
Development of Biomarkers and Molecular Therapy Based on Inflammatory Genes in Diabetic Nephropathy


{kind=link}
Abstract
:1. Introduction
2. Brief Overview of Stages of Diabetic Nephropathy
- (a).
- Glomerular lesions
- Type 1: Glomerular Basement Membrane (GBM) Thickening.
- Types 2: Mild (IIa) or Severe (IIb) Mesangial Expansion.
- Types 3: Nodular Sclerosis (Kimmelstiel–Wilson lesions).
- Types 4: Advanced Diabetic Glomerulosclerosis.
- (b).
- Interstitial abnormalities
- Types 1: Interstitial Fibrosis and Tubular Atrophy (IFTA)
- Types 2: Interstitial inflammation
- (c).
- Vascular lesions
- Types 1: Arteriolar hyalinosis
- Types 2: Presence of large vessels
- Types 3: Arteriosclerosis
- (d).
- Other glomerular lesions
3. Specialized Cells in Kidney and Renal Systems
3.1. Specialized Cells
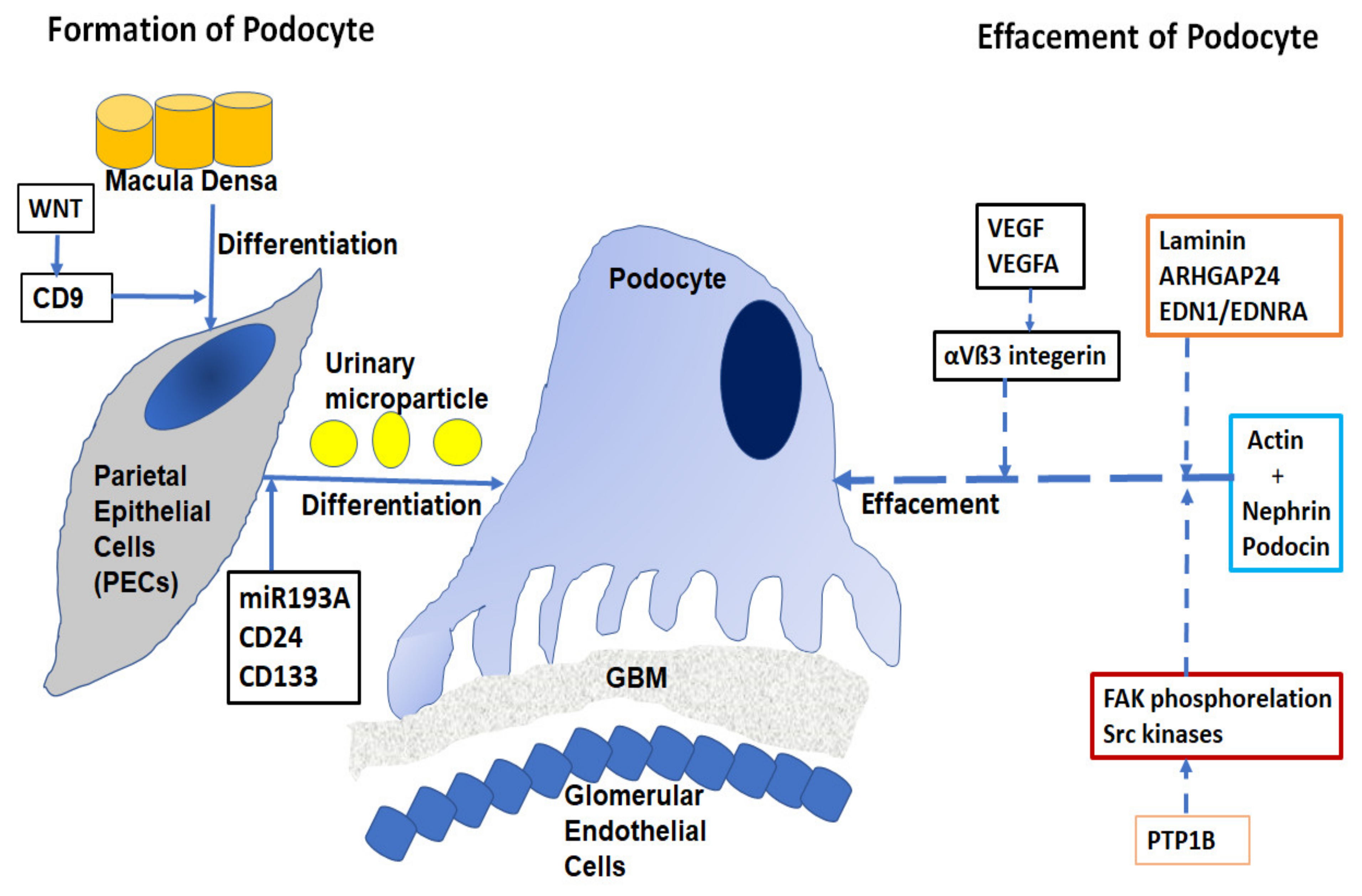
3.2. Structure and Function of Glomerulus Podocyte
4. Role of Autophagy in Developing DN
4.1. Mechanism
4.1.1. mTORC1 Pathway
4.1.2. AMPK Pathway
4.1.3. SIRT1 Pathway
4.2. Autophagy in Podocytes
4.3. Autophagy in Mesangial Cells
4.4. Autophagy in Glomerular Epithelial Cells
4.5. Mitophagy
5. Developing Biomarkers in DN
5.1. Biomarkers Using Biochemical Pathway
5.2. Using the Mass Spectrometry Method
5.3. Other Proteomics Method
5.4. miRNA Biomarker in DN
5.5. LncRNA Biomarker in DN
5.6. Developing Biomarkers Based on Autophagy
6. Epidemiology and Genetic Factors in DN
7. Developing Therapy Based on Mutations in Inflammatory Genes in DN
Inflammatory Genes for Fibrotic Changes in DN
8. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Afkarian, M.; Zelnick, L.R.; Hall, Y.N.; Heagerty, P.J.; Tuttle, K.; Weiss, N.S.; De Boer, I.H. Clinical Manifestations of Kidney Disease Among US Adults With Diabetes, 1988–2014. JAMA 2016, 316, 602–610. [Google Scholar] [CrossRef]
- Federation, I.D. International Diabetes Federation IDF Diabetes Atlas; International Diabetes Federation: Brussels, Belgium, 2015. [Google Scholar]
- Andersen, A.R.; Christiansen, J.S.; Andersen, J.K.; Kreiner, S.; Deckert, T. Diabetic nephropathy in type 1 (insulin-dependent) diabetes: An epidemiological study. Diabetologia 1983, 25, 496–501. [Google Scholar] [CrossRef]
- Sulaiman, M.K. Diabetic nephropathy: Recent advances in pathophysiology and challenges in dietary management. Diabetol. Metab. Syndr. 2019, 11, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballantyne, F.C.; Gibbons, J.; O’Reilly, D.S. Urine Albumin Should Replace Total Protein for the Assessment of Glomerular Proteinuria. Ann. Clin. Biochem. Int. J. Lab. Med. 1993, 30, 101–103. [Google Scholar] [CrossRef] [PubMed]
- Vitova, L.; Tuma, Z.; Moravec, J.; Kvapil, M.; Matejovic, M.; Mares, J. Early urinary biomarkers of diabetic nephropathy in type 1 diabetes mellitus show involvement of kallikrein-kinin system. BMC Nephrol. 2017, 18, 112. [Google Scholar] [CrossRef] [Green Version]
- Matoba, K.; Takeda, Y.; Nagai, Y.; Kawanami, D.; Utsunomiya, K.; Nishimura, R. Unraveling the Role of Inflammation in the Pathogenesis of Diabetic Kidney Disease. Int. J. Mol. Sci. 2019, 20, 3393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, S.; Chen, S.Y. Macrophage polarization in kidney diseases. Macrophage Houst 2015, 2, e679. [Google Scholar] [PubMed] [Green Version]
- Tervaert, T.W.C.; Mooyaart, A.; Amann, K.; Cohen, A.H.; Cook, H.T.; Drachenberg, C.B.; Ferrario, F.; Fogo, A.B.; Haas, M.; De Heer, E.; et al. Pathologic Classification of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2010, 21, 556–563. [Google Scholar] [CrossRef] [Green Version]
- Osterby, R.; Asplund, J.; Bangstad, H.J.; Nyberg, G.; Rudberg, S.; Viberti, G.; Walker, J.D. Neovascularization at the vascular pole region in diabetic glomerulopathy. Nephrol. Dial. Transplant. 1999, 14, 348–352. [Google Scholar] [CrossRef] [Green Version]
- Stout, L.C.; Kumar, S.; Whorton, E.B. Focal mesangiolysis and the pathogenesis of the Kimmelstiel-Wilson nodule. Hum. Pathol. 1993, 24, 77–89. [Google Scholar] [CrossRef]
- Shankland, S.J. The podocyte’s response to injury: Role in proteinuria and glomerulosclerosis. Kidney Int. 2006, 69, 2131–2147. [Google Scholar] [CrossRef] [Green Version]
