CrossRef]
- Münzel, T.; Feil, R.; Mülsch, A.; Lohmann, S.M.; Hofmann, F.; Walter, U. Physiology and Pathophysiology of Vascular Signaling Controlled by Cyclic Guanosine 3′,5′-Cyclic Monophosphate–Dependent Protein Kinase. Circulation 2003, 108, 2172–2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, K.G. Calcium and vascular smooth muscle tone. Am. J. Med. 1987, 82, 9–15. [Google Scholar] [CrossRef]
- Butler, T.; Paul, J.; Europe-Finner, N.; Smith, R.; Chan, E.-C. Role of serine-threonine phosphoprotein phosphatases in smooth muscle contractility. Am. J. Physiol. Physiol. 2013, 304, C485–C504. [Google Scholar] [CrossRef] [Green Version]
- Kadrmas, J.L.; Beckerle, M.C. The LIM domain: From the cytoskeleton to the nucleus. Nat. Rev. Mol. Cell Biol. 2004, 5, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Schmeichel, K.L.; Beckerle, M.C. Molecular dissection of a LIM domain. Mol. Biol. Cell. 1997, 8, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Bach, I. The LIM domain: Regulation by association. Mech. Dev. 2000, 91, 5–17. [Google Scholar] [CrossRef]
- Dawid, I.B.; Breen, J.J.; Toyama, R. LIM domains: Multiple roles as adapters and functional modifiers in protein interactions. Trends Genet. 1998, 14, 156–162. [Google Scholar] [CrossRef]
- Chen, L.; Kurokawa, J.; Kass, R.S. Phosphorylation of the A-kinase-anchoring Protein Yotiao Contributes to Protein Kinase A Regulation of a Heart Potassium Channel. J. Biol. Chem. 2005, 280, 31347–31352. [Google Scholar] [CrossRef] [Green Version]
- Sinnaeve, P.; Chiche, J.-D.; Gillijns, H.; Van Pelt, N.; Wirthlin, D.; Van de Werf, F.; Collen, D.; Bloch, K.D.; Janssens, S. Overexpression of a Constitutively Active Protein Kinase G Mutant Reduces Neointima Formation and In-Stent Restenosis. Circulation 2002, 105, 2911–2916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornwell, T.L.; Soff, G.A.; Traynor, A.E.; Lincoln, T.M. Regulation of the Expression of Cyclic GMP-Dependent Protein Kinase by Cell Density in Vascular Smooth Muscle Cells. J. Vasc. Res. 1994, 31, 330–337. [Google Scholar] [CrossRef]
- Lincoln, T.M.; Dey, N.; Sellak, H. Invited Review: cGMP-dependent protein kinase signaling mechanisms in smooth muscle: From the regulation of tone to gene expression. J. Appl. Physiol. 2001, 91, 1421–1430. [Google Scholar] [CrossRef]
- Kimura, K.; Ito, M.; Amano, M.; Chihara, K.; Fukata, Y.; Nakafuku, M.; Yamamori, B.; Feng, J.; Nakano, T.; Okawa, K.; et al. Regulation of Myosin Phosphatase by Rho and Rho-Associated Kinase (Rho-Kinase). Science 1996, 273, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Kiss, A.; Erdődi, F.; Lontay, B. Myosin phosphatase: Unexpected functions of a long-known enzyme. Biochim. Biophys. Acta-Mol. Cell Res. 2019, 1866, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Kamm, K.E.A.; Stull, J.T. The Function of Myosin and Myosin Light Chain Kinase Phosphorylation in Smooth Muscle. Annu. Rev. Pharmacol. Toxicol. 1985, 25, 593–620. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.-N.; He, W.; Chen, C.-P.; Zhang, C.-H.; Zhao, W.; Wang, P.; Zhang, L.; Wu, Y.-Z.; Yang, X.; Peng, Y.-J.; et al. Myosin Phosphatase Target Subunit 1 (MYPT1) Regulates the Contraction and Relaxation of Vascular Smooth Muscle and Maintains Blood Pressure. J. Biol. Chem. 2014, 289, 22512–22523. [Google Scholar] [CrossRef] [Green Version]
