
Insulin and Insulin Resistance in Alzheimer’s Disease


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Role of Insulin in CNS
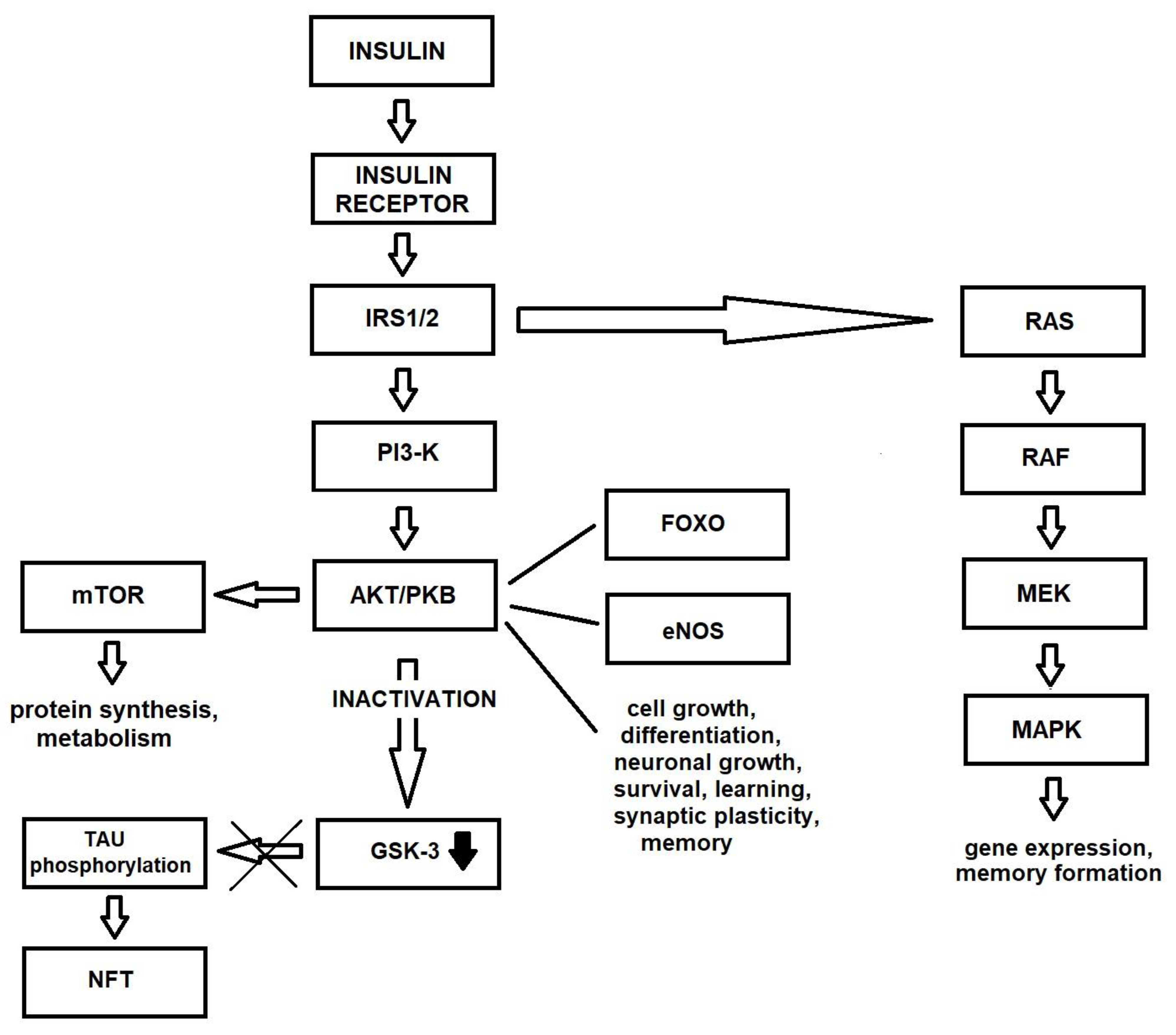
Insulin Signaling in the Brain
3. The Role of Insulin in the Brain
3.1. Insulin and Brain Glucose Metabolism
3.2. Insulin and Cognition
3.3. The Effects of Insulin in Neurons
3.4. Effects of Insulin in Glial Cells
3.5. Insulin and Hippocampal Adult Neurogenesis
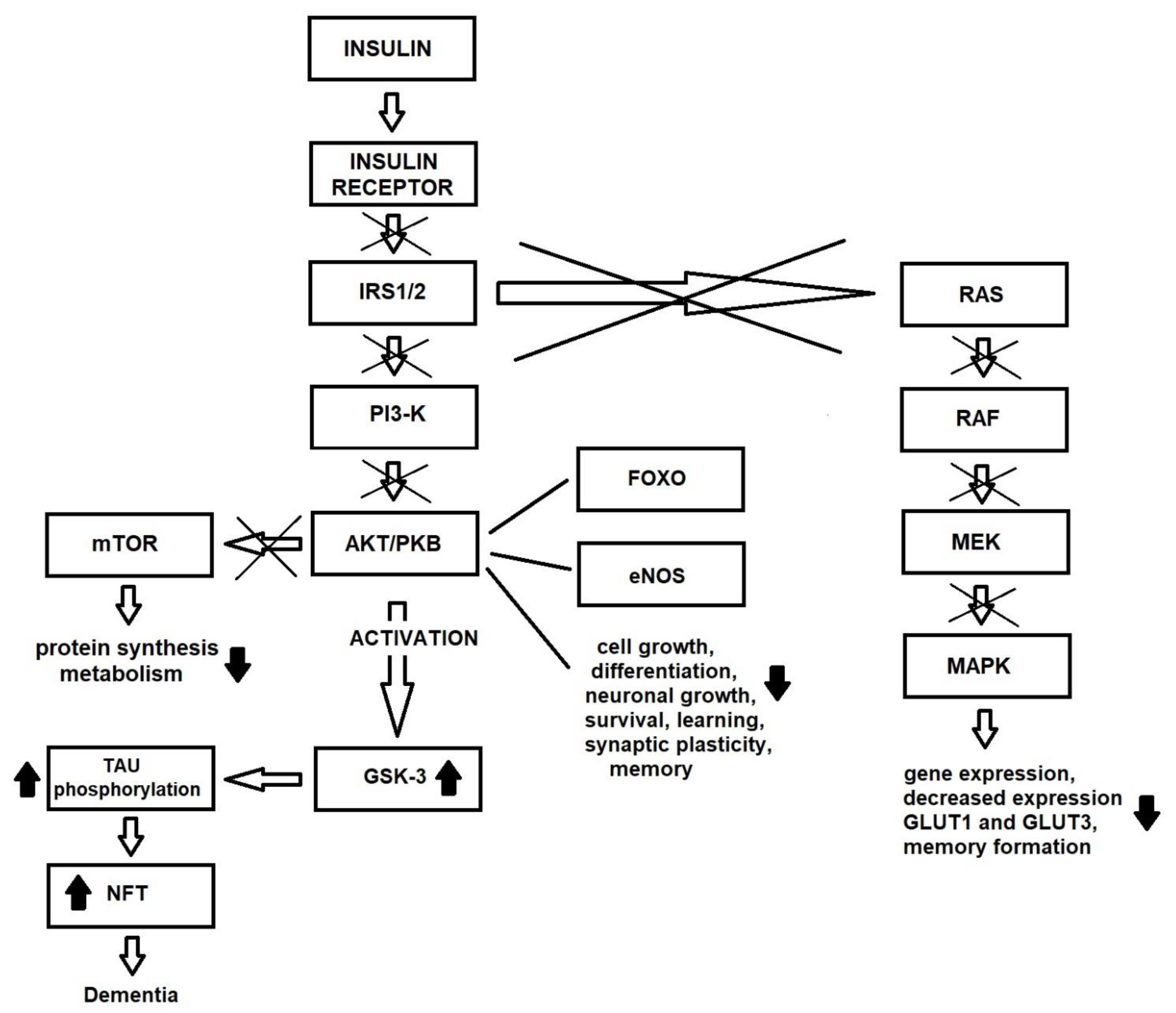
4. Insulin Resistance and Alzheimer’s Disease
4.1. Insulin Resistance and Tau Phosphorylation
4.2. Insulin Resistance and Amyloid-β Pathology
4.3. Insulin Resistance and Inflammation
4.4. Insulin Resistance and Oxidative Stress
4.5. Insulin Resistance and Cognitive Impairment
5. Therapy
5.1. Insulin Therapy
5.2. Insulin Sensitizers Therapy
5.3. Insulin Stimulating/Releasing Hormones Therapy
5.4. Other Antidiabetic Drug Therapies
5.5. Non-Pharmacological Approaches
6. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bromander, S.; Anckarsäter, R.; Ahrén, B.; Kristiannson, M.; Blennow, K.; Holmäng, A.; Zetterberg, H.; Anckarsäter, H.; Wass, C.E. Cerebrospinal fluid insulin during non-neurological surgery. J. Neurol. Transm. 2010, 117, 1167–1170. [Google Scholar] [CrossRef]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.-Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef]
- Banks, W.A. The source of cerebral insulin. Eur. J. Pharmacol. 2004, 490, 5–12. [Google Scholar] [CrossRef]
- Gray, S.M.; Aylor, K.W.; Barrett, E.J. Unravelling the regulation of insulin transport across the brain endothelial cells. Diabetologia 2017, 60, 1512–1521. [Google Scholar] [CrossRef] [Green Version]
- Banks, W.A.; Owen, J.B.; Erickson, M.A. Insulin in the brain. Pharmacol. Ther. 2012, 136, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.J.; Hope, I.D.; Ader, M.; Bergman, R.N. Insulin transport across capillaries is rate limiting for insulin action in dogs. J. Clin. Invest. 1989, 84, 1620–1628. [Google Scholar] [CrossRef]
- Struble, J.H.; Porte, D., Jr.; Woods, S.C. Insulin responses and glucose levels in plasma and cerebrospinal fluid during fasting and refeeding in rat. Physiol. Behav. 1988, 44, 205–208. [Google Scholar] [CrossRef]
- Baskin, D.G.; Figlewicz, D.P.; Woods, D., Jr.; Dorsa, D.M. Insulin in the brain. Annu. Rev. Physiol. 1987, 49, 335–347. [Google Scholar] [CrossRef]
- Duarte, A.I.; Moreira, P.I.; Oliveira, C.R. Insulin in central nervous system: More than just a peripheral hormone. J. Aging Res. 2012, 2012, 384017. [Google Scholar] [CrossRef] [Green Version]
- Blázquez, E.; Velázquez, E.; Huartado-Carneiro, V.; Ruiz-Albusac, J.M. Insulin in the brain: Its pathophysiological implications for states related with central insulin resistance, type 2 diabetes and Alzheimer’s disease. Front. Endocrinol. (Lausanne) 2014, 5, 161. [Google Scholar] [CrossRef] [Green Version]
- Marks, J.L.; Porte, D., Jr.; Stahl, W.L.; Baskin, D.G. Localization of insulin receptor mRNA in rat brain by in situ hybridization. Endocrinology 1990, 127, 3234–3236. [Google Scholar] [CrossRef]
- Unger, J.W.; Betz, M. Insulin receptors and signal transduction proteins in the hypothalamo-hypophyseal system: A review on morphological findings and functional implications. Histol. Histopathol. 1998, 13, 1215–1224. [Google Scholar]
- Werther, G.A.; Hogg, A.; Oldfield, B.J.; McKinley, M.J.; Figdor, R.; Allen, A.M.; Mendelsohn, F.A. Localization and characterization of insulin receptors in rat brain and pituitary gland using in vitro autoradiography and computerized densitometry. Endocrinology 1987, 121, 1562–1570. [Google Scholar] [CrossRef]
- Adamo, M.; Raizada, M.K.; LeRoith, D. Insulin and insulin-like growth factor receptors in the nervous system. Mol. Neurobiol. 1989, 3, 71–100. [Google Scholar] [CrossRef]
- Unger, J.; McNeil, T.H.; Moxley, R.T., III; White, M.; Moss, A.; Livingston, J.N. Distribution of insulin receptor-like immunoreactivity in the rat forebrain. Neuroscience 1989, 31, 143–157. [Google Scholar] [CrossRef]
- Roth, R.; Morgan, D.O.; Beaudoin, J.; Sara, V. Purification and characterization of the human brain insulin receptor. J. Biol. Chem. 1986, 261, 3753–3757. [Google Scholar] [CrossRef]
- Kotzke, G.; Schutt, M.; Missler, U.; Moller, D.E.; Fehm, H.L.; Klein, H.H. Binding of human, porcine and bovine insulin to insulin receptors from human brain, muscle and adipocytes and to expressed recombinant alternatively spliced insulin receptor isoforms. Diabetologia 1995, 38, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Kullmann, S.; Heni, M.; Hallschmid, M.; Fritsche, A.; Preissl, H.; Haring, H.U. Brain insulin resistance at the crossroad of metabolic and cognitive disorders in humans. Physiol. Rev. 2016, 96, 1169–1209. [Google Scholar] [CrossRef] [Green Version]
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocrinol. Rev. 2009, 30, 586–623. [Google Scholar] [CrossRef] [Green Version]
- Bruning, J.C.; Gautam, D.; Burks, D.J.; Gillette, J.; Schubert, M.; Orban, P.C.; Klein, R.; Krone, W.; Müller-Wieland, D.; Kahn, C.R. Role of brain insulin receptor in control of body weight and reproduction. Science 2000, 289, 2122–2125. [Google Scholar] [CrossRef]
- Fernandez, A.M.; Torres-Aleman, I. The many faces of insulin-like peptide signaling in the brain. Nat. Rev. Neurosci. 2012, 13, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Bailes, E.M.; Nave, B.T.; Soos, M.A.; Orr, S.R.; Hayward, A.C.; Siddle, K. Insulin receptor/IGF-1 receptor hybrids are widely distributed in mammalian tissues: Quantitation of individual receptor species by selective immunoprecipitation and immunoblotting. Biochem. J. 1997, 327, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Ghasenmi, R.; Haeri, A.; Dargahi, L.; Mohamed, Z.; Ahmadiani, A. Insulin in the brain: Sources, localization and functions. Mol. Neurobiol. 2013, 47, 145–171. [Google Scholar] [CrossRef]