- Quaggin, S.E.; Kreidberg, J.A. Development of the renal glomerulus: Good neighbors and good fences. Development 2008, 135, 609–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiggins, R.-C. The spectrum of podocytopathies: A unifying view of glomerular diseases. Kidney Int. 2007, 71, 1205–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, G.; Gbadegesin, R.A. Translating genetic findings in hereditary nephrotic syndrome: The missing loops. Am. J. Physiol. Renal. Physiol. 2015, 309, F24–F28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, P. A Review of Podocyte Biology. Am. J. Nephrol. 2018, 47, 3–13. [Google Scholar] [CrossRef]
- Ronco, P. Proteinuria: Is it all in the foot? J. Clin. Investig. 2007, 117, 2079–2082. [Google Scholar] [CrossRef]
- Kestilä, M.; Lenkkeri, U.; Männikkö, M.; Lamerdin, J.; McCready, P.; Putaala, H.; Ruotsalainen, V.; Morita, T.; Nissinen, M.; Herva, R.; et al. Positionally Cloned Gene for a Novel Glomerular Protein—Nephrin—Is Mutated in Congenital Nephrotic Syndrome. Mol. Cell 1998, 1, 575–582. [Google Scholar] [CrossRef]
- Ma, H.; Togawa, A.; Soda, K.; Zhang, J.; Lee, S.; Ma, M.; Yu, Z.; Ardito, T.; Czyzyk, J.; Diggs, L.; et al. Inhibition of Podocyte FAK Protects against Proteinuria and Foot Process Effacement. J. Am. Soc. Nephrol. 2010, 21, 1145–1156. [Google Scholar] [CrossRef] [Green Version]
- Lovric, S.; Ashraf, S.; Tan, W.; Hildebrandt, F. Genetic testing in steroid-resistant nephrotic syndrome: When and how? Nephrol. Dial. Transplant. 2016, 31, 1802–1813. [Google Scholar] [CrossRef]
- Daehn, I.; Casalena, G.; Zhang, T.; Shi, S.; Fenninger, F.; Barasch, N.; Yu, L.; D’Agati, V.; Schlondorff, D.; Kriz, W.; et al. Endothelial mitochondrial oxidative stress determines podocyte depletion in segmental glomerulosclerosis. J. Clin. Investig. 2014, 124, 1608–1621. [Google Scholar] [CrossRef] [Green Version]
- Eremina, V.; Sood, M.; Haigh, J.; Nagy, A.; Lajoie, G.; Ferrara, N.; Gerber, H.-P.; Kikkawa, Y.; Miner, J.H.; Quaggin, S.E. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J. Clin. Investig. 2003, 111, 707–716. [Google Scholar] [CrossRef] [Green Version]
- Garg, P.; Rabelink, T. Glomerular proteinuria: A complex interplay between unique players. Adv. Chronic Kidney Dis. 2011, 18, 233–242. [Google Scholar] [CrossRef] [Green Version]
- Han, K.; Zhou, H.; Pfeifer, U. Inhibition and Restimulation by Insulin of Cellular Autophagy in Distal Tubular Cells of the Kidney in Early Diabetic Rats. Kidney Blood Press. Res. 1997, 20, 258–263. [Google Scholar] [CrossRef]
- Yang, D.; Livingston, M.J.; Liu, Z.; Dong, G.; Zhang, M.; Chen, J.-K.; Dong, Z. Autophagy in diabetic kidney disease: Regulation, pathological role and therapeutic potential. Cell. Mol. Life Sci. 2018, 75, 669–688. [Google Scholar] [CrossRef]
- Mori, T.; Nishimura, H.; Ueyama, M.; Kubota, J.; Kawamura, K. Comparable Effects of Angiotensin II and Converting Enzyme Blockade on Hemodynamics and Cardiac Hypertrophy in Spontaneously Hypertensive Rats. Jpn. Circ. J. 1995, 59, 624–630. [Google Scholar] [CrossRef]
- Sooparb, S.; Price, S.R.; Shaoguang, J.; Franch, H.A. Suppression of chaperone-mediated autophagy in the renal cortex during acute diabetes mellitus. Kidney Int. 2004, 65, 2135–2144. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.J.; Shen, T.T.; Chen, R.H.; Wu, H.-L.; Wang, Y.J.; Deng, J.K.; Chen, Q.H.; Pan, Q.; Fu, C.-M.H.; Tao, J.-L.; et al. Autophagy-Lysosome Pathway in Renal Tubular Epithelial Cells Is Disrupted by Advanced Glycation End Products in Diabetic Nephropathy. J. Biol. Chem. 2015, 290, 20499–20510. [Google Scholar] [CrossRef] [Green Version]
- Alers, S.; Löffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kume, S.; Uzu, T.; Horiike, K.; Chin-Kanasaki, M.; Isshiki, K.; Araki, S.-I.; Sugimoto, T.; Haneda, M.; Kashiwagi, A.; Koya, D. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J. Clin. Investig. 2010, 120, 1043–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartleben, B.; Gödel, M.; Meyer-Schwesinger, C.; Liu, S.; Ulrich, T.; Köbler, S.; Wiech, T.; Grahammer, F.; Arnold, S.J.; Lindenmeyer, M.T.; et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J. Clin. Investig. 2010, 120, 1084–1096. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Kim, J.K.; Kim, S.I.; Na, H.J.; Jun, S.Y.; Lee, S.J.; Choi, M.E. TGF-{beta}1 protects against mesangial cell apoptosis via induction of autophagy. J. Biol. Chem. 2010, 285, 37909–37919. [Google Scholar] [CrossRef] [Green Version]