- Schermuly, R.; Stasch, J.-P.; Pullamsetti, S.S.; Middendorff, R.; Muller, D.; Schluter, K.-D.; Dingendorf, A.; Hackemack, S.; Kolosionek, E.; Kaulen, C.; et al. Expression and function of soluble guanylate cyclase in pulmonary arterial hypertension. Eur. Respir. J. 2008, 32, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Chester, M.; Seedorf, G.; Tourneux, P.; Gien, J.; Tseng, N.; Grover, T.; Wright, J.; Stasch, J.-P.; Abman, S.H. Cinaciguat, a soluble guanylate cyclase activator, augments cGMP after oxidative stress and causes pulmonary vasodilation in neonatal pulmonary hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2011, 301, L755–L764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friebe, A.; Mergia, E.; Dangel, O.; Lange, A.; Koesling, D. Fatal gastrointestinal obstruction and hypertension in mice lacking nitric oxide-sensitive guanylyl cyclase. Proc. Natl. Acad. Sci. USA 2007, 104, 7699–7704. [Google Scholar] [CrossRef] [Green Version]
- Münzel, T.; Sayegh, H.; Freeman, B.A.; Tarpey, M.M.; Harrison, D.G. Evidence for enhanced vascular superoxide anion production in nitrate tolerance. A novel mechanism underlying tolerance and cross-tolerance. J. Clin. Investig. 1995, 95, 187–194. [Google Scholar] [CrossRef] [Green Version]
- Patrucco, E.; Domes, K.; Sbroggió, M.; Blaich, A.; Schlossmann, J.; Desch, M.; Rybalkin, S.D.; Beavo, J.A.; Lukowski, R.; Hofmann, F. Roles of cGMP-dependent protein kinase I (cGKI) and PDE5 in the regulation of Ang II-induced cardiac hypertrophy and fibrosis. Proc. Natl. Acad. Sci. USA 2014, 111, 12925–12929. [Google Scholar] [CrossRef] [Green Version]
- Joshua, J.; Schwaerzer, G.K.; Kalyanaraman, H.; Cory, E.; Sah, R.L.; Li, M.; Vaida, F.; Boss, G.R.; Pilz, R.B. Soluble Guanylate Cyclase as a Novel Treatment Target for Osteoporosis. Endocrinology 2014, 155, 4720–4730. [Google Scholar] [CrossRef] [Green Version]
- Burke, D.H.; O’Hara, G.P. Sodium Nitroprusside-Induced Hypothermia in Mice. J. Pharm. Sci. 1977, 66, 1658–1660. [Google Scholar] [CrossRef] [PubMed]
- Gehrmann, J.; Meister, M.; Maguire, C.T.; Martins, D.C.; Hammer, P.E.; Neer, E.J.; Berul, C.I.; Mende, U. Impaired parasympathetic heart rate control in mice with a reduction of functional G protein betagamma-subunits. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H445–H456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweigert, O.; Adler, J.; Längst, N.; Aïssi, D.; Escobar, J.D.; Tong, T.; Müller, C.; Trégouët, D.-A.; Lukowski, R.; Zeller, T. CRIP1 expression in monocytes related to hypertension. Clin. Sci. 2021, 135, 911–924. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Längst, N.; Adler, J.; Schweigert, O.; Kleusberg, F.; Cruz Santos, M.; Knauer, A.; Sausbier, M.; Zeller, T.; Ruth, P.; Lukowski, R. Cyclic GMP-Dependent Regulation of Vascular Tone and Blood Pressure Involves Cysteine-Rich LIM-Only Protein 4 (CRP4). Int. J. Mol. Sci. 2021, 22, 9925. https://doi.org/10.3390/ijms22189925
Längst N, Adler J, Schweigert O, Kleusberg F, Cruz Santos M, Knauer A, Sausbier M, Zeller T, Ruth P, Lukowski R. Cyclic GMP-Dependent Regulation of Vascular Tone and Blood Pressure Involves Cysteine-Rich LIM-Only Protein 4 (CRP4). International Journal of Molecular Sciences. 2021; 22(18):9925. https://doi.org/10.3390/ijms22189925
Chicago/Turabian StyleLängst, Natalie, Julia Adler, Olga Schweigert, Felicia Kleusberg, Melanie Cruz Santos, Amelie Knauer, Matthias Sausbier, Tanja Zeller, Peter Ruth, and Robert Lukowski. 2021. "Cyclic GMP-Dependent Regulation of Vascular Tone and Blood Pressure Involves Cysteine-Rich LIM-Only Protein 4 (CRP4)" International Journal of Molecular Sciences 22, no. 18: 9925. https://doi.org/10.3390/ijms22189925
APA StyleLängst, N., Adler, J., Schweigert, O., Kleusberg, F., Cruz Santos, M., Knauer, A., Sausbier, M., Zeller, T., Ruth, P., & Lukowski, R. (2021). Cyclic GMP-Dependent Regulation of Vascular Tone and Blood Pressure Involves Cysteine-Rich LIM-Only Protein 4 (CRP4). International Journal of Molecular Sciences, 22(18), 9925. https://doi.org/10.3390/ijms22189925