- Garwood, C.J.; Ratcliffe, L.E.; Morgan, S.V.; Simpson, J.E.; Owens, H.; Vazquez-Villaseñor, I.; Heath, P.R.; Romero, I.A.; Ince, P.G.; Wharton, S.B. Insulin and IGF1 signaling pathways in human astrocytes in vitro and in vivo: Characterisation, subcellular localisation and modulation of the receptors. Mol. Brain. 2015, 8, 51. [Google Scholar] [CrossRef] [Green Version]
- Pomytkin, I.; Pinelis, V. Brain insulin resistance: Focus on insulin receptor-mitochondria interactions. Life 2021, 11, 262. [Google Scholar] [CrossRef]
- Kleinridders, A.; Ferris, H.A.; Cai, W.; Kahn, C.R. Insulin action in brain regulates systemic metabolism and brain function. Diabetes 2014, 63, 2232–2243. [Google Scholar] [CrossRef] [Green Version]
- De Felice, F.G.; Ferreira, S.T. Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes 2014, 63, 2262–2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyer, A.H.; Vahdatpour, C.; Sanfeliu, A.; Tropea, D. The role of insulin-like growth factor 1 (IGF-1) in brain development, maturation and neuroplasticity. Neuroscience 2016, 325, 89–99. [Google Scholar] [CrossRef]
- Cheng, Z.; Tseng, Y.; White, M.F. Insulin signaling meets mitochondria in metabolism. Trends. Endocrinol. Metab. 2010, 21, 589–598. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, B.J.; Bittner-Kowalczyk, A.; White, M.F.; Harbeck, M. Tyrosine dephosphorylation and deactivation of insulin receptor substrate-1 by protein tyrosine phosphatase 1B. Possible facilitation by the formation of a ternary complex with the GRB2 adaptor protein. J. Biol. Chem. 2000, 275, 4283–4289. [Google Scholar] [CrossRef] [Green Version]
- Khezri, N.; Yaghoubnezhadzanganeh, G.; Attarzadeh, A. The links between brain insulin resistance and Alzheimer’s disease. Int. J. Med. Health Sci. 2019, 13, 41–49. [Google Scholar]
- Grillo, C.A.; Piroli, G.G.; Hendry, R.M.; Reagan, L.P. Insulin-stimulated translocation of GLUT4 to the plasma membrane in rat hippocampus is PI3-kinase dependent. Brain. Res. 2009, 1296, 35–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condorelli, F.; Salomoni, P.; Cotteret, S.; Cesi, V.; Srinivasula, S.M.; Alnemri, E.S.; Calabretta, S. Caspase cleavage enhances the apoptosis-inducing effects of BAD. Mol. Cell Biol. 2001, 21, 3025–3036. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P.; Doran, E.; Gillespie, J.P.; O’Toole, A. Mitochondria and cell death. Biochem. Soc. Trans. 2000, 28, 170–177. [Google Scholar] [CrossRef]
- Roith, D.L.; Zick, Y. Recent advances in our understanding of insulin action and insulin resistance. Diabetes Care 2001, 24, 588–597. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, C.; Cardoso, S.M.; Correira, S.C.; Moreira, P.I. Tortuous paths of insulin signaling and mitochondria in Alzheimer’s disease. Adv. Exp. Med. Biol. 2019, 1128, 161–183. [Google Scholar]
- Annott, M.A.; Wells, D.G.; Fallon, J.R. The insulin receptor kinase substrate p58/53 and the insulin receptor are components of CNS synapses. J. Neurosci. 1999, 19, 7300–7308. [Google Scholar]
- Neth, B.J.; Craft, S. Insulin resistance and Alzheimer’s disease: Bioenergetics linkages. Front. Aging Neurosci. 2017, 9, 345. [Google Scholar] [CrossRef]
- De la Monte, S.M. Brain insulin resistance and deficiency as therapeutic targets in Alzheimer’s disease. Curr. Alzheimer’s Res. 2012, 9, 35–66. [Google Scholar] [CrossRef] [PubMed]
- Pearson-Leary, J.; McNay, E.C. Novel roles for the insulin-regulated glucose trnsporter-4 in hippocampal dependent memory. J. Neurosci. 2016, 36, 11851–11864. [Google Scholar] [CrossRef] [PubMed]
- Surguchov, A. Caveolin: A new link between diabetes and AD. Cell Mol. Neurobiol. 2020, 40, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- McNay, E.C.; Ong, C.T.; McCrimmon, R.J.; Creswell, J.; Bogan, J.S.; Sherwin, R.S. Hippocampal memory processes are modulated by insulin and high-fat induced insulin resistance. Neurobiol. Learn. Mem. 2010, 93, 546–553. [Google Scholar] [CrossRef] [Green Version]
- Heni, M.; Hennige, A.M.; Peter, A.; Siegel-Axel, D.; Ordelheide, A.M.; Krebs, N.; Machicao, F.; Fritsche, A.; Häring, H.-U.; Staiger, H. Insulin promotes glycogen storage and cells proliferation in primary human astrocytes. PLoS ONE 2011, 6, e21594. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, M.; Fusco, S.; Grassi, C. Brain insulin resistance and hippocampal plasticity: Mechanisms and biomarkers of cognitive decline. Front. Neurosci. 2019, 13, 788. [Google Scholar] [CrossRef] [Green Version]
- Piroli, G.G.; Grillo, C.A.; Hoskin, E.K.; Znamensky, V.; Katz, E.B.; Milner, T.A.; McEwen, B.S.; Charron, M.J.; Reagan, L.P. Peripheral glucose administration stimulates the translocation of GLUT8 glucose transporter to the endoplasmic reticulum in the rat hippocampus. J. Compar. Neurol. 2002, 452, 103–114. [Google Scholar] [CrossRef]
- Reagan, L.P.; Rosell, D.R.; Alves, S.E.; Hoskin, E.K.; McCall, A.L.; Charron, M.J.; McEwen, B.S. GLUT8 glucose transporter is localized to excitatory and inhibitory neurons in the rat hippocampus. Brain Res. 2002, 932, 129–134. [Google Scholar] [CrossRef]
- Moult, P.R.; Harvey, J. Hormonal regulation of hippocampal dendritic morphology and synaptic plasticity. Cell. Adhes. Migr. 2008, 2, 269–275. [Google Scholar] [CrossRef] [Green Version]
- Van der Heide, L.P.; Kamal, A.; Artola, A.; Gispen, W.H.; Ramakers, G.M. Insulin modulates hippocampal activity-dependent, synaptic plasticity in a N-methyl-D-aspartate receptor and phosphatidyl-inositol-3-kinase dependent manner. J. Neurochem. 2005, 94, 1158–1166. [Google Scholar] [CrossRef]