- Xavier, S.; Gilbert, V.; Rastaldi, M.P.; Krick, S.; Kollins, D.; Reddy, A.; Böttinger, E.; Cohen, C.D.; Schlöndorff, D. BAMBI Is Expressed in Endothelial Cells and Is Regulated by Lysosomal/Autolysosomal Degradation. PLoS ONE 2010, 5, e12995. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nat. Cell Biol. 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Tan, A.L.Y.; Sourris, K.C.; Harcourt, B.E.; Thallas-Bonke, V.; Penfold, S.; Andrikopoulos, S.; Thomas, M.C.; O’Brien, R.C.; Bierhaus, A.; Cooper, M.E.; et al. Disparate effects on renal and oxidative parameters following RAGE deletion, AGE accumulation inhibition, or dietary AGE control in experimental diabetic nephropathy. Am. J. Physiol. Physiol. 2010, 298, F763–F770. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Hara, S.; Hattori, M.; Sakai, K.; Onishi, Y.; Yoshida, Y.; Kawazu, S.; Kushiyama, A. Role of elevated serum uric acid levels at the onset of overt nephropathy in the risk for renal function decline in patients with type 2 diabetes. J. Diabetes Investig. 2015, 6, 98–104. [Google Scholar] [CrossRef]
- Hovind, P.; Rossing, P.; Tarnow, L.; Smidt, U.M.; Parving, H.-H. Progression of diabetic nephropathy. Kidney Int. 2001, 59, 702–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colhoun, H.M.; Marcovecchio, M.L. Biomarkers of diabetic kidney disease. Diabetologia 2018, 61, 996–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkins, B.A.; Ficociello, L.H.; Roshan, B.; Warram, J.H.; Krolewski, A.S. In patients with type 1 diabetes and new-onset microalbuminuria the development of advanced chronic kidney disease may not require progression to proteinuria. Kidney Int. 2010, 77, 57–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gluck, C.; Ko, Y.-A.; Susztak, K. Precision Medicine Approaches to Diabetic Kidney Disease: Tissue as an Issue. Curr. Diabetes Rep. 2017, 17, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathan, D.M.; Cleary, P.A.; Backlund, J.-Y.C.; Genuth, S.M.; Lachin, J.; Orchard, T.; Raskin, P.; Zinman, B.; Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study Research Group. Intensive Diabetes Treatment and Cardiovascular Disease in Patients with Type 1 Diabetes. N. Engl. J. Med. 2005, 353, 2643–2653. [Google Scholar] [CrossRef] [PubMed]
- Ekinci, E.I.; Jerums, G.; Skene, A.; Crammer, P.; Power, D.; Cheong, K.Y.; Panagiotopoulos, S.; McNeil, K.; Baker, S.T.; Fioretto, P.; et al. Renal Structure in Normoalbuminuric and Albuminuric Patients With Type 2 Diabetes and Impaired Renal Function. Diabetes Care 2013, 36, 3620–3626. [Google Scholar] [CrossRef] [Green Version]
- Burns, K.D.; Lytvyn, Y.; Mahmud, F.; Daneman, D.; Deda, L.; Dunger, P.D.; Deanfield, J.; Dalton, R.N.; Elia, Y.; Har, R.; et al. The relationship between urinary renin-angiotensin system markers, renal function, and blood pressure in adolescents with type 1 diabetes. Am. J. Physiol. Physiol. 2017, 312, F335–F342. [Google Scholar] [CrossRef] [PubMed]
- Velho, G.; El Boustany, R.; Lefèvre, G.; Mohammedi, K.; Fumeron, F.; Potier, L.; Bankir, L.; Bouby, N.; Hadjadj, S.; Marre, M.; et al. Plasma Copeptin, Kidney Outcomes, Ischemic Heart Disease, and All-Cause Mortality in People With Long-standing Type 1 Diabetes. Diabetes Care 2016, 39, 2288–2295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dieter, B.P.; McPherson, S.M.; Afkarian, M.; De Boer, I.H.; Mehrotra, R.; Short, R.; Barbosa-Leiker, C.; Alicic, R.Z.; Meek, R.L.; Tuttle, K. Serum amyloid a and risk of death and end-stage renal disease in diabetic kidney disease. J. Diabetes Its Complicat. 2016, 30, 1467–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, V.; Kumar, M.; Mahapatra, H.S.; Chitkara, A.; Gadpayle, A.K.; Sekhar, V. Novel urinary biomarkers in pre-diabetic nephropathy. Clin. Exp. Nephrol. 2015, 19, 895–900. [Google Scholar] [CrossRef]
- Agarwal, R.; Duffin, K.L.; Laska, D.A.; Voelker, J.R.; Breyer, M.; Mitchell, P.G. A prospective study of multiple protein biomarkers to predict progression in diabetic chronic kidney disease. Nephrol. Dial. Transplant. 2014, 29, 2293–2302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argiles, A.; Siwy, J.; Duranton, F.; Gayrard, N.; Dakna, M.; Lundin, U.; Osaba, L.; Delles, C.; Mourad, G.; Weinberger, K.M.; et al. CKD273, a New Proteomics Classifier Assessing CKD and Its Prognosis. PLoS ONE 2013, 8, e62837. [Google Scholar] [CrossRef] [Green Version]
- Schlatzer, D.; Maahs, D.M.; Chance, M.R.; Dazard, J.-E.; Li, X.; Hazlett, F.; Rewers, M.; Snell-Bergeon, J.K. Novel Urinary Protein Biomarkers Predicting the Development of Microalbuminuria and Renal Function Decline in Type 1 Diabetes. Diabetes Care 2012, 35, 549–555. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, K.; Kato, M.; Natarajan, R. Mini-review: Emerging roles of microRNAs in the pathophysiology of renal diseases. Am. J. Physiol. Physiol. 2016, 310, F109–F118. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.; Arce, L.; Wang, M.; Putta, S.; Lanting, L.; Natarajan, R. A microRNA circuit mediates transforming growth factor-β1 autoregulation in renal glomerular mesangial cells. Kidney Int. 2011, 80, 358–368. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Gan, H.; Zhang, H.; Tang, W.; Sun, Y.; Tang, X.; Kong, D.; Zhou, J.; Wang, Y.; Zhu, Y. MicroRNA-21 inhibits SMAD7 expression through a target sequence in the 3′ untranslated region and inhibits proliferation of renal tubular epithelial cells. Mol. Med. Rep. 2014, 10, 707–712. [Google Scholar] [CrossRef]
- Yu, Y.; Bai, F.; Qin, N.; Liu, W.; Sun, Q.; Zhou, Y.; Yang, J. Non-Proximal Renal Tubule-Derived Urinary Exosomal miR-200b as a Biomarker of Renal Fibrosis. Nephron 2018, 139, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Assmann, T.S.; Recamonde-Mendoza, M.; de Souza, B.M.; Bauer, A.C.; Crispim, D. MicroRNAs and diabetic kidney disease: Systematic review and bioinformatic analysis. Mol. Cell. Endocrinol. 2018, 477, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, S.D.; Putta, S.; Wang, M.; Lai, J.Y.; Bitzer, M.; Nelson, R.G.; Lanting, L.L.; Kato, M.; Natarajan, R. Transforming Growth Factor-β–Induced Cross Talk Between p53 and a MicroRNA in the Pathogenesis of Diabetic Nephropathy. Diabetes 2013, 62, 3151–3162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Liu, Z.; Gong, R. Long noncoding RNA: An emerging player in diabetes and diabetic kidney disease. Clin. Sci. 2019, 133, 1321–1339. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, F.C.; Werner, A.; John, D.; Boeckel, J.-N.; Melissari, M.T.; Grote, P.; Glaser, S.F.; Demolli, S.; Uchida, S.; Michalik, K.M.; et al. Identification and Functional Characterization of Hypoxia-Induced Endoplasmic Reticulum Stress Regulating lncRNA (HypERlnc) in Pericytes. Circ. Res. 2017, 121, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Das, B.; Xiao, W.; Li, Z.; Li, H.; Lee, K.; He, J.C. A Novel Inhibitor of Homeodomain Interacting Protein Kinase 2 Mitigates Kidney Fibrosis through Inhibition of the TGF-β1/Smad3 Pathway. J. Am. Soc. Nephrol. 2017, 28, 2133–2143. [Google Scholar] [CrossRef] [Green Version]
- Long, J.; Badal, S.S.; Ye, Z.; Wang, Y.; Ayanga, B.A.; Galvan, D.; Green, N.H.; Chang, B.H.; Overbeek, P.A.; Danesh, F.R. Long noncoding RNA Tug1 regulates mitochondrial bioenergetics in diabetic nephropathy. J. Clin. Investig. 2016, 126, 4205–4218. [Google Scholar] [CrossRef] [Green Version]
- Kato, M. Noncoding RNAs as therapeutic targets in early stage diabetic kidney disease. Kidney Res. Clin. Pract. 2018, 37, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoki, K.; Mori, H.; Wang, J.; Suzuki, T.; Hong, S.; Yoshida, S.; Blattner, S.M.; Ikenoue, T.; Rüegg, M.A.; Hall, M.N.; et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J. Clin. Investig. 2011, 121, 2181–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.C.; Chang, C.Y.; Wu, Y.T.; Huang, J.P.; Yen, T.H.; Hung, L.M. Resveratrol retards progression of diabetic nephropathy through modulations of oxidative stress, proinflammatory cytokines, and AMP-activated protein kinase. J. Biomed. Sci. 2011, 18, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freedman, B.I.; Bostrom, M.; Daeihagh, P.; Bowden, D.W. Genetic Factors in Diabetic Nephropathy. Clin. J. Am. Soc. Nephrol. 2007, 2, 1306–1316. [Google Scholar] [CrossRef] [Green Version]