- Gratuze, M.; Joly-Amado, A.; Vieau, D.; Buée, L.; Blum, D. Mutual relationship between Tau and central insulin signaling: Consequences for AD and tauopathies. Neuroendocrinology 2018, 107, 181–195. [Google Scholar] [CrossRef]
- Skeberdis, V.A.; Lan, J.; Zheng, X.; Zukin, R.S.; Bennett, M.V. Insulin promotes rapid delivery of N-methyl-D-aspartate receptors to the cell surface by exocytosis. Proc. Natl. Acad. Sci. USA 2001, 98, 3561–3566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.P.; Lu, K.T.; Chang, W.C.; Gean, P.W. Involvement of mitogen-activated protein kinase in hippocampal long-term potentiation. J. Biomed. Sci. 1999, 6, 409–417. [Google Scholar] [CrossRef]
- Sanna, P.P.; Cammalleri, M.; Berton, F.; Simpson, C.; Lutjens, R.; Bloom, E.E.; Francesconi, W. Phosphatidylinositol 3-kinase is required for the expression but not for the induction or the maintenance of long-term potentiation in the hippocampal CA1 region. J. Neurosci. 2002, 22, 3359–3365. [Google Scholar] [CrossRef] [Green Version]
- Spinelli, M.; Fusco, S.; Mainardi, M.; Scala, F.; Natale, F.; Lapenta, R.; Mattera, A.; Rinaudo, M.; Li Puma, D.D.; Ripoli, C.; et al. Brain insulin resistance impairs hippocampal synaptic plasticity and memory by increasing GluA1 palmitoylation through FoxO3a. Nat. Commun. 2017, 8, 2009. [Google Scholar] [CrossRef]
- Zhao, W.; Chen, H.; Xu, H.; Moore, E.; Meiri, N.; Quon, M.J.; Alkon, D.L. Brain insulin resistance and spatial memory. Correlated changes in gene expression, tyrosine phosphorylation, and signal molecules in the hippocampus of water maze trained rats. J. Biol. Chem. 1999, 274, 34893–34902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grillo, C.A.; Piroli, G.G.; Kaigler, K.F.; Wilson, M.A.; Reagan, L.P. Downregulation of hypothalamic insulin receptor expression elicits depressive-like behaviors in rats. Behav. Brain Res. 2011, 222, 230–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benedict, C.; Hallschmid, M.; Hatke, A.; Schultes, B.; Fehm, H.L.; Born, J.; Kern, W. Intranasal insulin improves memory in humans. Psychoneuroendocrinology 2004, 29, 1326–1334. [Google Scholar] [CrossRef]
- Folch, J.; Ettcheto, M.; Busquets, O.; Sánchez-López, E.; Castro-Torres, R.D.; Vardaguer, E.; Manzine, P.R.; Poor, S.R.; Garcia, M.L.; Olloquequi, J.; et al. The implication of the brain insulin receptor in late onset Alzheimer’s disease dementia. Pharmaceuticals 2018, 11, 11. [Google Scholar] [CrossRef] [Green Version]
- Nelson, T.J.; Sun, M.K.; Hongpaisan, J.; Alkon, D.L. Insulin, PKC signaling pathways and synaptic remodeling during memory storage and neuronal repair. Eur. J. Pharmacol. 2008, 585, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Van der Heide, L.P.; Ramakers, G.M.; Smidt, M.P. Insulin signaling in the central nervous system: Learning to survive. Prog. Neurobiol. 2006, 79, 205–221. [Google Scholar] [CrossRef]
- Kaur, S.; Singh, S.; Arora, A.; Ram, P.; Kumar, S.; Kumar, P.; Abed, S.N. Pharmacology of GABA and its receptors. Front. Pharmacol. Neurotransm. 2020, 241–292. [Google Scholar] [CrossRef]
- Sandoval-Salazar, C.; Ramirez-Emiliano, J.; Trejo-Bahena, A.; Oviedo-Solis, C.I.; Solis-Ortiz, M.S. A high-fat diet decreases GABA concentration in the frontal cortex and hippocampal of rats. Biol. Res. 2016, 49, 15. [Google Scholar] [CrossRef] [Green Version]
- Chiu, S.L.; Chen, C.M.; Cline, H.T. Insulin receptor signaling regulates synapse number, dendritic plasticity and circuit function in vivo. Neuron 2008, 58, 708–719. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.C.; Huang, C.C.; Hsu, K.S. Insulin promotes dendritic spine and synapse formation by the PI3K/Akt/mTOR and Rac1 signaling pathways. Neuropharmacology 2011, 61, 867–879. [Google Scholar] [CrossRef]
- Peineau, S.; Taghibiglou, C.; Bradley, C.; Wong, T.P.; Liu, L.; Lu, J.; Lo, E.; Wu, D.; Saule, E.; Bouschet, T.; et al. LTP inhibits LTD in the hippocampus via regulation of GSK3β. Neuron 2007, 53, 703–717. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Han, Y. Insulin inhibits AMPA-induced neuronal damage via stimulation of protein kinase B (Akt). J. Neural. Transm. (Vienna) 2005, 112, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Moosavi, M.; Naghdi, N.; Maghsoudi, N.; Zahedi, S. The effect of intrahippocampal insulin microinjection on spatial learning and memory. Horm. Behav. 2006, 50, 748–752. [Google Scholar] [CrossRef]
- Pelvig, D.P.; Pakkenberg, H.; Stark, A.K.; Pakkenberg, B. Neocortical glial cell members in human brains. Neurobiol. Aging 2008, 29, 1754–1762. [Google Scholar] [CrossRef]
- Wender, R.; Brown, A.M.; Fern, R.; Swanson, R.A.; Farrell, K.; Ransom, B.R. Astrocytic glycogen influences axon function and survival during glucose deprivation in central white matter. J. Neurosci. 2000, 20, 6804–6810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellerin, L.; Pellegri, G.; Bittar, P.G.; Charnay, Y.; Bouras, C.; Martin, J.L.; Stella, N.; Magistretti, P.J. Evidence supporting the existence of an activity-different astrocyte-neuron lactate shuttle. Dev. Neurosci. 1998, 20, 291–299. [Google Scholar] [CrossRef]
- Spilman, L.J.; Bahniwal, M.; Little, J.P.; Walker, D.G.; Klegeris, A. Insulin modulates in vitro secretion of cytokines and cytotoxins by human glial cells. Curr. Alzheimer’s Res. 2015, 12, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.L.; Fragoso, G.; Miron, V.E.; Darlington, P.J.; Mushynski, W.E.; Antel, J.; Almazan, G. Response of human oligodendrocyte progenitors to growth factors and axon signals. J. Neuropathol. Exp. Neurol. 2010, 69, 930–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, S.M.; Jessberger, S. Adult neurogenesis: Mechanisms and functional significance. Development 2014, 141, 1983–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eriksson, P.S.; Perfilieva, E.; Bjork-Eriksson, T.; Alborn, A.M.; Nordborg, C.; Peterson, D.A.; Gage, F.H. Neurogenesis in the adult human hippocampus. Nat. Med. 1998, 4, 1313–1317. [Google Scholar] [CrossRef] [PubMed]
- Kempermann, G.; Gast, D.; Kronenberg, G.; Yamaguchi, M.; Gage, F.H. Early determination and long-term persistence of adult-generated new neurons in the hippocampus of mice. Development 2003, 130, 391–399. [Google Scholar] [CrossRef] [Green Version]
- Taylor, C.J.; Jhaveri, D.J.; Barlett, P.F. The therapeutic potential stem cells for the treatment of neurological disorders. Front. Cell Neurosci. 2013, 7, 5. [Google Scholar] [CrossRef] [Green Version]
- Chell, J.M.; Brand, A.H. Nutrition-response glia control exit of neural stem cells from quiescence. Cell 2010, 143, 1161–1173. [Google Scholar] [CrossRef] [Green Version]
- Sousa-Nunes, R.; Yee, L.L.; Gould, A.P. Fat cells reactivate quiescent neuroblasts via TOR and glial insulin relays in Drosophila. Nature 2011, 471, 508–512. [Google Scholar] [CrossRef] [Green Version]
- Ǻberg, M.A.; Ǻberg, N.D.; Palmer, T.D.; Alborn, A.M.; Carlsson-Skwirut, C.; Bang, P.; Rosengren, L.E.; Olsson, T.; Gage, F.H.; Eriksson, P.S. IGF-I has a direct proliferative effect in adult hippocampal progenitor cells. Mol. Cell Neurosci. 2003, 24, 23–40. [Google Scholar] [CrossRef]
- Brooker, G.J.; Kalloniatis, M.; Russo, V.C.; Murphy, M.; Werther, G.A.; Barlett, P.F. Endogenous IGF-1 regulates the neuronal differentiation of adult stem cells. J. Neurosci. Res. 2000, 59, 332–341. [Google Scholar] [CrossRef]
- Sun, L.Y. Hippocampal IGF-1 expression, neurogenesis and slowed aging: Clues to longevity from mutant mice. Age 2006, 28, 181–189. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, B.J. Insulin resistance as the core defect of type 2 diabetes mellitus. Am. J. Cardiol. 2002, 90, 3G–10G. [Google Scholar] [CrossRef]
- Mielke, J.G.; Taghibiglou, C.; Liu, L.; Zhang, Y.; Jia, J.; Adeli, K.; Wang, Y.T. A biochemical and functional characterization of diet-induced brain insulin resistance. J. Neurochem. 2005, 93, 1568–1578. [Google Scholar] [CrossRef]
- Burillo, J.; Marqués, P.; Jiménez, B.; González-Blanco, C.; Benito, M.; Guillén, C. Insulin resistance and diabetes mellitus in Alzheimer’s disease. Cells 2021, 10, 1236. [Google Scholar] [CrossRef]
- Hayer, S. The aging brain. Changes in the neuronal/insulin receptor signal transduction cascade trigger late-onset sporadic Alzheimer disease (SAD). A mini review. J. Neural. Transm. 2002, 109, 991–1002. [Google Scholar] [CrossRef]
- De la Monte, S.M.; Wands, J.R. Alzheimer’s disease is type 3 diabetes: Evidence reviewed. J. Diabetes Sci. Tech. 2008, 2, 1101–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weingarten, M.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brion, J.P. Immunological demonstration of tau protein in neurofibrillary tangles of Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 177–185. [Google Scholar] [CrossRef]
- Caillet-Boudin, M.L.; Buee, L.; Sergeant, N.; Lefebvre, B. Regulation of human MAPT gene expression. Mol. Neurodegener. 2015, 10, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassimeris, L.; Spittle, C. Regulation of microtubule-associated proteins. Int. Rev. Cytol. 2001, 210, 163–226. [Google Scholar] [PubMed]
- Arendt, T.; Stieler, J.T.; Holzer, M. Tau and tauopathies. Brain Res. Bull. 2016, 126, 238–292. [Google Scholar] [CrossRef] [PubMed]
- Pooler, A.M.; Phillips, E.C.; Lau, D.H.; Noble, W.; Hanger, D.P. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013, 14, 389–394. [Google Scholar] [CrossRef]
- Soitropoulos, I.; Galas, M.C.; Silva, J.M.; Skoulakis, E.; Wegmann, S.; Maina, M.B.; Blum, D.; Sayas, C.L.; Mandelkow, E.M.; Mandelkow, E.; et al. Atypical, non-standard functions of the microtubule associated Tau protein. Acta Neuropathol. Commun. 2017, 5, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sergeant, N.; Bretterville, A.; Hamdane, M.; Caillet-Boudin, M.L.; Grognet, P.; Bombois, S.; Blum, D.; Delacourte, A.; Pasquier, F.; Vanmechelen, E.; et al. Biochemistry of Tau in Alzheimer’s disease and related neurological disorders. Expert. Rev. Proteom. 2008, 5, 207–224. [Google Scholar] [CrossRef]
- Schubert, M.; Brazil, D.P.; Burks, D.J.; Kushner, J.A.; Ye, J.; Flint, C.L.; Farhang-Fallah, J.; Dikkers, P.; Warot, X.M.; Rio, C.; et al. Insulin receptor substrate-2 deficiency impairs brain growth and promotes tau phosphorylation. J. Neurosci. 2003, 23, 7084–7092. [Google Scholar] [CrossRef] [Green Version]
- Rad, S.K.; Arya, A.; Karimian, H.; Madhavan, P.; Rizwan, F.; Koshy, S.; Prabhu, G. Mechanism involved in insulin resistance via accumulation of β-amyloid and neurofibrillary tangles: Link between type 2 diabetes and Alzheimer’s disease. Drug Des. Dev. Ther. 2018, 12, 3999–4021. [Google Scholar]
- Avila, J.; Leon-Espinosa, G.; Garcia, E.; Garcia-Escudero, V.; Hernandez, F.; Defelipe, J. Tau phosphorylation by GSK3 in different conditions. Int. J. Alzheimer’s Dis. 2012, 2012, 578373. [Google Scholar] [CrossRef] [Green Version]
- Doble, B.W.; Woodgett, J.R. GSK-3: Tricks of the trade for a multi-tasking kinase. J. Cell. Sci. 2003, 116, 1175–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuzawa, S.A.; Vocadlo, D.J. O-GlcNAc and neurodegeneration: Biochemical mechanisms and potential roles in Alzheimer’s disease and beyond. Chem. Soc. Rev. 2014, 43, 6839–6858. [Google Scholar] [CrossRef] [Green Version]
- De la Monte, S. Insulin resistance and Alzheimer’s disease. BMB Rep. 2009, 42, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, E.; Leboucher, A.; Caron, E.; Ahmed, T.; Tailleux, A.; Dumont, J.; Issad, T.; Gerhardt, E.; Pagesy, P.; Vilano, M.; et al. Tau deletion promotes brain insulin resistance. J. Exp. Med. 2017, 214, 2257–2269. [Google Scholar] [CrossRef]