- Tziastoudi, M.; Stefanidis, I.; Zintzaras, E. The genetic map of diabetic nephropathy: Evidence from a systematic review and meta-analysis of genetic association studies. Clin. Kidney J. 2020, 13, 768–781. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Xiao, Y.; Li, L.; Xiong, X.; Han, Y.; Zhu, X.; Sun, L. The Susceptibility Genes in Diabetic Nephropathy. Kidney Dis. 2018, 4, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Tziastoudi, M.; Stefanidis, I.; Hadjigeorgiou, G.M.; Stravodimos, K.; Zintzaras, E. A systematic review and meta-analysis of genetic association studies for the role of inflammation and the immune system in diabetic nephropathy. Clin. Kidney J. 2017, 10, 293–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, T.G.; De Souza, M.L.; Da Silva, V.D.; dos Santos, M.; Da Silva, W.I.C.; Itaquy, T.P.; Garbin, H.I.; Veronese, F.V. Markers of renal fibrosis: How do they correlate with podocyte damage in glomerular diseases? PLoS ONE 2019, 14, e0217585. [Google Scholar] [CrossRef]
- Lee, H.S. Mechanisms and consequences of TGF-ß overexpression by podocytes in progressive podocyte disease. Cell Tissue Res. 2012, 347, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Liu, S.; Zhao, Z.; Liu, Y.; Li, T. The effect of connective tissue growth factor on renal fibrosis and podocyte injury in hypertensive rats. Ren. Fail. 2014, 36, 1420–1427. [Google Scholar] [CrossRef] [Green Version]
- Cho, M.E.; Smith, D.C.; Branton, M.H.; Penzak, S.R.; Kopp, J. Pirfenidone Slows Renal Function Decline in Patients with Focal Segmental Glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2007, 2, 906–913. [Google Scholar] [CrossRef] [Green Version]
- Vincenti, F.; Fervenza, F.C.; Campbell, K.N.; Diaz, M.; Gesualdo, L.; Nelson, P.; Praga, M.; Radhakrishnan, J.; Sellin, L.; Singh, A.; et al. A Phase 2, Double-Blind, Placebo-Controlled, Randomized Study of Fresolimumab in Patients with Steroid-Resistant Primary Focal Segmental Glomerulosclerosis. Kidney Int. Rep. 2017, 2, 800–810. [Google Scholar] [CrossRef] [Green Version]
- Adler, S.G.; Schwartz, S.; Williams, M.E.; Arauz-Pacheco, C.; Bolton, W.K.; Lee, T.; Li, N.; Neff, T.B.; Urquilla, P.R.; Sewell, K.L. Phase 1 Study of Anti-CTGF Monoclonal Antibody in Patients with Diabetes and Microalbuminuria. Clin. J. Am. Soc. Nephrol. 2010, 5, 1420–1428. [Google Scholar] [CrossRef]
- Hathaway, C.K.; Chang, A.S.; Grant, R.; Kim, H.-S.; Madden, V.J.; Bagnell, C.R.; Jennette, J.C.; Smithies, O.; Kakoki, M. High Elmo1 expression aggravates and low Elmo1 expression prevents diabetic nephropathy. Proc. Natl. Acad. Sci. USA 2016, 113, 2218–2222. [Google Scholar] [CrossRef] [Green Version]
- Hills, C.E.; Squires, P.E. The role of TGF-β and epithelial-to mesenchymal transition in diabetic nephropathy. Cytokine Growth Factor Rev. 2011, 22, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Shimazaki, A.; Kawamura, Y.; Kanazawa, A.; Sekine, A.; Saito, S.; Tsunoda, T.; Koya, D.; Babazono, T.; Tanaka, Y.; Matsuda, M.; et al. Genetic Variations in the Gene Encoding ELMO1 Are Associated With Susceptibility to Diabetic Nephropathy. Diabetes 2005, 54, 1171–1178. [Google Scholar] [CrossRef] [Green Version]
- Gu, H.F. Biomarkers of Adiponectin: Plasma Protein Variation and Genomic DNA Polymorphisms. Biomark. Insights 2009, 4, 123–133. [Google Scholar] [CrossRef]
- Lindström, T.; Frystyk, J.; Hedman, C.A.; Flyvbjerg, A.; Arnqvist, H.J. Elevated circulating adiponectin in type 1 diabetes is associated with long diabetes duration. Clin. Endocrinol. 2006, 65, 776–782. [Google Scholar] [CrossRef]
- Ohashi, K.; Iwatani, H.; Kihara, S.; Nakagawa, Y.; Komura, N.; Fujita, K.; Maeda, N.; Nishida, M.; Katsube, F.; Shimomura, I.; et al. Exacerbation of Albuminuria and Renal Fibrosis in Subtotal Renal Ablation Model of Adiponectin-Knockout Mice. Arter. Thromb. Vasc. Biol. 2007, 27, 1910–1917. [Google Scholar] [CrossRef] [PubMed]