- Rodriguez-Rodriguez, A.; Sandebring-Matton, A.; Merino-Serrais, P.; Parrado-Fernandez, C.; Rabano, A.; Winblad, B.; Ávila, J.; Ferrer, I.; Cedazo-Minguez, A. Tau hyperphosphorylation induces oligomeric insulin accumulation and insulin resistance in neurons. Brain 2017, 140, 3269–3285. [Google Scholar] [CrossRef] [Green Version]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Lei, P.; Ayton, S.; Finkelstein, D.I.; Spoerri, L.; Ciccotosto, G.D.; Wright, D.K.; Wong, B.X.W.; Adlard, P.A.; Cherny, R.A.; Lam, L.Q.; et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat. Med. 2012, 18, 291–295. [Google Scholar] [CrossRef]
- Pandini, G.; Pace, V.; Copani, A.; Squatrito, S.; Milardi, D.; Vigneri, R. Insulin has multiple antiamyloidogenic effects on human neuronal cells. Endocrinology 2013, 154, 375–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.Q.; Lacor, P.N.; Chen, H.; Lambert, M.P.; Quon, M.J.; Krafft, G.A.; Klein, W.L. Insulin receptor dysfunction impairs cellular clearance of neurotic oligomeric Aβ. J. Biol. Chem. 2009, 284, 18742–18753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef] [PubMed]
- De la Monte, S. Contributions of brain insulin resistance and deficiency in amyloid-related neurodegeneration in Alzheimer’s disease. Drugs 2012, 72, 49–66. [Google Scholar] [CrossRef]
- Hoe, H.S.; Lee, H.K.; Pak, D.T. The upside of APP at synapses. CNS Neurosci. Ther. 2012, 18, 47–56. [Google Scholar] [CrossRef]
- Berlanga-Acosta, J.; Guillén-Nieto, G.; Rodriguez-Rodriguez, N.; Bringas-Vegá, M.L.; Garcia-del-Barcó-Herrera, D.; Berlanga-Saez, J.O.; Garcia-Ojàlvo, A.; Valdés-Sosa, M.J.; Valdés-Sosa, P.A. Insulin resistance at the crossroad of Alzheimer disease pathology: A review. Front. Endocrinol. (Lausanne) 2020, 11, 560375. [Google Scholar] [CrossRef]
- Klyubin, I.; Cullen, W.K.; Hu, N.W.; Rowan, M.J. Alzheimer’s disease Aβ assemblies mediating rapid disruption of synaptic plasticity and memory. Mol. Brain. 2012, 5, 25. [Google Scholar] [CrossRef] [Green Version]
- Rajendran, L.; Paolicelli, R.C. Microglia-mediated synapse loss in Alzheimer’s disease. J. Neurosci. 2018, 38, 2911–2919. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhou, B.; Deng, B.; Zhang, F.; Wu, J.; Wang, Y.; Le, Y.; Zhai, Q. Amyloid-ε reduces hepatic insulin resistance in vivo via JAK2. Diabetes 2013, 62, 1159–1166. [Google Scholar] [CrossRef] [Green Version]
- Solke, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Forny-Germano, L.; Lyra e Silva, N.M.; Batista, A.F.; Brito-Moreira, J.; Gralle, M.; Boehnke, S.E.; Coe, B.C.; Lablans, A.; Marques, S.A.; Martinez, A.M.; et al. Alzheimer’s-like pathology induced by amyloid-ε oligomers in nonhuman primates. J. Neurosci. 2014, 34, 13629–13643. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.R.; Lyra, E.; Silvo, N.M.; Figueiredo, C.P.; Frozza, R.L.; Ledo, J.H.; Beckman, D.; Katashima, C.K.; Razoli, D.; Carvalho, B.M.; et al. Alzheimer-associated Aε oligomers impact the central nervous system to induce peripheral metabolic deregulation. EMBO Mol. Med. 2015, 7, 190–210. [Google Scholar] [CrossRef]
- Morales-Corraliza, J.; Wong, H.; Mozzella, M.J.; Che, S.; Lee, S.H.; Pethova, E.; Wagner, J.D.; Hemby, S.E.; Ginsberg, S.D.; Mathews, P.M. Brain-wide insulin resistance, Tau phosphorylation changes, and hippocampal neprilysin and amyloid-β alterations in a monkey model of type 1 diabetes. J. Neurosci. 2016, 36, 4248–4258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakabayashi, T.; Yamaguchi, K.; Matsui, K.; Sano, T.; Kubota, T.; Hashimoto, T.; Mano, A.; Yamada, K.; Matsuo, Y.; Kubota, N.; et al. Differential effects of diet- and genetically-induced brain insulin resistance on amyloid pathology in a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 15. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, L.; Netzer, W.J.; Greengard, P.; Hu, H. Does insulin dysfunction play a role in Alzheimer’s disease? Trends. Pharmacol. Sci. 2002, 23, 288–293. [Google Scholar] [CrossRef]
- Xie, L.; Helmerhorst, E.; Taddei, K.; Plewright, B.; van Bronswijk, W.; Martins, R. Alzheimer’s beta-amyloid peptides compete for insulin binding to the insulin receptor. J. Neurosci. 2002, 22, RC221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dore, S.; Kar, S.; Quirion, R. Insulin-like growth factor 1 protects and rescues hippocampal neurons against beta-amyloid- and human amylin-induced toxicity. Proc. Natl. Acad. Sci. USA 1997, 94, 4772–4777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phiel, C.J.; Wilson, C.A.; Lee, V.M.; Klein, P.S. GSK-3α regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 2003, 423, 435–439. [Google Scholar] [CrossRef]
- Gupta, S.; Nair, A.; Jhawat, V.; Mustaq, N.; Sharma, A.; Dhanawat, M.; Khan, S.A. Unwinding complexities of diabetic Alzheimer by potent novel molecules. Am. J. Alzheimer’s Dis. Other Dement. 2020, 35, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Swardfager, W.; Lanctot, K.; Rothenburg, L.; Wong, A.; Cappell, J.; Herrmann, N. A meta-analysis of cytokines in Alzheimer’s disease. Biol. Psychiatry 2010, 68, 930–941. [Google Scholar] [CrossRef]
- De la Monte, S.M. Insulin resistance and neurodegeneration: Progress towards the development of new therapeutics for Alzheimer’s disease. Drugs 2017, 77, 47–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dandrea, M.R.; Reiser, P.A.; Gumula, N.A.; Hertzog, B.M.; Andrade-Gordon, P. Application of triple immunochemistry to characterize amyloid plaque-associated inflammation in brains with Alzheimer’s disease. Biotech. Histochem. 2001, 76, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, M.G.; Scali, C.; Prosperi, C.; Bellucci, A.; Vannucchi, M.G.; Rosi, S.; Pepeu, G.; Casamenti, F. Beta-amyloid-induced inflammation and cholinergic hypofunction in the rat brain in vivo: Involvement of the p38MAPK pathway. Neurobiol. Dis. 2002, 11, 257–274. [Google Scholar] [CrossRef] [Green Version]
- Agostino, P.; Cunha, R.A.; Oliveira, C. Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer’s disease. Curr. Pharm. Des. 2010, 16, 2766–2778. [Google Scholar] [CrossRef]
- Femuninella, G.D.; Livingston, N.R.; Raza, S.; van der Doef, T.; Frangou, E.; Love, S.; Busza, G.; Calsolaro, V.; Carver, S.; Holmes, C.; et al. Does insulin resistance influence neurodegeneration in non-diabetic Alzheimer’s subjects? Alzheimer’s Res. Ther. 2021, 13, 47. [Google Scholar] [CrossRef]
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced glycation end-products: A review. Diabetologia 2001, 44, 129–146. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, S.; Maeda, S.; Matsui, T.; Ueda, S.; Fukami, K.; Okuda, S. Role of advanced glycation end products (AGEs) and oxidative stress on vascular complications in diabetes. Biochim. Biophys. Acta 2012, 1820, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Ramsamy, R.; Vannucci, S.J.; Yan, S.S.; Herold, K.; Yan, S.F.; Schmidt, A.M. Advanced glycation end products and RAGE: A common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology 2005, 15, 16R–28R. [Google Scholar] [CrossRef] [PubMed]
- Verdile, G.; Keane, K.N.; Cruzat, V.F.; Medic, S.; Sabale, M.; Rowles, J.; Wijesekara, N.; Martins, R.N.; Fraser, P.E.; Newsholme, P. Inflammation and oxidative stress: The molecular connectivity between insulin resistance, obesity, and Alzheimer’s disease. Mediat. Inflam. 2015, 2015, 105828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced glycation and products: Sparking the development of diabetic vascular injury. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Liu, N.; Wang, C.; Qin, B.; Zhou, Y.; Xiao, M.; Chang, L.; Yan, L.-J.; Zhao, B. Role of RAGE in Alzheimer’s disease. Cell. Mol. Neurobiol. 2016, 36, 483–495. [Google Scholar] [CrossRef]
- Deane, R.; Yan, S.D.; Submamaryan, K.R.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Matrone, C.; Djelloul, M.; Tagliatela, G.; Perrone, L. Inflammatory risk factors and pathologies promoting Alzheimer’s disease progression: Is RAGE the key? Histol. Histopathol. 2015, 30, 125–139. [Google Scholar]
- Hartlage-Rübsamen, M.; Zeitschel, U.; Apelt, J.; Gärtner, U.; Franke, H.; Stahl, T.; Günther, A.; Schliebs, R.; Penkowa, M.; Bigl, V.; et al. Astrocytic expression of the Alzheimer’s disease β-secretase (BACE1) is stimulus-dependent. Glia 2003, 41, 169–179. [Google Scholar] [CrossRef]
- Giri, S.; Selvaraj, S.; Miller, C.A.; Hofman, F.; Yan, S.D.; Stern, D.; Zlokovic, B.V.; Kalra, V.K. On beta-amyloid-induced migration of monocytes across normal and AD endothelium. Am. J. Physiol. Cell. Physiol. 2002, 283, C895–C904. [Google Scholar] [CrossRef] [Green Version]
- De Felice, F.G.; Lourenco, M.V.; Ferreira, S.T. How does brain insulin resistance develop in Alzheimer’s disease? Alzheimer’s Dement. 2014, 10, S26–S32. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.-L.; Du, Y.-F.; Du, H.; Shao, P. Insulin resistance in Alzheimer’s disease (AD) mouse intestinal macrophages is mediated by activation of JNK. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 1787–1794. [Google Scholar]
- Robbinson, J.; Busquets, O.; Tong, M.; de la Monte, S. Dysregulation of insulin-linked metabolic pathways in Alzheimer’s disease: C0-factor role of apolipoprotein E ε4. J. Alzheimer’s Dis. Rep. 2020, 4, 479–493. [Google Scholar] [CrossRef] [PubMed]
- De la Monte, S.M.; Tong, M. Brain metabolic dysfunction at the core of Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 548–559. [Google Scholar] [CrossRef] [Green Version]
- De la Monte, S.M.; Tong, M.; Bowling, N.; Moskal, P. si-RNA inhibition of brain insulin or insulin-like growth factor receptors causes developmental cerebellar abnormalities: Relevance to fetal alcohol spectrum disorder. Mol. Brain. 2011, 4, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, E.; di Domenico, F.; Manusco, C.; Butterfield, D.A. The Janus face of the heme oxygen/biliverdin reductase system in Alzheimer disease: It’s time for reconciliation. Neurobiol. Dis. 2014, 62, 144–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunomura, A.; Moreira, P.I.; Castellani, R.J.; Lee, H.G.; Zhu, X.; Smith, M.A.; Perry, G. Oxidative damage to RNA in aging and neurodegenerative disorders. Neurotox. Res. 2012, 22, 231–248. [Google Scholar] [CrossRef]
- Bonda, D.J.; Wang, X.; Lee, H.G.; Smith, M.A.; Perry, G.; Zhu, X. Neuronal failure in Alzheimer’s disease: A view through the oxidative stress looking-glass. Neurosci. Bull. 2014, 30, 243–252. [Google Scholar] [CrossRef] [PubMed]
- De la Monte, S.M.; Luong, T.; Neely, T.R.; Robinson, D.; Wands, J.R. Mitochondrial DNA damage as a mechanism of cell loss in Alzheimer’s disease. Lab. Investig. 2000, 80, 1323–1335. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Ma, C.; Un, H.; Wang, H.; Peng, W.; Zhou, Z.; Wang, H.; Pi, C.; Shi, Y.; He, X. Metabolism: A novel shared link between diabetes mellitus and Alzheimer’s disease. J. Diabetes Res. 2020, 2020, 4981814. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhong, C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Prog. Neurobiol. 2013, 108, 21–43. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.A.; di Domenico, F.; Barone, E. Elevated risk of type 2 diabetes for development of Alzheimer disease: A key role for oxidative stress in brain. Biochem. Biophys. Acta 2014, 1842, 1693–1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Binton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.H.; Tripathi, R.; Troung, Q.; Tirumala, K.; Reddy, T.P.; Anekonda, V.; Shirendeb, U.P.; Calkins, M.J.; Reddy, A.P.; Mao, P.; et al. Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer’s disease: Implications to mitochondria-targeted antioxidant therapeutics. Biochim. Biophys. Acta Mol. Bas. Dis. 2012, 1822, 639–649. [Google Scholar] [CrossRef] [Green Version]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; et al. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—Is this type 3 diabetes? Alzheimer’s Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef] [Green Version]