- Cammisotto, P.G.; Bendayan, M. Adiponectin stimulates phosphorylation of AMP-activated protein kinase alpha in renal glomeruli. J. Mol. Histol. 2008, 39, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Okada-Iwabu, M.; Yamauchi, T.; Iwabu, M.; Honma, T.; Hamagami, K.I.; Matsuda, K.; Yamaguchi, M.; Tanabe, H.; Kimura-Someya, T.; Shirouzu, M.; et al. A small-molecule AdipoR agonist for type 2 diabetes and short life in obesity. Nature 2013, 503, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Yang, J.W.; Han, B.G.; Choi, S.O.; Kim, J.S. Adiponectin for the treatment of diabetic nephropathy. Korean J. Intern. Med. 2019, 34, 480–491. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Li, Q.; Wang, J.; Chen, P.; Jiang, S. Ellagic acid attenuates streptozocin induced diabetic nephropathy via the regulation of oxidative stress and inflammatory signaling. Food Chem. Toxicol. 2019, 123, 16–27. [Google Scholar] [CrossRef]
- Ahad, A.; Ganai, A.A.; Mujeeb, M.; Siddiqui, W.A. Ellagic acid, an NF-κB inhibitor, ameliorates renal function in experimental diabetic nephropathy. Chem. Interact. 2014, 219, 64–75. [Google Scholar] [CrossRef]
- Nangaku, M.; Eckardt, K.U. Pathogenesis of renal anemia. Semin. Nephrol. 2006, 26, 261–268. [Google Scholar] [CrossRef]
- Asada, N.; Takase, M.; Nakamura, J.; Oguchi, A.; Asada, M.; Suzuki, N.; Yamamura, K.-I.; Nagoshi, N.; Shibata, S.; Rao, T.N.; et al. Dysfunction of fibroblasts of extrarenal origin underlies renal fibrosis and renal anemia in mice. J. Clin. Investig. 2011, 121, 3981–3990. [Google Scholar] [CrossRef] [Green Version]
- Ji, Z.Z.; Xu, Y.C. Melatonin protects podocytes from angiotensin II-induced injury in an in vitro diabetic nephropathy model. Mol. Med. Rep. 2016, 14, 920–926. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Wu, H.; Zhang, H.; Li, F.; Chen, S.; Hou, B.; Shi, Y.; Zhao, L.; Duan, H. Anthocyanins inhibit high glucose-induced renal tubular cell apoptosis caused by oxidative stress in db/db mice. Int. J. Mol. Med. 2018, 41, 1608–1618. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Y.; Ning, G.; Deb, D.K.; Kong, J.; Li, Y.C. Combination therapy with AT1 blocker and vitamin D analog markedly ameliorates diabetic nephropathy: Blockade of compensatory renin increase. Proc. Natl. Acad. Sci. USA 2008, 105, 15896–15901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Yuan, W.; Sun, L.; Szeto, F.L.; Wong, K.E.; Li, X.; Kong, J.; Li, Y.C. 1,25-Dihydroxyvitamin D3 targeting of NF-kappaB suppresses high glucose-induced MCP-1 expression in mesangial cells. Kidney Int. 2007, 72, 193–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanasaki, K.; Taduri, G.; Koya, D. Diabetic nephropathy: The role of inflammation in fibroblast activation and kidney fibrosis. Front. Endocrinol. 2013, 4, 7. [Google Scholar] [CrossRef] [Green Version]
- Lan, H.Y.; Yang, N.; Nikolic-Paterson, D.J.; Xue, Q.Y.; Mu, W.; Isbel, N.M.; Metz, C.N.; Bucala, R.; Atkins, R.C. Expression of macrophage migration inhibitory factor in human glomerulonephritis. Kidney Int. 2000, 57, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Chow, F.Y.; Nikolic-Paterson, D.J.; Ma, F.Y.; Ozols, E.; Rollins, B.J.; Tesch, G.H. Monocyte chemoattractant protein-1-induced tissue inflammation is critical for the development of renal injury but not type 2 diabetes in obese db/db mice. Diabetologia 2007, 50, 471–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezzano, S.; Aros, C.; Droguett, A.; Burgos, M.E.; Ardiles, L.; Flores, C.; Schneider, H.; Ruiz-Ortega, M.; Egido, J. NF-kappaB activation and overexpression of regulated genes in human diabetic nephropathy. Nephrol. Dial. Transplant. 2004, 19, 2505–2512. [Google Scholar] [CrossRef]
- Wada, T.; Furuichi, K.; Sakai, N.; Iwata, Y.; Yoshimoto, K.; Shimizu, M.; Takeda, S.-I.; Takasawa, K.; Yoshimura, M.; Kida, H.; et al. Up-regulation of monocyte chemoattractant protein-1 in tubulointerstitial lesions of human diabetic nephropathy. Kidney Int. 2000, 58, 1492–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panee, J. Monocyte Chemoattractant Protein 1 (MCP-1) in obesity and diabetes. Cytokine 2012, 60, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galkina, E.; Ley, K. Leukocyte recruitment and vascular injury in diabetic nephropathy. J. Am. Soc. Nephrol. 2006, 17, 368–377. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Yuan, Y.; Zhang, T.; Xu, B.; Gao, Q.; Guan, T. Pentosan polysulfate ameliorates apoptosis and inflammation by suppressing activation of the p38 MAPK pathway in high glucose-treated HK-2 cells. Int. J. Mol. Med. 2017, 41, 908–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Li, N.; Yan, S.; Liu, M.; Sun, B.; Lu, Y.; Shao, Y. Ursolic acid alleviates inflammation and against diabetes-induced nephropathy through TLR4-mediated inflammatory pathway. Mol. Med. Rep. 2018, 18, 4675–4681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, R.; Banerjee, S.; Tripathi, Y.B. Pueraria tuberosa extract inhibits iNOS and IL-6 through suppression of PKC-α and NF-kB pathway in diabetes-induced nephropathy. J. Pharm. Pharmacol. 2018, 70, 1102–1112. [Google Scholar] [CrossRef] [PubMed]