- Ghasemi, R.; Dargahi, L.; Haeri, A.; Moosavi, M.; Mohamed, Z.; Ahmadiani, A. Brain insulin dysregulation: Implication for neurological and neuropsychiatric disorders. Mol. Neurobiol. 2013, 47, 1045–1065. [Google Scholar] [CrossRef]
- Schrijvers, E.M.; Witteman, J.C.; Sijbrands, E.J.; Hofman, A.; Koudstaal, P.J.; Breteler, M.M.B. Insulin metabolism and the risk of Alzheimer disease: The Rotterdam Study. Neurology 2010, 75, 1982–1986. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Wang, J.; Li, Y. Insulin resistance and cognitive dysfunction. Clin. Chim. Acta 2015, 444, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Grillo, C.A.; Piroli, G.G.; Lawrence, R.C.; Wrighten, S.A.; Green, A.J.; Wilson, S.P.; Sakai, R.R.; Kelly, S.L.; Wilson, M.A.; Mott, D.D.; et al. Hippocampal insulin resistance impairs spatial learning and synaptic plasticity. Diabetes 2015, 64, 3927–3936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willmann, C.; Brockmann, K.; Wagner, R.; Kullmann, S.; Preissl, H.; Schnauder, G.; Maetzler, W.; Gasser, T.; Berg, D.; Eschweiler, G.W.; et al. Insulin sensitivity predicts cognitive decline in individuals with prediabetes. BMJ Open. Diabetes Res. Care 2020, 8, e001741. [Google Scholar] [CrossRef]
- Talbot, K.; Wang, H.-Y.; Kazi, H.; Han, E.Y.; Bakshi, K.R.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [Green Version]
- Rebelos, E.; Bucci, M.; Karjalainen, T.; Oikonen, V.; Bertoldo, A.; Hannukainen, J.C.; Hirvonen, R.; Heinonen, J.; Parkkola, R.; Laakso, M.; et al. Insulin resistance is associated with enhanced brain glucose uptake during euglycemic hyperinsulinemia: Large-scale PET cohort. Diabetes Care 2021, 44, 788–794. [Google Scholar] [CrossRef]
- Hoscheidt, S.M.; Kellawan, J.M.; Berman, S.E.; River-Rivera, L.A.; Krause, R.A.; Oh, J.M.; Beeri, M.S.; Rowley, H.A.; Wieben, O.; Carlsson, C.M.; et al. Insulin resistance is associated with lower arterial blood flow and reduced cortical perfusion in cognitively asymptomatic middle-aged adults. J. Cereb. Blood. Flow. Metabol. 2017, 37, 2249–2261. [Google Scholar] [CrossRef] [Green Version]
- Willette, A.A.; Bendlin, B.B.; Starks, E.J.; Birdsill, A.C.; Johnson, S.C.; Christian, B.T.; Okonkwo, O.C.; La Rue, A.; Hermann, B.P.; Koscik, R.L.; et al. Association of insulin resistance with cerebral glucose uptake in late middle-aged adults at risk for Alzheimer disease. JAMA Neurol. 2015, 72, 1013–1020. [Google Scholar] [CrossRef]
- Hohman, T.J.; Beason-Held, L.L.; Lamar, M.; Resnick, S.M. Subjective cognitive complaints and longitudinal changes in memory and brain function. Neuropsychology 2011, 25, 125–130. [Google Scholar] [CrossRef] [Green Version]
- Kellar, D.; Craft, S. Brain insulin resistance in Alzheimer’s disease and related disorders: Mechanisms and therapeutic approaches. Lancet Neurol. 2020, 19, 758–766. [Google Scholar] [CrossRef]
- Claxton, A.; Baker, L.D.; Hanson, A.; Trittschuh, E.H.; Cholerton, B.; Morgan, A.; Callaghan, M.; Arbuckle, M.; Behl, C.; Craft, S. Long acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s disease dementia. J. Alzheimer’s Dis. 2015, 45, 1269–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Wilkinson, C.W.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Breitner, J.C.S.; DeGroodt, W.; et al. Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology 2008, 70, 440–448. [Google Scholar] [CrossRef]
- Craft, S.; Claxton, A.; Baker, L.D.; Hanson, A.J.; Cholerton, B.; Trittschuh, E.H.; Dahl, D.; Caulder, C.; Neth, B.; Montine, T.J.; et al. Effects of regular and long-acting insulin on cognition and Alzheimer’s diseases biomarkers: A pilot clinical trials. J. Alzheimer’s Dis. 2017, 57, 1325–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R.; et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: A pilot clinical trial. Arch. Neurol. 2012, 69, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Reger, M.A.; Watson, G.S.; Frey, W.H.; Baker, L.D.; Cholerton, B.; Keeling, M.L.; Belongia, D.A.; Fishel, M.A.; Plymate, S.R.; Schellenberg, G.D.; et al. Effects of intranasal insulin on cognition in memory-impaired older adults: Modulation by APOE genotype. Neurobiol. Aging 2006, 27, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Janelidze, S.; Zetterberg, H.; Mattson, N.; Palmqvist, S.; Vanderstichele, H.; Lindberg, O.; Westen, D.; Stomrud, E.; Minthon, L.; Blennow, K.; et al. CSF Aβ42/Aβ40 and Aβ42/Aβ38 ratios: Better diagnostic markers of Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2016, 3, 154–165. [Google Scholar] [CrossRef] [Green Version]
- Holscher, C. Brain insulin resistance: Role in neurodegenerative disease and potential for targeting. Exp. Opin. Investig. Drugs 2020, 29, 333–348. [Google Scholar] [CrossRef]
- Fereira, L.S.S.; Fernandes, C.S.; Vieira, M.N.N.; de Felice, F.G. Insulin resistance in Alzheimer’s disease. Front. Neurosci. 2018, 12, 830. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Hanyu, H.; Hirao, K.; Kanetaka, H.; Sakurai, H.; Iwamoto, T. Efficacy of PPAR gamma agonist pioglitazone in mild Alzheimer disease. Neurobiol. Aging 2011, 32, 1626–1633. [Google Scholar] [CrossRef]
- Watson, G.; Cholerton, B.A.; Reger, M.A.; Baker, L.D.; Plymate, S.R.; Asthana, S.; Fishel, M.A.; Kulstad, J.J.; Green, P.S.; Cook, D.G.; et al. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: A preliminary study. Am. J. Geriatr. Psychiatry 2005, 13, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Risner, M.E.; Saunders, A.M.; Altman, J.F.; Ormandy, G.C.; Craft, S.; Foley, I.M.; Zvartau-Hind, M.E.; Hosford, D.A.; Roses, A.D. Rosiglitazone in Alzheimer’s Disease Study Group Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharm. J. 2006, 6, 246–254. [Google Scholar]
- Harrington, C.; Sawchak, S.; Chiang, C.; Davies, J.; Donovan, C.; Saunders, A.M.; Irizarry, M.; Jeter, B.; Zvartau-Hind, M.; van Dyck, C.H.; et al. Rosiglitazone does not improve cognition or global function when used as adjunctive therapy to AChE inhibitors in mild-to-moderate Alzheimer’s disease: Two phase 3 studies. Curr. Alzheimer’s Res. 2011, 8, 592–606. [Google Scholar] [CrossRef] [PubMed]
- Holscher, C. Incretin analogues that have been developed to treat type 2 diabetes hold promise as a novel treatment strategy for Alzheimer’s disease. Recent Pat. CNS Drug Discov. 2010, 5, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Gejl, M.; Gjedde, A.; Egefjord, L.; Moller, A.; Hansen, S.B.; Vang, K.; Rodell, A.; Brændgaard, H.; Gottrup, H.; Schacht, A.; et al. In Alzheimer’s disease, 6-month treatment with GLP-1 analog prevents decline of brain glucose metabolism: Randomized placebo-controlled, double-blind clinical trials. Front. Aging Neurosci. 2016, 8, 108. [Google Scholar] [CrossRef] [PubMed]
- Kornelius, E.; Lin, C.L.; Chang, H.H.; Li, H.H.; Huang, W.N.; Yang, Y.S.; Lu, Y.-L.; Peng, C.-H.; Huang, C.-N. DPP-4 inhibitor linagliptin attenuates Aβ-induced cytotoxicity through activation of AMPK in neuronal cells. CNS Neurosci. Ther. 2015, 21, 549–557. [Google Scholar] [CrossRef] [Green Version]
- Infeld, P.; Bodmer, M.; Jick, S.S.; Meier, C.R. Metformin, other antidiabetic drugs, and risk of Alzheimer’s disease: A population-based case-control study. J. Am. Geriatr. Soc. 2012, 60, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Mueller, J.L.; Vitori, A.C.; Misaghi, S.; Fedorova, A.; Deshayes, K.; Lee, W.P.; Hoffman, H.M.; Dixit, V.M. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J. Cell. Biol. 2009, 187, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohyagi, Y.; Takei, S. Insulin signaling as a therapeutic target in Alzheimer’s disease: Efficacy of apomorphine. Neurol. Clin. Neurosci. 2020, 8, 146–154. [Google Scholar] [CrossRef]
- Wang, P.; Su, C.; Feng, H.; Chen, X.; Dong, Y.; Rao, Y.; Ren, Y.; Yang, J.; Shi, J.; Tian, J.; et al. Curcumin regulates insulin pathways and glucose metabolism in the brain of APPswe/PSIdE9 mice. Int. J. Immunopathol. Pharmacol. 2017, 30, 25–43. [Google Scholar] [CrossRef] [Green Version]
- Craft, S.; Asthana, S.; Newcomer, J.W.; Wilkinson, C.W.; Matos, I.T.; Baker, L.D.; Cherrier, M.; Lofgreen, C.; Latendress, S.; Petrova, A.; et al. Enhancement of memory in Alzheimer disease with insulin and somatostatin, but not glucose. Arch. Gen. Psychiatry 1999, 56, 1135–1140. [Google Scholar] [CrossRef] [Green Version]
- Bayer-Carter, J.L.; Green, P.S.; Montine, T.J.; VanFossen, B.; Baker, L.D.; Watson, G.S.; Bonner, L.M.; Callaghan, M.; Leverenz, J.B.; Walter, B.K.; et al. Diet intervention and cerebrospinal fluid biomarkers in amnestic mild cognitive impairment. Arch. Neurol. 2011, 68, 743–752. [Google Scholar] [CrossRef] [Green Version]
- Jicha, G.A.; Markesbery, W.R. Omega-3 fatty acids: Potential role in the management of early Alzheimer’s disease. Clin. Interv. Aging 2010, 5, 45–61. [Google Scholar] [CrossRef] [Green Version]
- Wurtman, R.J.; Cansev, M.; Ulus, I.H. Synapse formation is enhanced by oral administration of uridine and DHA, the circulating precursors of brain phosphatides. J. Nutr. Health. Aging 2009, 13, 189–197. [Google Scholar] [CrossRef]
- Nash, D.T.; Fillit, H. Cardiovascular disease risk factors and cognitive impairment. Am. J. Cardiol. 2006, 97, 1262–1265. [Google Scholar] [CrossRef]
- Stephen, R.; Hongisto, K.; Solomon, A.; Lönnroos, E. Physical activity and Alzheimer’s disease: A systematic review. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 733–739. [Google Scholar] [CrossRef] [Green Version]
- Ruegsegger, G.N.; Vanderboom, P.M.; Dasari, S.; Klaus, K.A.; Kabiraj, P.; McCarthy, C.B.; Lucchinetti, C.F.; Nair, K.S. Exercise and metformin counteract altered mitochondrial function in the insulin-resistant brain. JCI Insight 2019, 4, e130681. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.H.; Koo, J.H.; Kang, E.B. Neuroprotective effect of treadmill exercise against blunted brain insulin signaling, NADPH oxidase, and tau hyperphosphorylation in rats fed a high-fat diet. Brain Res. Bull. 2018, 142, 374–386. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sędzikowska, A.; Szablewski, L. Insulin and Insulin Resistance in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 9987. https://doi.org/10.3390/ijms22189987
Sędzikowska A, Szablewski L. Insulin and Insulin Resistance in Alzheimer’s Disease. International Journal of Molecular Sciences. 2021; 22(18):9987. https://doi.org/10.3390/ijms22189987
Chicago/Turabian StyleSędzikowska, Aleksandra, and Leszek Szablewski. 2021. "Insulin and Insulin Resistance in Alzheimer’s Disease" International Journal of Molecular Sciences 22, no. 18: 9987. https://doi.org/10.3390/ijms22189987
APA StyleSędzikowska, A., & Szablewski, L. (2021). Insulin and Insulin Resistance in Alzheimer’s Disease. International Journal of Molecular Sciences, 22(18), 9987. https://doi.org/10.3390/ijms22189987