- Tesch, G.H. Macrophages and Diabetic Nephropathy. Semin. Nephrol. 2010, 30, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Chow, F.; Ozols, E.; Nikolic-Paterson, D.; Atkins, R.C.; Tesch, G.H. Macrophages in mouse type 2 diabetic nephropathy: Correlation with diabetic state and progressive renal injury. Kidney Int. 2004, 65, 116–128. [Google Scholar] [CrossRef] [Green Version]
- Ji, L.; Chen, Y.; Wang, H.; Zhang, W.; He, L.; Wu, J.; Liu, Y. Overexpression of Sirt6 promotes M2 macrophage transformation, alleviating renal injury in diabetic nephropathy. Int. J. Oncol. 2019, 55, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Yin, N.; Liu, W.; Cui, X.; Chen, S.; Wang, E. Curcumin Ameliorates Diabetic Nephropathy by Suppressing NLRP3 Inflammasome Signaling. BioMed. Res. Int. 2017, 2017, 1516985. [Google Scholar] [CrossRef] [PubMed]
- Corrêa-Costa, M.; Braga, T.T.; Semedo, P.; Hayashida, C.Y.; Bechara, L.R.G.; Elias, R.M.; Barreto, C.R.; Silva-Cunha, C.; Hyane, M.I.; Gonçalves, G.M.; et al. Pivotal Role of Toll-Like Receptors 2 and 4, Its Adaptor Molecule MyD88, and Inflammasome Complex in Experimental Tubule-Interstitial Nephritis. PLoS ONE 2011, 6, e29004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahzad, K.; Bock, F.; Dong, W.; Wang, H.; Kopf, S.; Kohli, S.; Al-Dabet, M.M.; Ranjan, S.; Wolter, J.; Wacker, C.; et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015, 87, 74–84. [Google Scholar] [CrossRef] [Green Version]
- Diwan, V.; Gobe, G.; Brown, L. Glibenclamide improves kidney and heart structure and function in the adenine-diet model of chronic kidney disease. Pharmacol. Res. 2014, 79, 104–110. [Google Scholar] [CrossRef]
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.L.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A.V.; et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 2019, 15, 556–559. [Google Scholar] [CrossRef]
- Hsu, B.; Fu, S.-H.; Hsu, S.; Chen, S.-T. Interleukin-1 Receptor Antagonist Enhances Islet Engraftment Without Impacting Serum Levels of Nitrite or Osteopontin. Transplant. Proc. 2009, 41, 1781–1785. [Google Scholar] [CrossRef]
- Tashiro, K.; Koyanagi, I.; Saitoh, A.; Shimizu, A.; Shike, T.; Ishiguro, C.; Koizumi, M.; Funabiki, K.; Horikoshi, S.; Shirato, I.; et al. Urinary levels of monocyte chemoattractant protein-1 (MCP-1) and interleukin-8 (IL-8), and renal injuries in patients with type 2 diabetic nephropathy. J. Clin. Lab. Anal. 2002, 16, 1–4. [Google Scholar] [CrossRef]
- Sanchez-Niño, M.D.; Martin, A.B.; Gonçalves, S.; Sanz, A.B.; Ucero, A.; Izquierdo, M.C.; Ramos, A.M.; Berzal, S.; Selgas, R.; Ruiz-Ortega, M.; et al. TNF Superfamily: A Growing Saga of Kidney Injury Modulators. Mediat. Inflamm. 2010, 2010, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Le Hir, M.; Haas, C.; Marino, M.; Ryffel, B. Prevention of crescentic glomerulonephritis induced by anti-glomerular membrane antibody in tumor necrosis factor-deficient mice. Lab. Investig. 1998, 78, 1625–1631. [Google Scholar]
- Khan, S.B.; Cook, H.T.; Bhangal, G.; Smith, J.; Tam, F.W.; Pusey, C.D. Antibody blockade of TNF-α reduces inflammation and scarring in experimental crescentic glomerulonephritis. Kidney Int. 2005, 67, 1812–1820. [Google Scholar] [CrossRef] [Green Version]
- Hehlgans, T.; Pfeffer, K. The intriguing biology of the tumour necrosis factor/tumour necrosis factor receptor superfamily: Players, rules and the games. Immunology 2005, 115, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Nosratabadi, R.; Arababadi, M.K.; Hassanshahi, G.; Yaghini, N.; Pooladvand, V.; Shamsizadeh, A.; Zarandi, E.R.; Hakimi, H. Evaluation of IFN-gamma serum level in nephropatic type 2 diabetic patients. Pak. J. Biol. Sci. 2009, 12, 746–749. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.H.; Ding, X.M.; Li, Y.; Liu, H.B.; Xue, W.J.; Tian, X.H.; Feng, X.S.; Jiao, F.M.; Zheng, J. Simultaneous blockade of the CD40/CD40L and NF-κB pathways prolonged islet allograft survival. Transpl. Int. 2012, 25, 118–126. [Google Scholar] [CrossRef]
- Bai, Y.; Wang, J.; He, Z.; Yang, M.; Li, L.; Jiang, H. Mesenchymal Stem Cells Reverse Diabetic Nephropathy Disease via Lipoxin A4 by Targeting Transforming Growth Factor β (TGF-β)/smad Pathway and Pro-Inflammatory Cytokines. Med. Sci. Monit. 2019, 25, 3069–3076. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Tian, Z.; Zhang, L.; Liu, R.; Guan, Q.; Jiang, J. Effects of CCR5 59029G/A polymorphism on the risk to diabetic nephropathy. Oncotarget 2017, 8, 106926–106934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro-González, J.F.; Mora-Fernández, C.; De Fuentes, M.M.; García-Pérez, J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat. Rev. Nephrol. 2011, 7, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Sayyed, S.G.; Ryu, M.; Kulkarni, O.P.; Schmid, H.; Lichtnekert, J.; Grüner, S.; Green, L.; Mattei, P.; Hartmann, G.; Anders, H.-J. An orally active chemokine receptor CCR2 antagonist prevents glomerulosclerosis and renal failure in type 2 diabetes. Kidney Int. 2011, 80, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Gale, J.D.; Gilbert, S.; Blumenthal, S.; Elliott, T.; Pergola, P.E.; Goteti, K.; Scheele, W.; Perros-Huguet, C. Effect of PF-04634817, an Oral CCR2/5 Chemokine Receptor Antagonist, on Albuminuria in Adults with Overt Diabetic Nephropathy. Kidney Int. Rep. 2018, 3, 1316–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.-Y.; Huang, P.-H.; Yang, A.-H.; Tarng, D.-C.; Yang, W.-C.; Lin, C.-C.; Chen, J.-W.; Schmid-Schönbein, G.; Lin, S.-J. Matrix metalloproteinase-9 deficiency attenuates diabetic nephropathy by modulation of podocyte functions and dedifferentiation. Kidney Int. 2014, 86, 358–369. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Kang, Y.S.; Dai, C.; Kiss, L.P.; Wen, X.; Liu, Y. Epithelial-to-Mesenchymal Transition Is a Potential Pathway Leading to Podocyte Dysfunction and Proteinuria. Am. J. Pathol. 2008, 172, 299–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kundu, S.; Pushpakumar, S.B.; Tyagi, A.; Coley, D.; Sen, U. Hydrogen sulfide deficiency and diabetic renal remodeling: Role of matrix metalloproteinase-9. Am. J. Physiol. Metab. 2013, 304, E1365–E1378. [Google Scholar] [CrossRef] [Green Version]
- Masola, V.; Onisto, M.; Zaza, G.; Lupo, A.; Gambaro, G. A new mechanism of action of sulodexide in diabetic nephropathy: Inhibits heparanase-1 and prevents FGF-2-induced renal epithelial-mesenchymal transition. J. Transl. Med. 2012, 10, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, A.I.; Chan, L.Y.; Lai, K.N.; Tang, S.C.W.; Chow, C.W.; Lam, M.F.; Leung, J.C. Distinct role of matrix metalloproteinase-3 in kidney injury molecule-1 shedding by kidney proximal tubular epithelial cells. Int. J. Biochem. Cell Biol. 2012, 44, 1040–1050. [Google Scholar] [CrossRef] [PubMed]
- Amanzadeh, M.; Mota, A.; Zarghami, N.; Abedi-Azar, S.; Abroon, S.; Akbarian, N.; Mihanfar, A.; Rahmati-Yamchi, M. Association Between Matrix Metalloproteinase-3 Activity and Glomerular Filtration Rate and Albuminuria Status in Patients With Type 2 Diabetes Mellitus. Iran. J. Kidney Dis. 2018, 12, 40–47. [Google Scholar] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maiti, A.K. Development of Biomarkers and Molecular Therapy Based on Inflammatory Genes in Diabetic Nephropathy. Int. J. Mol. Sci. 2021, 22, 9985. https://doi.org/10.3390/ijms22189985
Maiti AK. Development of Biomarkers and Molecular Therapy Based on Inflammatory Genes in Diabetic Nephropathy. International Journal of Molecular Sciences. 2021; 22(18):9985. https://doi.org/10.3390/ijms22189985
Chicago/Turabian StyleMaiti, Amit K. 2021. "Development of Biomarkers and Molecular Therapy Based on Inflammatory Genes in Diabetic Nephropathy" International Journal of Molecular Sciences 22, no. 18: 9985. https://doi.org/10.3390/ijms22189985
APA StyleMaiti, A. K. (2021). Development of Biomarkers and Molecular Therapy Based on Inflammatory Genes in Diabetic Nephropathy. International Journal of Molecular Sciences, 22(18), 9985. https://doi.org/10.3390/ijms22189985